


















































































































































































































































































































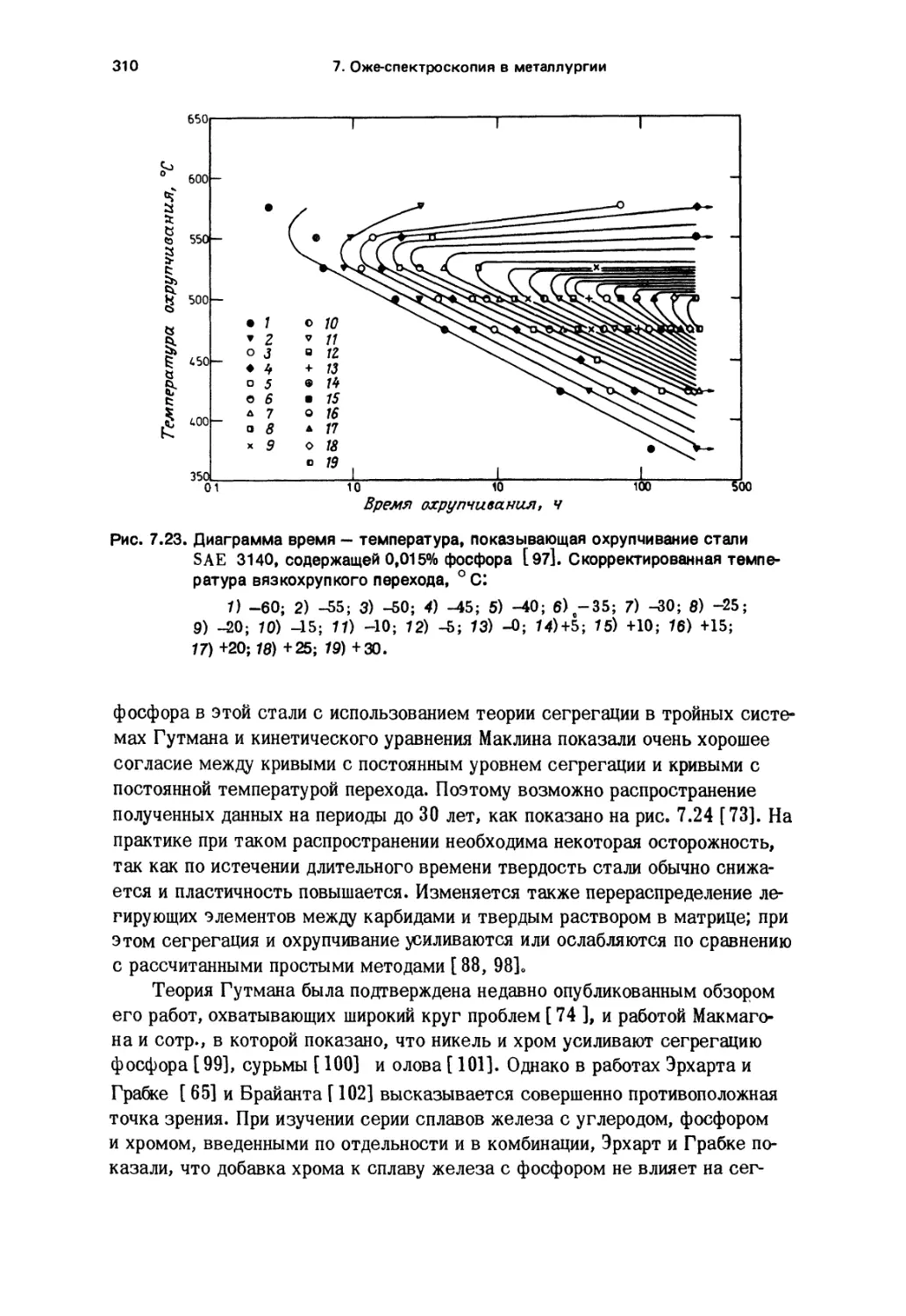

































































































































































































































































































Tags: распространение световых лучей отражение преломление поглощение излучение физика химия спектроскопия
Year: 1987




Text
Анализ поверхности
методами оже-
и рентгеновской
фотоэлектронной
спектроскопии
Д. Бриггс и М.П. Сих возле современного оже/ЭСХА-спектрометра.
Анализ поверхности
методами оже-
и рентгеновской
фотоэлектронной
спектроскопииПод редакцией
Д. Бриггса и М. П. СихаПеревод с английского
под редакциейд-ра физ.-мат. наук В.И. Раховского
и д-ра хим. наук И. С. РезаМосква «Мир» 1987
PRACTICAL SURFACE ANALYSIS
BY AUGER AND X-RAY
PHOTOELECTRON SPECTROSCOPY
Edited byD. BRIGGSICIPLC, Petrochemicals and Plastics Division,Wilton. Middlesbrough, Cleveland, UK
andMP.SEACHDivision of Materials Applications,National Physical Laboratory,Teddington, Middlesex, UK
John Wiley & SonsChichester • New York • Brisbane • Toronto • Singapore 1983ББК 22.344
A64
УДК 535.33Переводчики: A.M.Гофман, О.В.Раховская, канд. физ.-мат. наук В.М.Шустря ков,С.Е.Егоров, канд. техн. наук А.3.Пименова, канд. физ.-мат. наук А.Н. ШибановСих М.П., Бриггс Д., Ривьер Дж.К., Хофман С., Олсон P.P., Палмберг П., Хов-
ланд С.Т., Бреди Т.Е., Барр Т.Л., Макинтайр Н.С., Энтони М.Т., Свифт П., Шат-
луорс Д., Шервуд П.М.А., Вагнер К.Д.Анализ поверхности методами оже- и рентгеновской фотоэлектронной спек-
А64 троскопии: Пер. с англ./Под ред. Д.Бриггса, М.П.Сиха. — М.: Мир, 1987. - 600 с., илКнига известных специалистов из Великобритании, США, ФРГ и Канады пред¬
ставляет собой детальное и конкретное руководства по применению оже-спектро-
скопии и рентгеновской фотоэлектронной спектроскопии для анализа поверхности.
Она является первой учебно-справочной монографией, в которой обобщены резуль¬
таты огромного числа публикаций по этим вопросам. В первых пяти главах изложе¬
ны необходимые научные основы методов, в гл. 6—10 рассмотрены их основные
применения в важнейших областях современной техники.Для физиков и химиков, занимающихся исследованиями поверхности, инжене¬
ров, связанных с технологией работ, аспирантов и студентов.Д 1704050000.-170 66 _87 ч1 ББК 22.344
041 (01)-87Редакция литературы по физике и астрономииQ 1983 John Wiley & Sons Ltd.All rights reservedAuthorised translation from the English langua
edition published by John Wiley & Sons Ltd.© перевод на русский язык, "Мир", 1987
ПРЕДИСЛОВИЕ
РЕДАКТОРОВ ПЕРЕВОДАВопросам анализа поверхност и и приповерхностных слоев, изучению ка¬
чественного и количественного состава и микроморфологии поверхностей
оаздела фаз, которые стали предельно актуальными с совершенствованием
современной технологии в основных областях промышленности, на протяже¬
нии последних 20 лет посвящено огромное количество специализированных
статей и обзоров. По данным, представленным в предлагаемой вниманию чи¬
тателей коллективной монографии под редакцией Д. Бриггса и М. Сиха, в
1982 г. в мире насчитывалось уже более 1000 действующих аналитических и
'ЗСХА-спектрометров, использовавшихся в 30 индустриально развитых стра¬
нах для решения весьма широкого комплекса задач. Они включают как про¬
стейшие задачи - установление локального качественного и количественно¬
го элементного состава поверхности с переходом к изучению распределе¬
ния в глубину (в слоях толщиной до 10 нм), так и все более усложняющиеся -
изучение характеристик химической связи, определение точных параметров
мест вхождения изучаемых атомов или ионов в кристаллическую структуру
и получение данных по однородности и состоянию адсорбатов - монослоев и
островковых.Результаты, получаемые методами оже- и ЭСХА-сиектрометрии, в на¬
стоящее время необходимы для контроля производства в таких решающих от¬
раслях промышленности, как, например, электронная и электротехническая,
приборостроение, прецизионная металлургия, производство композитных ма¬
териалов - при анализе тонких пленок, распределения легирующих примесей,
однородности адгезии, при выявлении природы центров коррозии и т. п. Дан¬
ные, получаемые с помощью рассматриваемых в монографии методов, приоб¬
ретают все большее значение и в ряде других областей науки и техники, обес¬
печивая пространственное разрешение до 1 нм при чувствительности 1 • 10~6
и предоставляя количественную информацию как об элементном составе по
6Предисловие редакторов переводаповерхности и глубине, так и по значительному числу химических характерис¬
тик.Роль и значение рассматриваемых методов заведомо будут возрастать
в связи с ожидаемым в 1990-х годах достижением предельной плотности ин¬
теграции СБИС и микропроцессоров, допускаемой в господствующей сейчас
планарной технологии. Дальнейшее повышение плотности интеграции и быст¬
родействия возможно в первую очередь за счет создания и совершенствова¬
ния трехмерной вертикальной технологии, включая разработку и реализацию
устройств молекулярной электроники. Во всех этих случаях определяющим
явится точное знание требуемых особенностей локальной структуры в про¬
цессе создания сверхбыстродействующих сверхплотноупакованных супермик¬
ромодулей грядущей нанометровой интегральной микрооптоэлектроники, обес¬
печиваемой, в частности, методами, рассматриваемыми в данной книге.К сожалению, несмотря на высокую информативность и неуклонно расши¬
ряющееся применение этих методов, в литературе до сего времени не имелось
монографии учебно-справочного характера, рассчитанной на инженера-физика
или химика, не получившего специальной подготовки. Сказанное сделало соз¬
дание подобного руководства необходимым, а его появление - весьма свое¬
временным.Редакторы коллективной монографии и авторы большинства статей пред¬
лагаемой советскому читателю учебно-справочной коллективной монографии,
д-р Д. Бриггс из исследовательского центра фирмы "Импириэл кемикл инда-
стриз" и д-р. М. Сих из Национальной физической лаборатории в Теддингто-
не, хорошо известны широкому кругу специалистов как высокоавторитетные
ученые, давно и успешно работающие в данной области. Им вполне удалось
создать руководство, содержащее совокупность методических и справочных
данных, использование которых обеспечивает возможность результативной
самостоятельной работы.В первых пяти главах излагаются необходимые методические основы,
гл. 6 - 10 посвящены практическому использованию разработанных методов,
а в приложениях содержатся необходимые для работы дополнительные дан¬
ные, в том числе протабулированы все известные данные по энергиям линий
оже-спектров и аналитической чувствительности ЭСХА для отдельных эле¬
ментов и соединений. По полноте информации соответствующие таблицы, кото¬
рые содержат данные, опубликованные вплоть до 1982 г., уникальны.Книга будет полезна физикам и химикам, занимающимся исследования¬
ми поверхности с применением ЭОС и ЭСХА, как практическое руководство
по применению этих методов.
Предисловие редакторов перевода7Перевод выполнили: А.М. Гофман (предисловие, гл. 1, 4, 5), 0„Во Рахов-ская (гл. 2), канд.физ.-мат. наук В.М. Шустриков (гл. 3, 8, приложения), С.Е. Егоров(гл0 6, 9), кащи техн. наук А.З. Пименова (гл. 7), канд. физ.-мат. наукА.Н. Шибанов (гл. 10)-В Л. Раховский
ИХ. РезЛитература1. Немошкаленко В,В., Алешин В,Г, Электронная спектроскопия кристаллов.
2-е изд. - Киев: Наукова Думка, 1983,2 о Нефедов В М., Черепин В*Т, Физические методы исследования поверхно¬
сти твердых тел. - М.: Наука, 1983.30 Нефедов ВРентгеноэлектронная спектроскопия химических соединений
Справочник. - М„: Химия, 1984.4. Ченг Р., Фургпак Т» Гигантское комбинационное рассеяние. Пер. с англ, -
М.: Мир, 1984о5. Поверхностные явления на границах конденсированных фаз/ Под ред.Х.В. Хоконова, - Нальчик: изд-во Кабардино-Балкарского гос0 универси¬
тета, 1983об о Физика межфазных явлений/ Под редэ Х,В. Хоконова» — Нальчик: изд-во
Кабардино-Балкарского гос. университета, 1984.7. Ecole d’hiver sur PESCA Les Arcs. Coptes rendus, Suppl. Revue " Le vide,
les couches minces", N 215 (1983).8. Springer Tracts in Modern Physics, Vol. 104 (ed. I. Pockrand), Surface Enhanc¬
ed Raman Vibrationalstudies of Solid/Gas Interfaces, Springer-Verlagy Berlin,
New York, 1984.9. Springer Series in Chemical Physics, Vol. 35 (eds. R. Vanselow, R. How),
Chemistry and Physics of Surfaces, Springer-Verlag., Berlin, New York, 1984.10. Advances in Surface Treatments.Technology, Applications, Effects (ed.A. Niku-Lari), Pergamon Press, Oxford, 1984.11. Surface research. Proceeding of l-8th Seminar of Surface Physics, Wroclaw,
1976 - 1984.12. Thompson М., Bake* М., Christie A,, Tyson J. Auger Electron Spectroscopy,
Wiley, New York, 1985.l3o Springer Series in Surface Sciences. Vol. 3. Dynamical Phenomena at Sur¬
faces, Interfaces and Superlattices (eds. F. Nizzoli, K. Rieder, R. Willis),
Springer-Verlag, Berlin, New York, 1985.
8Предисловие редакторов перевода14. Ion Implantation Science and Technology (ed. J. W. Ziegler), Academic
Press, New York, 1984.15. Кулешов В.Ф; Кухаренко Ю.А., Запорожченко В.И. и др. Спектроскопия
и дифракция электронов при исследовании поверхностей твердых тел. -
М.: Наука, 1985.
ПРЕДИСЛОВИЕВ 1960-х годах развитие электронной спектроскопии вызвало бурный
рост разнообразных методов анализа поверхности. Вначале появилась элект¬
ронная оже-спектроскопия (ЭОС), вскоре затем - рентгеновская фотоэлект¬
ронная спектроскопия (РФЭС). В настоящее время эти два взаимодополняю¬
щих метода сохраняют свою роль при анализе поверхности. Существует свы¬
ше тысячи установок, работающих с использованием этих методов. Посколь¬
ку в основе обоих методов лежит анализатор энергии электронов, ЭОС и
РФЭС часто используют совместно; при этом в современных приборах чет¬
вертого поколения не наблюдается ухудшения параметров. ЭОС в виде оже-
микроскопии высокого разрешения и сканирующей оже-спектроскопии позво¬
ляет получить данные о распределении примесей по поверхности, дополняю¬
щие результаты исследований на сканирующем электронном микроскопе. При¬
менение дополнительного источника ионов для удаления поверхностных слоев
распылением позволяет применять ЭОС и РФЭС для послойного анализа.В последние годы эти методы быстро совершенствовались, о чем свиде¬
тельствует появление большого количества публикаций, рост групп пользова¬
телей в Европе, США и Японии, систематическое повышение точности прибо¬
ров и улучшение методов количественной оценки и т. д. В настоящее время
ЭОС и РФЭС широко используются во многих отраслях промышленности; бо¬
лее того, в ряде случаев их развитие было связано с ростом этих новых от¬
раслей (например, ЭОС и микроэлектронная промышленность). Появилось боль¬
шое количество центров, заключающих контракты на проведение исследований.
С каждым годом все большее число людей имеет дело с этими методами в
процессе решения проблем, связанных с составом поверхностных или гранич¬
ных областей, причем многим приходится применять эти методы на практике,
не имея соответствующей теоретической подготовки. Хотя опубликовано мно¬
жество работ, в которых приводится обзор различных аспектов ЭОС и РФЭС,
10Предисловиедо сих пор не существовало монографий, в которых описаны последние дости¬
жения в области практического применения этих методов, ориентированных
специально на новичков в прикладном анализе поверхности.Цель данной книги заключается в том, чтобы исправить это упущение и
представить все основные положения и таблицы справочных данных. В крат¬
ком введении описаны история развития ЭОС и РФЭС и их перспективное мес¬
то среди методов анализа поверхности в целом. Физико-химические основы
методов ЭОС и РФЭС описаны в главах, посвященных оборудованию, интерпре¬
тации спектров, послойному анализу и количественной оценке. Остальные
главы призваны осветить основные области применения методов с точки зре¬
ния особенностей ЭОС и РФЭС и связанных с ними достижений. Этими облас¬
тями являются электроника, металлургия, катализ, полимерная технология
и наука о коррозии. В книге постоянно подчеркивается внутренняя связь меж¬
ду ЭОС и РФЭС как методами электронной спектроскопии. В приложениях об¬
суждаются практические вопросы, имеющие важнейшее значение для повсед¬
невной работы - калибровка приборов, получение значений энергии связи при
РФЭС и обработка данных РФЭС (в особенности связанных с анализом слож¬
ных спектров). Кроме того, приведены подробные таблицы распределения пи¬
ков ЭОС и РФЭС, коэффициенты чувствительности для РФЭС и данные о со¬
отношении энергии связи и оже-параметра для элементов и соединений.Уилтон Д. БриггсТеддингтон М.П. СихФевраль 1983
Глэвэ. 1
ПЕРСПЕКТИВЫ АНАЛИЗА ПОВЕРХНОСТИ
И ГРАНИЦ РАЗДЕЛАМ.П. Сих’, Д. Бриггс21.1. ВведениеАнализ поверхности и наука о поверхности постоянно развиваются. Сво¬
им современным состоянием они обязаны большому числу исследователей,
каждый из которых внес в их развитие свой посильный вклад. Это развитие
происходило неравномерно по всему фронту: в одних областях прогресс был
весьма значительным, в других отсутствовал вовсе. Этот прогресс был обус¬
ловлен как промышленными прикладными исследованиями, так и чисто научными ис¬
следованиями ученых, удовлетворяющих собственное любопытство. Эти побуждающие
мотивы обусловили появление множества методик анализа поверхности (каждая из ко¬
торых имеет свое официальное название) и большого количества исследований и ре¬
зультатов. Не все из этих методик и работ могут быть использованы исследовате¬
лями для решения различных проблем прикладной науки, связанных с изуче¬
нием поверхности. За последние годы наиболее широкое применение нашли
электронная оже-спектроскопия (ЭОС) и рентгеновская фотоэлектронная спект¬
роскопия (РФЭС). В соответствии с этим в данной главе приведена хроноло¬
гия развития указанных методов среди современных методов спектроскопи¬
ческого исследования поверхности.Мы уже применили термин "анализ поверхности", не определив его пред¬
варительно. В простейшем случае его можно характеризовать как определе¬
ние элементного состава поверхностного слоя атомов твердого тела. После
проведения такого анализа немедленно встает вопрос о состоянии химиче¬
ских связей, расположении атомов в кристаллической решетке, однородности
поверхности и состоянии адсорбатов. Для одних исследователей наука о по¬
верхности связана с получением полных характеристик чистых металличе¬
ских поверхностей с малыми кристаллографическими индексами в вакууме,
для других к этому может добавиться рассмотрение некоторого количестваМ.Р» Seah, Division of Materials Applications, National Physical Laboratory, Ted-
dington, Middlesex, UK.2)D, Briggs, ICI PLC, Petrochemicals and Plastics Division, Wilton, Middlesbrough,
Cleveland, UK.
121. Перспективы анализа поверхности и границ разделаадсорбированных молекул моноокиси углерода, оказавшихся на поверхности.
В прикладных же исследованиях состояние поверхности является гораздо бо¬
лее сложным и в общем случае не может быть охарактеризовано полностью,
его характеристика просто будет адекватна поставленной задаче. При прове¬
дении прикладных исследований необходимо получить все перечисленные вы¬
ше данные, однако, кроме того, потребуются аналогичные данные для слоев
атомов, прилегающих к поверхности приблизительно до глубины 1 мкм, в виде
функции глубины, как это показано на рис. 1.1. Каждый из множества мето¬
дов поверхностного анализа позволяет решить одну или несколько из этих
проблем лучше, чем остальные, так что в принципе каждый метод имеет свои
преимущества. Однако ценность какого-то данного метода для исследователя
зависит не только от его теоретических преимуществ, но также и от накоплен¬
ного опыта по использованию этого метода, существующих справочных дан¬
ных и примеров проведения аналогичных исследований в прошлом. Эта книга
призвана предоставить такие сведения для двух основных методов анализа
поверхности - ЭОС и РФЭС.Рис. 1.1. Характерные области анализа поверхности, анализа тонких пленок и объем¬
ного анализа.В заключительных главах приведены примеры использования ЭОС и РФЭС
в основных областях микроэлектроники, металлургии, катализа, технологии
полимеоов и коррозии. На рис. 1.2 изображены э'Т’и и другие области, в разви¬
тии котооых значительную роль сыграл анализ поверхности, начиная от иссле¬
дования износа режущих кромок в производстве листового металла и разра¬
ботки смазывающих присадок до оптоэлектроники и разработки интегральных
схем. Многообразию сфер применения анализа поверхности соответствует
М.П. Сих, Д. Бриггс13РиСс 1о2. Области применения анализа поверхности на примере промышленности Вели¬
кобритании (по данным Сиха [1]). Секторная диаграмма показывает пропор¬
ции производящих отраслей Великобритании, за исключением нефтяной; штри»
ховка характеризует частоту использования анализа поверхности в мировой
практике: I — частое использование; и — использование время от време¬
ни; ш — редкое использование.1 - окисление жаропрочных сплавов, усталость материалов, адгезия;2 - отделка, доводка, соединение, сварка, пайка, адгезия; 3 — анодирова¬
ние, адгезия, лазеры и оптоэлектроника, научные приборы, зубопротезные
материалы; 4 - волокна, пластмассы, печать на пластмассах; 5 - флота¬
ция, износ буров; 6 - разрушения стали: от отпускной хрупкости, охруп¬
чивания, коррозии, при сварке; плазма токамак/стенка, солнечные бата¬
реи, топливные элементы; 7 - коррозия, специальное стекло; в - консер¬
вирование, коррозия; 9 ^ катализ, фармацевтика, пластмассы, пигменты,
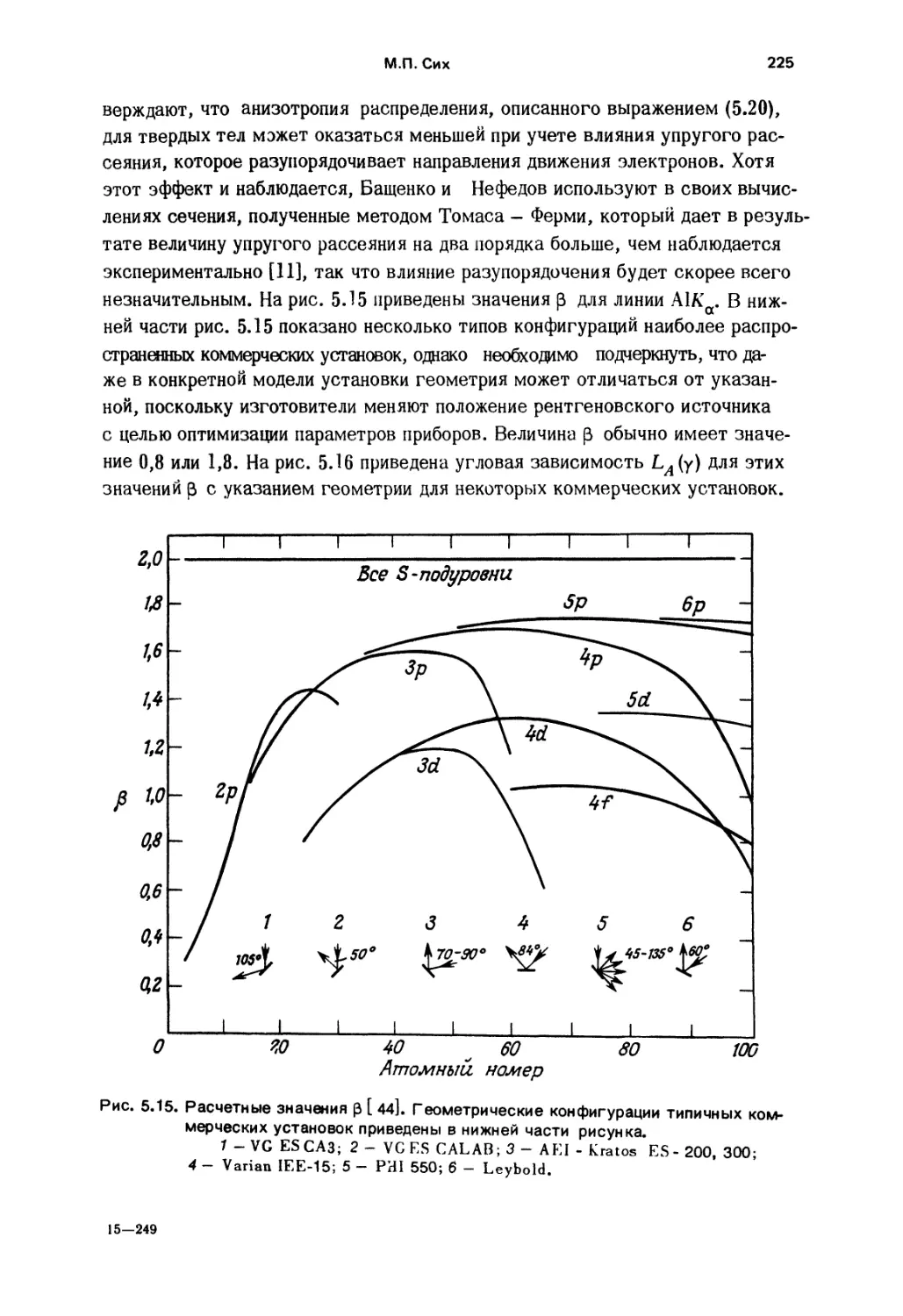
моющие средства, удобрения; 10 - нитрирование, прокатка, сварка, шари»
коподшипники, очистка, механическая обработка, электрометаллургия; 11 -
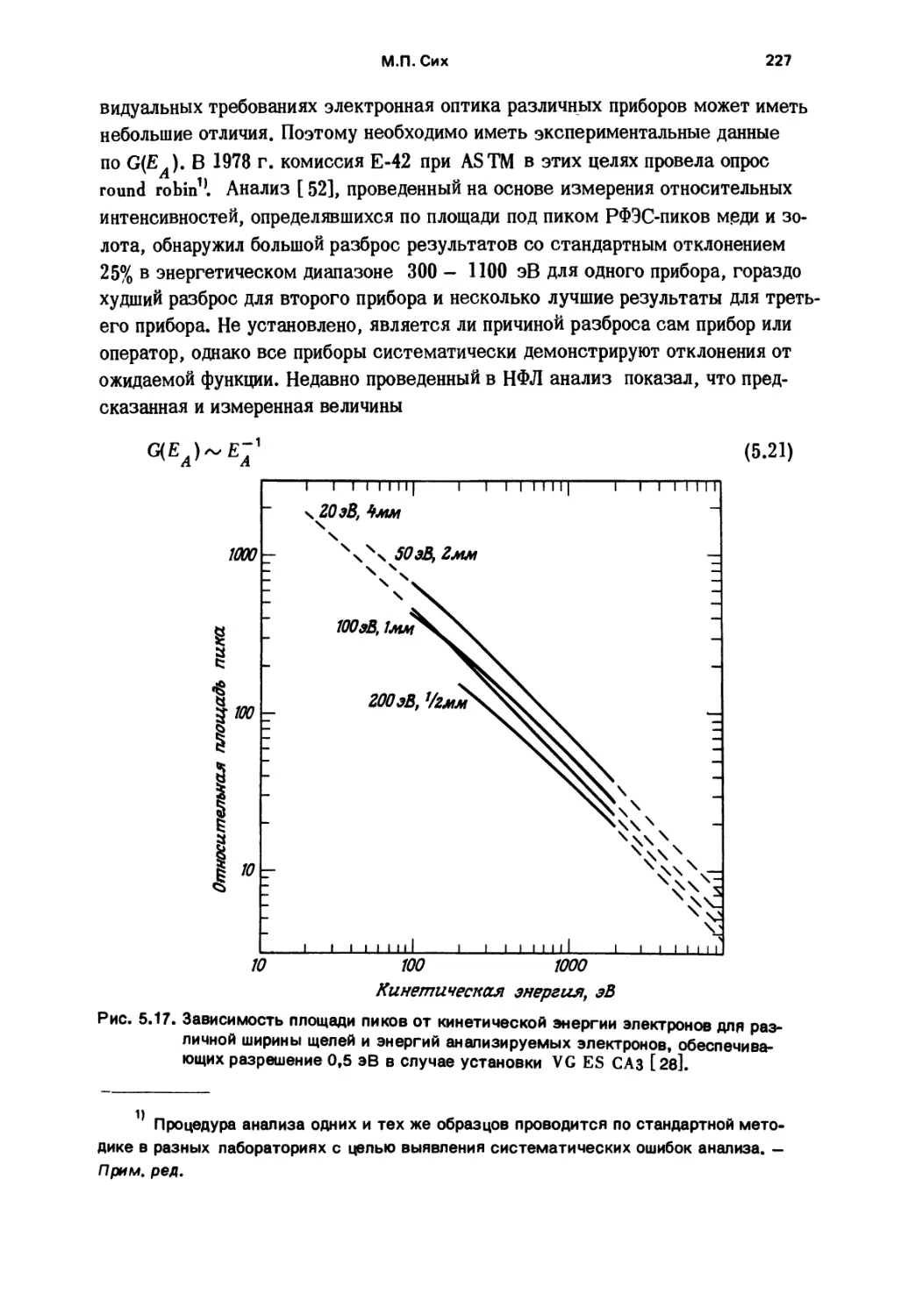
механическая обработка, твердые сплавы, коррозия, окисление» триболо¬
гия; 12 — катоды, контакты, лампы, люминофоры;73 - микросхемы: ста¬
бильность тонких пленок, смачиваемость, распределение примесей, запира¬
ющие слои, диагностика отказов; пайка и смачивание металлов, очистка;14 - смазка, износ, адгезия краски, аккумуляторы, коррозия.большое количество стран, в которых в настоящее время работают установки
для анализа поверхности (рис. 1.3). Этот существенный прогресс был достиг»
нут после 1970 г. Некоторые проблемы были решены ранее, однако наиболь¬
шие успехи достигнуты в течение нескольких последних лет.
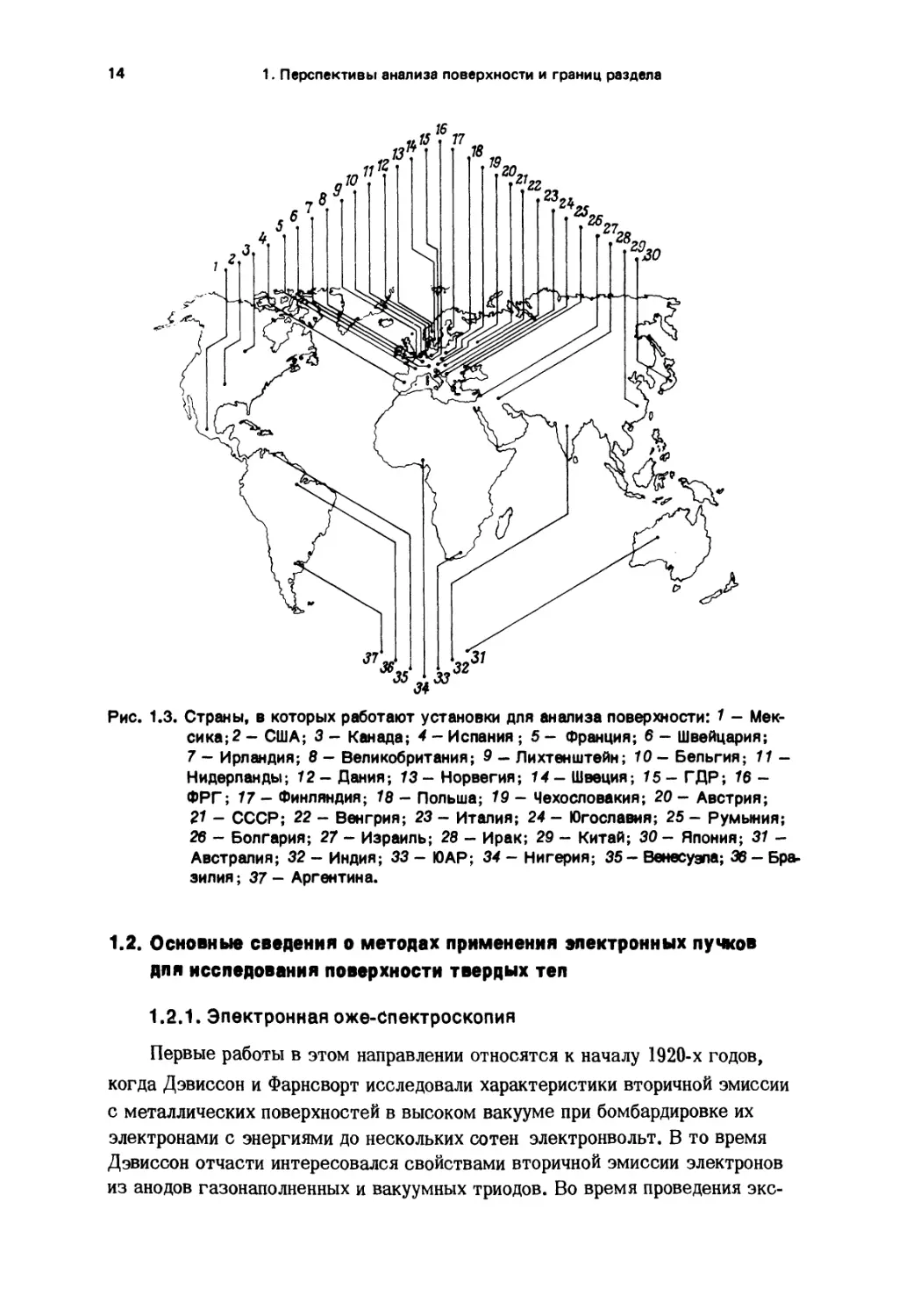
14 1. Перспективы анализа поверхности и границ разделаРис. 1.3. Страны, в которых работают установки для анализа поверхности: 7 - Мек¬
сика^ - США; 3 - Канада; 4 - Испания; 5 - Франция; 6 - Швейцария;7 - Ирландия; 8 - Великобритания; 9 - Лихтенштейн; 10- Бельгия; 11 -
Нидерланды; 72- Дания; 73- Норвегия; 14- Швеция; 75- ГДР; 16 -
ФРГ; 17 - Финляндия; 78 - Польша; 19 - Чехословакия; 20 - Австрия;21 - СССР; 22 - Венгрия; 23 - Италия; 24 - Югославия; 25 - Румыния;
26 - Болгария; 27 - Израиль; 28 - Ирак; 29 - Китай; 30- Япония; 37 -
Австралия; 32 - Индия; 33 - ЮАР; 34 - Нигерия; 35 - Венесуэла; 36 - Бра¬
зилия ; 37 - Аргентина.1.2. Основные сведения о методах применения электронных пучков
для исследования поверхности твердых тел1.2.1. Эпектронная оже-спектроскопияПервые работы в этом направлении относятся к началу 1920-х годов,
когда Дэвиссон и Фарнсворт исследовали характеристики вторичной эмиссии
с металлических поверхностей в высоком вакууме при бомбардировке их
электронами с энергиями до нескольких сотен электронвольт. В то время
Дэвиссон отчасти интересовался свойствами вторичной эмиссии электронов
из анодов газонаполненных и вакуумных триодов. Во время проведения экс-
М.П. Сих, Д. Бриггс15периментов (совместно с Джермером) произошла авария стеклянной вакуум¬
ной системы в процессе обезгаживания никелевой мишени при высокой тем¬
пературе. В результате произошло окисление мишени, которая была затем
подвергнута очистке путем продолжительного отжига в атмосфере водорода.
Это привело к тому, что первоначально мелкозернистая мишень, претерпев
перекристаллизацию, образовала несколько крупных кристаллов. В 1927 г.
Дэвиссон и Джермер [2] наблюдали дифракцию медленных электронов (ДМЭ)
на этих кристаллах, которую они правильно объяснили в свете публикаций
де Бройля по волновой механике. В 1930-х годах Дэвиссон, Джермер и Фарнс¬
ворт [3] продолжали исследования ДМЭ, используя стеклянные вакуумные ус¬
тановки и цилиндры Фарадея собственной конструкции для наблюдения за рас¬
пространением электронов малой энергии при дифракции. Эксперименты были
довольно сложны, однако этим ученым удалось исследовать большое количе¬
ство монокристаллических поверхностей с низкими кристаллографическими
индексами в условиях, очевидно соответствовавших сверхвысокому вакууму
(СВВ), в зависимости от температуры и степени покрытия исследуемой мише¬
ни газом. Эта работа не обратила на себя большого внимания, пока Джермер
[4] не возродил идею Эренберга [5] о получении изображений при ДМЭ, кото¬
рая была практически реализована фирмой "Вариан" в 1964 г. в металличе¬
ской СВВ-установке. Спустя несколько лет такие установки выпускались уже
и другими производителями. Было организовано большое количество лабора¬
торий для исследования монокристаллических металлических поверхностей
при помощи ДМЭ в связи с большим диапазоном прикладных проблем, поста¬
новка которых сегодня задним числом может показаться недостаточно обо¬
снованной. К сожалению, большое количество лабораторий при проведении
экспериментов не имело возможности удовлетворить требованиям, предъяв¬
ляемым теорией при реализации метода ДМЭ.В 1967 г. Харрис опубликовал два отчета фирмы "Дженерал электрик",
которые последовательно появились в периодической печати [6, 7 ] и которым
предшествовала идея Ландера [8] о том, что оже-электроны, испущенные твер¬
дым телом, могут быть использованы для анализа поверхности. Основная
идея метода заключается в облучении поверхности образца электронами с
энергией в диапазоне 1-10 кэВ, что приводит к эмиссии электронов с внут¬
ренних оболочек уровня Ех атомов образца приблизительно до глубины 1 мкм.
Затем вакансия в оболочке заполняется вследствие внутренних процессов,
протекающих в атоме, при которых электрон с уровня Е занимает эту вакан¬
сию, разность энергий поглощается третьим электроном, находящимся на уров¬
не Fz. Этот третий электрон, называемый оже-электроном в честь Пьера Оже,
который первым нзблюдал эти явления в камере Вильсона [9], затем эмити¬
руется за пределы атома с энергией Ел, которая может быть приближенно за-
161. Перспективы анализа поверхности и границ разделаписана какЕ = Е + Е -Е . (1.1)a z у х ' 'Таким образом, электроны в каждом атоме характеризуются определенным
значением энергии, и если измерения производятся в диапазоне от 0 до 2 кэВ,
то энергии пиков оже-электронов позволяют идентифицировать все присутст¬
вующие элементы, за исключением водорода и гелия. Причиной того, что ЭОС
является поверхностно-чувствительным методом, служит интенсивное неупру¬
гое рассеяние, имеющее место для электронов в этом диапазоне энергий, и,
таким образом, эмиссия за пределы твердого тела оказывается возможной
только для оже-электронов самых внешних слоев атомов1); эти электроны и
фиксируются при измерении спектра. Исследования оже-электронов произво¬
дились давно, однако не в этом диапазоне энергий и не в связи с анализом
поверхности [10]. Харрис убедился, что непосредственно полученный энерге¬
тический спектр, имеющий малые пики и большой уровень фона, значительно
лучше поддается анализу после дифференцирования. В соответствии с этим
он разработал метод модуляции потенциала анализатора, который и применя¬
ется с тех пор для получения дифференциальных спектров. Харрис продемон¬
стрировал также высокую чувствительность этого метода, исследовав поверх¬
ностные загрязнения при отношении сигнал/шум более 200.В 1967 г. Харрис обсудил свои исследования с Периа, который сразу по¬
нял, что стандартную исследовательскую установку для ДМЭ можно видоизме¬
нить путем добавления нескольких электронных блоков для работы по методу
ЭОС. Статья Вебера и Периа [11] была опубликована раньше статьи Харриса
в связи с проблемами, возникшими при рецензировании [12]. Первые восторги
по отношению к методу ДМЭ несколько улеглись, и стандартные установки,
работавшие с использованием этого метода, стали быстро преобразовывать¬
ся для работы по методу ЭОС. Таким образом, множество первых ЭОС-уста-
новок было идеально подготовлено для работы в условиях СВВ, однако в свя¬
зи с характерными особенностями ДМЭ-установок как электронных спектро¬
метров по сравнению со 127-градусным спектрометром-дефлектором Харриса
системы ДМЭ обычно работали с малым энергетическим разрешением. В ре¬
зультате малого разрешения многие исследователи считали, что пики оже-
электронов широки и содержат мало информации о химическом составе. Та¬
ким образом, ЭОС развивалась с самого начала как метод элементного ана¬
лиза, но успешно применялась только для хорошо подготовленных монокрис¬
таллических поверхностей в условиях СВВ. Это позволило Палмбергу и Роди¬
ну [13], нанося слои на поверхности кристаллов с малым кристаллографиче-1) Имеется в виду эмиссия оже-электронов с энергией, соответствующей е , т. е.
оже-электронов, которые не претерпевают неупругого рассеяния. — Прим. ред.
М.П. Сих, Д. Бриггс17ским индексом, показать, что ЭОС дает информацию лишь о поверхности об¬
разца до глубины 5-10 А. Следующим важным шагом явилось внедрение
Палмбергом, Бомом и Трейси [14] анализатора типа цилиндрическое зеркало,
что значительно повысило отношение сигнал/шум, допустимое при исследо¬
ваниях по методу ЭОС, и расширило сферу применения метода до современных
границ.1.2.2. Другие методыОдновременно с развитием этой области параллельно разрабатывались и
другие аналитические методы на основе использования электронного пучка,
которые, однако, не имели столь широкого применения, как ЭОС. Достаточно
широкое распространение получила спектроскопия потенциалов появления
(СПП) [15], при которой фиксировалось начало возбуждения основных уровней
при увеличении энергии пучка и которая была дешевле ЭОС, однако имела
малую чувствительность, и в настоящее время этот метод используется лишь
при исследованиях твердых тел. Спектроскопия потерь (СП) [16], при которой
измеряются потери электронов, связанные с началом возбуждения основных
уровней, также имеет малую чувствительность, но в настоящее время этот
метод возрождается как пригодный для локального химического анализа в
сканирующем просвечивающем электронном микроскопе (СЭМ) [17]. Существу¬
ют и другие варианты, однако лишь один из этих методов исследования элект¬
ронным пучком утвердил себя - это спектроскопия характеристических по¬
терь энергии электронов (СХПЭЭ) [18, 19]. При использовании этого метода
измеряются потери электронов в электронном пучке с высокой степенью мо¬
нохроматичности и энергетическим разрешением 5-20 МэВ. Эти потери связы¬
ваются с возбуждением колебаний атомов и молекул, расположенных в опре¬
деленных точках и определенным образом ориентированных на поверхности
металла. К настоящему времени накоплено значительное количество данных,
в связи с чем этот метод приобретает все более общее применение, однако
с его помощью нельзя исследовать полярные соединения. Это связано с тем,
что в суммарный спектр дает вклад большое количество спектров потерь в
связи с легкостью возбуждения оптических фононов. И наконец, метод, кото¬
рый позволяет анализировать структуру, а не химический состав, - это диф¬
ракция "быстрых” электронов (электронов больших энергий) (ДБЭ). Он нахо¬
дит в настоящее время широкое применение в связи с появлением большого
количества эпитаксиальных пленочных материалов. Как и ДМЭ, ДБЭ может
использоваться для исследования кристаллической структуры внешних слоев
твердого тела. Для достижения той же поверхностной чувствительности, что
и при ДМЭ с энергиями электронов 10-200 эВ, при ДБЭ пучок электронов с
энергией 10-30 кэВ направляется под острым углом к поверхности. Анализ2—249
181. Перспективы анализа поверхности и i раниц разделаамплитуд дифракционной картины в этом случае более прост, чем в случае
метода ДМЭ, что отчасти и объясняет популярность этого метода в настоящее
время.1.3. Основные сведения о метопах применения пучков фотоновдля исследования поверхности твердых тел1.3.1. Рентгеновская фотоэлектронная спектроскопия [20]История развития РФЭС весьма показательна, и эффект Оже сыграл в
ней свою роль. Эта длинная история тесно связана с развитием теории кор¬
пускулярно-волнового дуализма и с зарождением атомной физики. Более за¬
интересованные читатели, обладающие некоторым запасом времени, могут
ознакомиться с этой историей, превосходно и живо описанной в работах ис¬
следователей из Университета ла Троуб [20-23], где содержатся интерес¬
ные выдержки и исторические анекдоты.РФЭС зародилась при исследованиях фотоэлектрического эффекта (от¬
крытого Герцем в 1887 г.), в ходе которых рентгеновские лучи использова¬
лись в качестве источника возбуждения фотонов. Например, Иннес в 1907 г.
[24] описывал опыты, в которых использовалась рентгеновская трубка с пла¬
тиновым антикатодом и проводился анализ скоростей эмитированных элект¬
ронов по отклонению в магнитном поле, создававшемся при помощи двух ка¬
тушек Гельмгольца, с последующим фотодетектированием.В то время идеальными возможностями для дальнейших исследований
в этой области обладала лаборатория Резерфорда в Манчестере, которая за¬
нималась исследованием спектров (3 -лучей радиоактивных материалов мето¬
дом анализа в магнитном поле и находилась на переднем крае новой области
рентгеновской спектроскопии. Первые эксперименты были проведены Мозли,
Роулинсоном и Робинсоном до начала первой мировой войны. В 1914 г. Резер¬
форд [25] сделал первый шаг к выводу основного уравнения РФЭС, которое
впоследствии было приведено к видугде Ек - кинетическая энергия р -лучей (фотоэлектронов), /iv — энергия фо¬
тона, — энергия связи электрона. После войны Робинсон продолжил эту
работу, а Морис де Бройль начал исследования во Франции, причем оба ис¬
пользовали фотографическое детектирование. Были достигнуты значитель¬
ные успехи, и в начале 1920-х годов удалось получить фотоэлектронные спект¬
ры многих элементов, возбужденных рентгеновскими лучами высоких энер¬
гий. Было обнаружено, что аномальные "линии" (очевидно, не связанные с
основными уровнями) соответствуют флуоресценции исследуемых элементов
вследствие возбуждения рентгеновскими лучами. Подлинное понимание при-
М.П. Сих, Д. Бриггс19роды этих электронов пришло только после того, как Оже [9] продемонстри¬
ровал в камере Вильсона безызлучательный переход, названный его именем.В это время РФЭС рассматривалась как инструмент исследования структуры
атома за пределами, которых позволила достигнуть рентгеновская спектро¬
скопия (PC). Однако вскоре выяснилось, что ограничения, связанные с экс¬
периментальным применением метода РФЭС, с одной стороны, и новые дости¬
жения в области рентгеновской спектроскопии, с другой стороны, кардиналь¬
ным образом изменили ситуацию в пользу рентгеновской спектроскопии. Ис¬
следования продолжал лишь Робинсон, причем в течение 1930-х годов не было
достигнуто заметных успехов в повышении разрешающей способности или
чувствительности. С началом второй мировой войны эти исследования пре¬
рвались.После окончания войны Стейнхард и Серфас из Университета Лехай ре¬
шили возродить РФЭС в качестве аналитического метода, в частности для
исследования химических процессов на поверхности. Было разработано не¬
сколько установок, которые хотя и не обеспечивали улучшения характерис¬
тик, но уже использовали для регистрации метод Гейгера - Мюллера, а затем
и 127-градусный электростатический анализатор энергии электронов. Несмот¬
ря на применение рентгеновских лучей, обладавших очень высокой энергией,
влияние поверхностных эффектов на спектр оказалось вполне заметным [26].
Работа Стейнхарда на соискание степени доктора философии была озаглавле¬
на пророчески: ’’Рентгеновский фотоэлектронный спектрометр для химическо¬
го анализа’’.Тем временем решающие события происходили в Упсале, Швеция, Кай
Зигбан, развивая в течение 1940-х годов спектроскопию (5-излучения, достиг
очень высокой точности и сделал заключение о возможности реализации на
этой основе электронной спектроскопии с использованием рентгеновского воз¬
буждения, а не радиоактивных источников. Был специально разработан немаг¬
нитный спектрометр с двойной фокусировкой для точных магнитных измере¬
ний энергии с разрешающей способностью 10~5. В 1954 г. был получен первый
рентгеновский фотоэлектронный спектр очищенного хлорида натрия. До это¬
го все полученные спектры состояли из нескольких полос с более или менее
хорошо выраженным максимумом, за которым следовал спад. В упсальском
спектре впервые была получена полностью разрешенная линия со стороны
высоких энергий для каждого пика, которая соответствовала электронам, не
потерявшим энергии. Величина пика (Ек) позволила точно измерить энергию
связи электрона (Ев), и, таким образом, метод РФЭС внес свой вклад в изу¬
чение структуры атома. Впоследствии группа Зигбана наблюдала эффект хи¬
мического сдвига остовных уровней вследствие наличия химической связи и
в течение 1955 - 1970 гг. продолжала исследования в области электронной
201. Перспективы анализа поверхности и границ разделаспектроскопии. Зигбан ввел термин ЭСХА (электронная спектроскопия для
химического анализа), призванный подчеркнуть, что в РФЭС-спектре регист¬
рируются пики и фото- и оже-электроновЧ Под таким заглавием в 1967 г.
была выпущена полезная книга, касающаяся этих проблем и привлекшая все¬
общее внимание к всГзможностям РФЭС. В 1969 — 1970 гг. начали появляться
стандартные установки, хотя в этот период в результате работы Зигбана по
стеаратам [27] существовало мнение, что РФЭС характеризует лишь слои,
лежащие на глубине 100 А от поверхности. Только после публикации Брандла
и Робертса [28] об исследованиях в условиях сверхвысокого вакуума РФЭС
стала признанным методом анализа поверхности, а в 1972 г. Сих [29] показал,
что для РФЭС и ЭОС толщина исследуемого поверхностного слоя почти одина¬
кова.1.3.2. Другие методыВместо фотонов с энергиями порядка килоэлектронвольт для возбужде¬
ния электронов твердых тел могут быть использованы фотоны с гораздо бо¬
лее низкой энергией. В ультрафиолетовой фотоэлектронной спектроскопии
(УФЭС) источником фотонов является газонаполненная разрядная лампа на
инертном газе с дифференциальной накачкой. Она генерирует отдельные ре¬
зонансные линии низкой энергии (например, Не1 21,2 эВ, и Не11 40,8 эВ) с
шириной порядка нескольких миллиэлектронвольт. Поскольку такой энергии
хватает лишь на эмиссию электронов из валентной зоны, этот метод не явля¬
ется аналитическим (в смысле, который придается этому слову в данной книге). Тем
не менее УФЭС широко используется для проведения исследований электрон¬
ных зон в металлах, сплавах и полупроводниках, а также явлений адсорбции
[30]. Другим источником фотонов для фотоэлектронной спектроскопии явля¬
ется синхротронное излучение - излучение, испускаемое ускоряемыми элект¬
ронами. Электроны с энергией порядка 1 ГэВ, постоянно циркулирующие по
основному кольцу ускорителя, испускают непрерывный спектр фотонов с энер¬
гиями от нескольких единиц до нескольких тысяч электронвольт. Таким обра¬
зом, применяя монохроматор, можно получить источник фотонов переменной
энергии, что дает определенные преимущества для фотоэлектронной спектро¬
скопии [31, 32]. Синхротронное излучение является также идеальным для про¬
ведения исследований тонкой структуры края поглощения рентгеновских лучей
(EXAFS)2). В этом случае информацию о структуре и ближнем порядке полу¬
чают, анализируя характер колебаний на краю полосы рентгеновского погло¬
щения со стороны более высоких энергий. Этот метод пригоден для прозеде1) Siegbahn К, частное сообщение.От англ. Extended X-ray Absorption F in e-S true tare Spectroscopy.
М.П. Сих, Д. Бриггс21ния исследования массивных образцов и дополняет метод дифракции рентге¬
новских лучей (ДРЛ) в применении к аморфным материалам. Возможно, одна¬
ко, получение информации о поверхности образца при исследовании мелких
металлических кристаллитов, содержащихся в металлических катализаторах
'33]. Данные о собственно поверхностном слое могут быть получены множе¬
ством методов, например путем оценки выхода оже-электронов в одном и
том же диапазоне энергий фотонов в области края полосы поглощения. Такие
методы объединяются сокращением S-EXAFS [34].1.4. Основные сведения о методах применения ионных пучковдпя исследования поверхности твердых тепОдновременно с совершенствованием ЭОС шло развитие двух основных
методов, использующих ионные пучки: вторичной ионной масс-спектрометрии
(ВИМС) [35, 36] и спектроскопии рассеянных ионов (СИР) [37, 38]. В методе
ВИМС пучок отсортированных по массе ионов аргона, кислорода или цезия
направляется на поверхность образца и удаляет поверхностный слой атомов.
Некоторые из удаленных таким образом атомов положительно или отрицатель¬
но ионизуются, что может быть достаточно строго измерено при помощи масс-
спектрометра. Для анализа поверхности обычно используется в качестве пер¬
вичного пучок ионов аргона с плотностью порядка нескольких наноампер на
квадратный миллиметр и сечением порядка нескольких десятков квадратных
миллиметров. В таком режиме, называемом статическим, данный метод по¬
зволяет достичь очень высокой поверхностной чувствительности, особенно в
отношении электроположительных элементов, и в принципе дает возможность
получить подробную информацию о химическом состоянии поверхности, а це¬
ной некоторых усилий — и некоторые количественные результаты. Хотя метод
ВИМС существует в течение 12 лет, он не нашел еще столь широкого приме¬
нения, как ЭОС и РФЭС. Представляется, однако, весьма вероятным, что ин¬
терес к использованию статического метода ВИМС будет в ближайшем буду¬
щем расти при изучении образцов, уже исследованных с помощью ЭОС и РФЭС.
Последние методы позволяют достаточно быстро получить общую картину,
однако ВИМС иногда бывает незаменимым для получения детальной химиче¬
ской информации, обладает гораздо большей чувствительностью и, кроме то-
го, может быгь использован для выявления водорода. При использовании дру¬
гого метода, называемого динамическим, ионная оптика позволяет получать
изображения размером менее микрона для различных элементов. Высокая
плотность первичного ионного пучка, используемого при динамическом мето¬
де, вызывает эрозию поверхности в процессе измерения в отличие от стати¬
ческого метода, при котором верхний слой атомов удаляется лишь после мно-
гочасо^1 х измерений. При динамическом методе используются два принципи-
221. Перспективы анализа поверхности и границ разделаально различающихся источника ионов - кислородный и цезиевый, что позво¬
ляет повысить вылет соответственно положительных и отрицательных ионов
и улучшает условия детектирования. При использовании динамического мето¬
да ВИМС химическая информация теряется, установка при этом может рабо¬
тать в одном из двух режимов. В первом случае установка действует анало¬
гично электронно-зондовому рентгеновскому микроанализатору (ЭЗРМА), ис¬
пользуемому для анализа массивных образцов и получения карты распреде¬
ления элементов, а во втором - при сканировании первичным пучком площадки
размером приблизительно 50 х 50 мкм может быть произведен послойный ана¬
лиз содержания примесей в полупроводниковых материалах с высоким разре¬
шением по глубине.Второй метод, основанный на использовании ионных пучков (СИР), осно¬
ван на представлении, что столкновение ионов подчиняется тем же законам,
что и столкновение бильярдных шаров. Первичный ион подвергается рассея¬
нию и попадает в спектрометр с энергией, определяемой массой атома мише¬
ни, с которым произошло соударение. Этот метод не обладает широкой приме¬
нимостью, которая характерна для методов ЭОС, РФЭС и ВИМС, однако име¬
ет уникальное достоинство, заключающееся в том, что при таких исследова¬
ниях можно получить информацию исключительно о внешнем слое атомов, а
также, изменяя полярный и азимутальный углы падения ионного пучка в слу¬
чае монокристаллической мишени, о взаимном расположении атомов.Следует упомянуть еще два метода, использующие ионные пучки, - масс-
спектральный анализ рассеянных нейтральных частиц (МСАРН) [39] и спектро¬
скопию обратного резерфордовского рассеяния (СОРР) [40]. МСАРН использу¬
ет те же принципы, что и ВИМС, однако, поскольку в случае ВИМС большая
часть сигнала теряется за счет нейтрализации большого числа эмитируемых
с поверхности образца частиц, в МСАРН сделана попытка вновь ионизировать
их при помощи плазмы вблизи поверхности образца. Это должно значительно
повышать чувствительность по сравнению с ВИМС, но в связи со сложностью
контроля параметров плазмы и получения пространственного разрешения
этот метод еще не приобрел достаточной популярности. С другой стороны,
спектроскопия обратного резерфордовского рассеяния (СОРР), использую¬
щая концепцию соударения бильярдных шаров и энергетический анализ отра¬
женного пучка ионов, более близка к СИР. Путем использования ионов с энер¬
гией порядка нескольких мегаэлектронвольт может быть неразрушающим об¬
разом измерено содержание различных элементов по глубине образца до глу¬
бины 1 мкм с разрешением 20 нм. Тяжелые элементы с соседними атомными
номерами трудно различимы между собой, однако имеются сообщения о том,
что чувствительность метода составляет менее одного процента толщины мо¬
нослоя. Для реализации метода применяется простое и надежное оборудова-
М.П. Сих, Д. Бриггс23ние и могут быть использованы ускорители Ван-де-Граафа, удовлетворяющие
требованиям, предъявляемым к источникам питания, которые используются
при ядерных исследованиях на установках с более высокими энергиями. Ион-
но-нейтрализационная спектроскопия (ИНС) [41], при которой ионы низкой
энергии, нейтрализуемые у поверхности образца, генерируют электроны, ха¬
рактеризующие валентную зону, не описана в данной книге, поскольку, как и
У ФЭС, этот метод не применяется достаточно широко для анализа поверхности.1.5. ЗаключениеШирокий диапазон методов анализа поверхности1) проиллюстрирован в
табл. 1.1, где некоторые методы сгруппированы по видам возбуждающего и
регистрируемого излучений. Более детально эта таблица, а также описания
многих методов приведены в обзоре Кобурна и Кэя [42], в статьях в журнале
"Методы анализа поверхности" [43] и в обзоре Хонига [44]. Табл. 1.1 напоми¬
нает нам, что при каждом типе возбуждающего излучения в образце происхо¬
дит множество процессов. При этом к установкам ЭОС с высоким пространст¬
венным разрешением могут успешно добавляться детекторы рентгеновского
излучения, как это сделано в ЭЗРМА, однако при этом следует учитывать и де¬
сорбцию, обусловленную электронами (ЭД) [45]. Поскольку измерения по ме¬
тоду ЭОС требуют определенного количества электронов, а доля поверхност¬
ных атомов, удаленных за счет ЭД, зависит от количества электронов, падаю-
Таблица 1.1. Сводная таблица основных методов анализа поверхностиЭлект- Падающее излучение
роны Ионы Нейтраль- Рентге-ные частицы новские фотоныf Электроны ЭОС ИНС УФСДМЭ РФЭСсхпээРегистрируемые Ионы ЭД ВИМС МСБАчастицы СИРК СОРРНейтральные МСАРНчастицыРентгенов- (ЭЗРМА) (ПРИ) S-EXAFSские фото-
I ныПочти не упомянуты методы вибрационной спектроскопии, которые не могут
использоваться для анализа поверхности,, поскольку не позволяют выявлять элемент¬
ный состав. Однако при проведении исследований по адсорбции молекул или органи¬
ческих полимерных поверхностей эти методы играют важную роль. Некоторые из
них упомянуты в гл. 9.
241. Перспективы анализа поверхности и границ разделащих на единицу площади, ясно, что ЭД определяет предельную величину прост¬
ранственного разрешения для ЭОС [46]. В табл. 1.1 указано еще несколько ме¬
тодов, причем те из них, которые приведены в скобках, не относятся непосред¬
ственно к методам анализа поверхности, а представлены для полноты картины.В методе протонного возбуждения рентгеновского излучения (ПРИ) [47] для
облучения образца используются а -частицы, или протоны, с энергией поряд¬
ка мегаэлектронвольт; анализ испущенных рентгеновских лучей позволяет с
чувствительностью 10~6 определить элементы с Z > 12. Глубина анализа при
этом такая же, как при работе с электронно-зондовым рентгеновским микро¬
анализатором. В масс-спектрометрии с применением бомбардировки пучками
быстрых атомов (МСББА) [48] ионный пучок, используемый в ВИМС, заменен
пучком нейтральных высокоэнергетических атомов, что позволяет избежать
эффекта зарядки в случае использования диэлектрических образцов.Наиболее важные из этих методов несколько подробнее описаны в табл. 1.2,
где приведены и характеристики, хуже поддающиеся количественной оценке.
Стоимость не входит в эти характеристики, поскольку она часто определяется
не оптикой, а приспособлениями для крепления образца, средствами вычисли¬
тельной техники, незапланированными лабораторными расходами и платежа¬
ми в фонд социального страхования. Наиболее важными обычно являются про¬
стота подготовки образца и доступность процесса измерения. Простота приго¬
товления образца является главным ограничением в применении автоионного
микроскопа [49], хотя сомнительно, что другими методами можно будет до¬
стичь того же порядка пространственного разрешения при анализе граничных
слоев. Образцы должны представлять собой острия, изготовленные из иссле¬
дуемого материала; при этом количество исследуемых материалов постоянно
растет.В заключение этой главы следует отметить, что в области создания уста¬
новок для анализа поверхности наблюдается быстрый прогресс и, скорее всего,
данные по пространственному разрешению и чувствительности, приведенные в
табл. 1.2, устареют в течение ближайших нескольких лет. ЭОС и РФЭС наибо¬
лее распространены, поскольку это самостоятельные методы, доступные лю¬
бой лаборатории, причем имеется значительный опыт их применения и накопле¬
но большое количество данных. Применение обоих методов ограничено в том
смысле, что РФЭС обладает малым пространственным разрешением, а ЭОС
разрушает образцы, имеющие малую энергию связи* По мере совершенство¬
вания конструкции спектрометров и более широкого применения многоканаль¬
ных детекторов будет возрастать скорость регистрации. Это позволит увели¬
чить пространственное разрешение в случае РФЭС и уменьшить эффекты дест¬
рукции, присущие ЭОС. Для РФЭС с такими усовершенствованиями может
быть достигнута величина пространственного разрешения порядка 100 мкм,
Год нача¬
ла при¬
менения1968196819701967196719701968
19751969
1967Объем полу¬
ченной ин¬
формацииПростотапримене¬нияПодготовкаобразцов(П-простая)Популяр¬ностьНеопреде¬ляемыеэлементыКоличест¬
венный
анализ
(П-п ростой)Чувстви¬
тель¬
ность, %Глубинаанализа,монослойПространст¬
венное раз¬
решениеПолучаемаяинформация1’МетодЭОСАвтоэмис-
сионный
полевой
ионный
микроскоп
СХПЭЭ
СИР
СОРР
ВИМС
(стат.)
ВИМС (ди¬
нам )ВИМС (ди¬
нам., по¬
слойный
анализ)
УФС
РФЭСТаблица 1.2. Сводка наиболее популярных методов анализа поверхности и границ^ Е — информация по элементному составу, С — информация по химическому составу.Метод применим для получения данных о химическом составе, но с небольшим пространственным разрешением из-за ЭД [44]^ Данные по химическому составу могут быть получены4) Сообщалось о разрешающей способности 1 мкм на установке, использующей новые принципы с источником УФЭС на Не-лампе, разрешения менее 100 мкм в случае использова¬
ния РФЭС могут быть получены с помощью математической обработки экспериментальных данных [50].^ При сравнении со стандартными образцами
261. Перспективы анализа поверхности и границ разделахотя эта же величина может быть достигнута с использованием фотоэлект¬
ронного спектромикроскопа с аксиальным магнитным полем Бимсона, Порте¬
ра и Тернера [50]. В последнем приборе использованы принципиально новые
технические решения, в нем электроны движутся по орбитам, "вмороженным"
в поле сверхпроводящего магнита напряженностью 8 Тл.История ряда методов анализа поверхности измеряется десятилетиями,
однако из табл. 1.2 видно, что большинство наиболее эффективных современ¬
ных методов возникло в период с 1968 по 1970 г., когда они, как грибы, воз¬
никали во многих лабораториях. Это не случайное совпадение, а скорее след¬
ствие совершенствования техники СВВ и возросшей готовности к использова¬
нию сложных СВВ-систем в середине и конце 1960-х годов, что и послужило
стимулом к исследованию поверхностей. С тех пор до нашего времени уси¬
лиями исследователей техники СВВ разработаны системы с более высокой
надежностью, в которых образец может быть введен и удален при помощи
шлюзов в течение минут, а не дней, как это было раньше. Этими установками
могут управлять люди, не имеющие опыта работы с вакуумом, однако являю¬
щиеся специалистами в области анализа поверхностей.В последующих главах читатель найдет подробное описание установок, а
затем расшифровку спектров, полученных при изучении поверхностей твердых
тел методами РФЭС и ЭОС. Большая часть последних прикладных исследова¬
ний касается не просто внешних слоев, а сложной поверхности, состоящей из
чувствительных слоев или покрытий. Основные проблемы, связанные с полу¬
чением картины состава таких слоистых образцов по глубине, рассматрива¬
ются совместно с количественной интерпретацией спектров. Эта часть книги
вместе с таблицами данных и методов, приведенными в приложениях, обеспе¬
чивает читателя всем необходимым для максимального использования воз¬
можностей, представляемых различными методами. Вторая часть книги,
гл. 6-Ю, иллюстрирует возможности РФЭС и ЭОС в разрешении проблем,
возникающих в основных технологических областях, показанных на рис. 1.2.
В этих главах приведены примеры удачного использования характерных осо¬
бенностей описанных выше методов для решения различных задач.Литература1. Seah М.РSurf. Interface Anal., 2, 222 (1980).2. Davisson С., Germer L.H», Phys. Rev., 30, 705 (1927).3. Farnsworth Phys. Rev., 33, 1068 (1929).4. Scheibner Germer L.HHartman C.D., Rev. Sci. Ins., 31, 112 (1960).5. Ehrenberg WPhil. Mag., 18, 878 (1934).6. Harris L*A>, J. Appl. Phys., 39, 1419 (1968).7. Harris L.A., J. Appl. Phys., 39, 1428 (1968).
М.П. Сих, Д. Бриггс278. Lander J.J., Phys. Rev., 91# 1382 (1953).9. Auger P., J. Phys. Radium, 6, 205 (1925).10. Robinson H.R., Cassie A.M., Proc. Roy. Soc., A113, 282 (1926).11. Weber R.E., Peria W.T., J. Appl. Phys., 38, 4355 (1967).12. Harris L.A., J. Vac. Sci. Technol., 11, 23 (1974).13. PalmbergP.W., Rhodin T.N., J. Appl. Phys., 39, 2425 (1968).14. Palmberg P.W., Bohm G*K., Tracy J. СAppl. Phys. Lett., 16, 254 (1969).15. Webb С., Williams P.M., Surf. Sci., 53, 110 (1975).16. Gerlach R.L., J. Vac. Sci. Technol., 8, 599 (1971).17. Leapman R.D., Sanderson S.J., Whelan M.J., Metal Sci., 12, 215 (1978).18. Ibach H., Lehwald S., J. Vac. Sci. Technol., 15, 407 (1978).19. Ibach H., Lehwald S., J. Vac. Sci. Technol., 76, 1 (1978).20. Jenkin J.G., Leckey R.C.G., Liesgang J., J. Electron Speсtrosc., 12, 1(1977).21. Jenkin J.G., et al., J. Electron Spectrosc., 14, 477 (1978).22. Jenkin J.G., et al., J. Electron Spectrosc., 15, 307 (1979).23. Jenkin J.G., J. Electron Spectrosc., 23, 187 (1981).24. Innes J.G., Proc. Roy. Soc., A79, 442 (1907).25. Rutherford E., Phil. Mag., 28, 305 (1914).26. Steinhardt R.G%, Anal. Chem., 23, 1585 (1951).27. Siegbahn K., et al., ESC A: Atomic, Molecular and Solid State Structure
Studied by Means of Electron Spectroscopy, Almqvist and Wiksells, Uppsala,
1967.28. Brundle C.R., Roberts M.W., Proc. Roy. Soc., A331, 383 (1972).29. Seah M.P., Surf. Sci., 32, 703 (1972).30. Williams P.M., in: Handbook of X-ray and Ultraviolet Photoelectron
Spectroscopy (ed. D. Briggs), Heyden and Son, London, 1977, p. 313.31. Spicer W.E., in: Electron and Ion Spectroscopy of Solids, (eds. L. Fiermans,
J. Vennick, W. Dekeyser), Plenum, New York, 1978, p. 34.32. Carlson T.A., Surf. Interface Anal., 4, 125 (1982).33. KomingsbergerD.C., Prins R., Trend. Anal. Chem., 1, 16 (1981).34. Bianconi A., Appl. Surf. Sci., 6, 392 (1980).35. Benninghoven A., Z. Physik 230, 403 (1970).36. Werner H.W., Surf. Interface Anal., 2, 56 (1980).37. Baun W.L., Surf. Interface Anal., 2, 243 (1981).38. Haeussler E.N., Surf. Interface Anal., 2, 134 (1980).39. Oechsner H., Stumpe £., Appl. Phys., 14, 43 (1977).40. Mitchell L.V., Phys. Bull., 30, 23 (1970).41. Hagstrum H.D., in: Electron and Ion Spectroscopy of Solids (eds. L. Fier¬
mans, J. Vennick, W. Dekeyser), Plenum, New York, 1978, p. 273.42. Cobum J.W., Kay E., Solid State Science, 4, 561 (1974).
281. Перспективы анализа поверхности и границ раздела43. Methods of Surface Analysis (ed. A.W. Czandema), Elsevier, New York, 1975.
[Имеется перевод: Методы анализа поверхности/Под ред. А. Зандерны. -
М.: Мир, 1979.]44. Honig /£.£., Thin Solid Films, 31, 83 (1975).45. Pantano C.G., Madey 7\£., Appl. Surf. Sci., 7, 115 (1981).46. Seah M.P., in: Surface Analysis of High Temperature Materials — Chemistry
and Topography (ed. G. Kemeny), Applied Science (в печати).47. Campbell J»L•, et ah Anal, Chem., 47, 1542 (1975).48. Surman Van der Berg J.A., Vickerman J*C., Surf. Interface Anal., 4,
160 (1982).49. Miller M.K., Smith G.D.W., Metal Sci., 11, 249 (1977).50. Beamson G., Porter H*Q; Turner D.W., Nature, 290, 556 (1981).
Глава 2
ОБОРУДОВАНИЕДж. К. Ривьер12е1. ВведениеЭлектронная спектроскопия носила бы в значительной мере качествен¬
ный характер, если бы отсутствовали методы измерения и записи распреде¬
ления электронов по энергии. Для осуществления этих операций прежде всего
необходим спектрометр и потому этот прибор заслуживает более подробного
рассмотрения. В данном контексте понятием "спектрометр" охватываются
все виды приборов, которые имеют какое бы то ни было отношение к процес¬
су получения данных в электронной спектроскопии. Таким образом, следует
рассмотреть не только проектирование и изготовление самого анализатора
энергий электронов, но и конструкции источников электронов и рентгеновско¬
го излучения, свойства рабочей среды, используемой в спектрометре, и спо¬
собы ее получения, методы установки образца с помощью ввода перемещения
или манипулятора внутри спектрометра, способы подготовки и исследования
поверхности образца. Целью настоящей главы является описание современ¬
ного состояния практического применения спектрометров, используемых для
РФЭС и ЭОС, пояснение принципов действия и особенностей их применения, а
также получение некоторых практически полезных соотношений.2„2. Вакуумные условияСуществуют две причины, по которым электронные спектрометры, приме¬
няемые для анализа поверхности, должны работать в вакууме. Прежде всего
электроны, испущенные образцом, должны встретить на пути к анализатору
как можно меньше молекул газа, чтобы не претерпеть рассеяния и не быть утра¬
ченными для анализа. Другими словами, длина свободного пробега электро¬
нов должна быть намного больше размеров спектрометра. Это требование са¬
мо по себе не накладывает строгих ограничений на рабочий вакуум, так как
оно удовлетворяется уже при давлениях порядка 10~5 -10~6 мм рт. ст. Бо¬
лее жесткие требования к рабочему вакууму предъявляются по другой причи-Riviirei UK Atomic Energy Research Establishment, Harwell, Didcot,
Oxfordshire*
302. Оборудованиене. В гл. 5 будет показано [1] (см. изображенную на рис. 5.5 зависимость
длины свободного пробега относительно неупругих соударений от кинетиче¬
ской энергии электронов), что РФЭС и ЭОС являются методами, предназна¬
ченными специально для исследования поверхности, с глубиной зондирования
порядка нескольких атомных слоев. Большинство анализируемых электронов
образуется в одном или двух поверхностных атомных слоях. Такая возмож¬
ность получать информацию, относящуюся именно к поверхностным слоям об¬
разца, в сочетании с элементной чувствительностью порядка 0,5% атомного
слоя означает, что эти методы очень чувствительны к поверхностным загряз¬
нениям любого рода. Поскольку во многих экспериментах можно начинать ис¬
следования, лишь имея поверхность с хорошо известными свойствами, т. е.
чистую или находящуюся в каком-нибудь ином стабильном состоянии, и по¬
скольку очень малые количества загрязнений могут существенно воздейство¬
вать на ход эксперимента, очевидно, что необходимо работать в условиях,
когда скорость накопления загрязнений пренебрежимо мала по сравнению со
скоростью протекания процессов, обусловливающих изменение состояния по¬
верхности в ходе эксперимента. Основным источником загрязнений в вакуум¬
ной системе является остаточный газ. Как следует из любого учебника по мо¬
лекулярно-кинетической теории газов, с достаточной точностью можно счи¬
тать, что монослой газа образуется на поверхности за 1,5 с при давлении
10“6мм рт. ст. при комнатной температуре, если каждая молекула, сталки¬
вающаяся с поверхностью, остается на ней (т. е. если вероятность прилипания
равна единице). Если потребовать, как это делается обычно, чтобы за 30 мин
на поверхности накапливалось не более 0,05 моноатомного слоя примесей, то
для вероятности прилипания, равной единице, из кинетической теории следует,
что давление остаточного газа должно составлять 4 • Ю”11 мм рт. ст. На
самом деле вероятности прилипания обычно меньше единицы, и для подавляю¬
щего большинства экспериментов по изучению поверхности достаточно иметь
давление 10“1омм рт. ст. Такое давление соответствует условиям сверхвысо¬
кого вакуума (СВВ).2.2.1. Системы СВВ2.2.1.1. МатериалыУстановив, что для применения РФЭС и ЭОС необходим сверхвысокий вакуум,
следует кратко обсудить, какие требования при этом предъявляются к конст¬
рукционным материалам для спектрометров. Возвращаясь к кинетической те¬
ории газов, но не вдаваясь в детали, легко показать, что для имеющих реаль¬
ные размеры камер, вакуумных магистралей и насоса предельный достижи¬
мый вакуум всегда определяется скоростью, с которой адсорбированный газ
покидает внутренние поверхности при комнатной температуре. Эта так назы-
Дж. К. Ривьер31ваемая скорость газоотделения существенно меняется в зависимости от хи¬
мической природы поверхности и ее предварительной обработки или предысто¬
рии и природы адсорбированного газа. Однако даже в лучшем случае, т. е.
при минимальной скорости газоотделения, если температура внутренних по¬
верхностей не будет превосходить комнатную температуру, сверхвысокий
вакуум в спектрометре не будет получен или по крайней мере будет достиг¬
нут ненадолго. Чтобы получить сверхвысокий вакуум в течение разумного
времени, необходимо удалить адсорбированные газы со скоростью, сущест¬
венно превышающей скорость нормального газоотделения. Это достигается
путем повышения температуры спектрометра или отжига приблизительно при
200 °С в течение нескольких часов. При последующем охлаждении до комнат¬
ной температуры скорость газоотделения с внутренних поверхностей упадет
на несколько порядков, и тогда насосы смогут создавать и поддерживать
сверхвысокий вакуум.Необходимость отжига налагает ограничения на материалы, которые
могут быть использованы в спектрометре. В конструкции не должны приме¬
няться материалы, которые при нагреве разлагаются, дают сильное газо-
отделение или теряют прочность. Эти свойства материалов должны сохранять¬
ся в течение всей жизни спектрометра, так как отжиги регулярно повторяют¬
ся. Поэтому нельзя, например, использовать эластомеры, такие, как витон,
для уплотнений в сверхвысоковакуумных соединениях, так как они могут не
только окислиться и стать хрупкими, но при отжиге стать пористыми, что
способствует диффузии газов сквозь них. Таким образом, уплотнения, исполь¬
зуемые для соединения частей спектрометра, должны быть металлическими.
Температура отжига не позволяет использовать индий (температура плавле¬
ния 156°С), который мог бы служить полезным материалом для прокладок.
Практически наиболее часто для этой цели используются два материала: зо¬
лото и медь, причем последняя значительно чаще. Золото применяется в ви¬
де отожженной проволоки, зажимаемой между плоскими фланцами, однако та¬
кие прокладки, несмотря на полную надежность при использовании, обладают
следующими недостатками: трудно правильно установить проволоку на верти¬
кальном фланце, не всегда удается удалить золото с фланца после обжатия и
золото является дорогим материалом. Тип уплотнения, ставшего теперь по¬
чти универсальным, показан на рис. 2.1. Плоская кольцевая медная проклад¬
ка точно устанавливается в зазор, образуемый двумя идентичными фланца¬
ми при их сочленении. На поверхности каждого фланца выточен круговой кони¬
ческий нож. Когда фланцы прижимаются друг к другу при затягивании болтов,
расположенных в соответствующих отверстиях, направленные навстречу друг
Другу ножи одинакового диаметра вдавливаются в медную прокладку, застав¬
ляя медь обтекать их с обеих сторон. Внутренний диаметр прокладки не зада¬
ется, но внешний должен быть таким, чтобы не допустить разрушения проклад-
322. ОборудованиеРис. 2.1. Сверхвысоковакуумное металлическое ножевое уплотнение, в котором ис¬
пользуется круглая медная прокладка.а - два идентичных фланца с круговыми ножами и прокладка между
ними до уплотнения; б- фланцы, стянутые вместе, и прокладка, зажатая
между внутренними стенками углублений во фланцах под действием встреч¬
ных ножей. Уплотнение произведено на внешней (наклонной) поверхности
ножа. Этот вид уплотнений известен как уплотнение типа "конфлэт".ки стенками зазора, в котором она находится. Таким образом, в меди на этом
участке возникают огромные усилия, и создается очень эффективное уплот¬
нение между прокладкой и материалом фланца с внешней стороны ножей, спо¬
собное выдержать бесчисленное множество температурных циклов при отжи¬
гах.Мы рассматриваем здесь вакуумные уплотнения, поскольку теперь оче¬
видно, что фланцы, показанные на рис. 2.1 (называемые по-разному: "острие
ножа”, "медное уплотнение" или "конфлэт"1)-фланцы), должны быть сделаны
из твердого металла, полностью сохраняющего свою твердость при темпера¬
турах отжига и не подверженного окислению. Таким образом, алюминий и его
сплавы, которые в других случаях используются в качестве конструкционных
материалов в сверхвысоком вакууме, не могут быть использованы для изго¬
товления сверхвысоковакуумных камер, так как они слишком мягки для "кон-
флэтных" уплотнений на фланцах. Поэтому материалом, используемым для
производства подавляющего большинства сверхвысоковакуумных камер, яв¬
ляется нержавеющая сталь. Исключение составляют случаи, когда требуется
экранировка магнитного поля, например, вокруг энергоанализатора и в окрест¬
ности образца. В таких случаях некоторые изготовители используют в качест¬
ве материала для стенок камеры мю-металл вместо нержавеющей стали.Хотя, как отмечалось, такой эластомер, как витон, не следует применять
для сверхвысоковакуумных уплотнений, граничащих с атмосферой, его можно
использовать внутри сверхвысоковакуумной камеры, если принять определен¬
ные меры предосторожности. Иными словами, витоновые кольца должны бытьТорговый знак, запатентованный фирмой "Вариан".
Дж. К. Ривьер33очищены от жира и затем предварительно обезгажены во вспомогательной
вакуумной системе перед установкой в спектрометре; кроме того, эти кольца,
используемые в проходных вентилях, не следует подвергать отжигу, когда
вентиль закрыт. Сжатый витон фиксирует форму, существующую во время от¬
жига, что приведет впоследствии к невозможности уплотнения вентиля при за¬
крывании.Кроме нержавеющей стали и меди существуют и другие материалы, кото¬
рые могут надежно использоваться в сверхвысоковакуумных системах не в
качестве основных конструкционных материалов, а для изготовления допол¬
нительных частей, присоединяемых к спектрометру для решения различных задач.
До появления вакуумных систем из нержавеющей стали основным конструк¬
ционным материалом было боросиликатное стекло, которое и до сих пор широ¬
ко используется для изготовления окон, присоединяемых к фланцам с помощью
спаев металл - стекло. Для некоторых экспериментов используются также квар¬
цевые окна. Другим широко используемым керамическим материалом являет¬
ся алунд; он применяется всюду, где нужна электрическая изоляция для вводов
тока и напряжения, используемых в спектрометре.Внутри спектрометра может быть использован любой материал, который
не вызовет повышения остаточного давления ни при комнатной температуре,
ни при температуре отжига и который не содержит испаряющихся при этих
температурах компонентов, которые могут адсорбироваться на внутренних по¬
верхностях и загрязнить их. Например, латунь, содержащая большое количест¬
во легколетучего цинка, не должна применяться, так же как и любая из обыч¬
ных полимерных пластмасс. Однако разрешается использование проводников
с фторопластовой изоляцией для электрических цепей в сверхвысоком ваку¬
уме, если используется высококачественный фторопласт. В сверхвысоком ва¬
кууме для изготовления мелких узлов, т. е. устройств для крепления образ¬
цов и их нагрева, для электрических соединений и т. д., обычно используют¬
ся медь, никель, платина, молибден, тантал и вольфрам. Большинство специа¬
листов, работающих в области электронной спектроскопии, должны иметь за¬
пас по крайней мере некоторых из этих материалов в виде проволоки или
фольги.2.2.1.2. Проектирование и конструкцияЭти два аспекта взаимозависимы: проектирование любой составной ча-
'-ти, большой или малой, сверхвысоковакуумного прибора не может быть вы¬
полнено, если не известны точно ограничения и требования, налагаемые на
Инструкцию. К требованиям, о которых следует постоянно помнить, относят¬
ся, например, расположение фланцев относительно друг друга и внешней по¬
верхности вакуумной камеры, такое, чтобы постоянно были обеспечены дос-
342. Оборудованиетуп для установки и затягивания болтов и достаточное пространство как для
подготовки к сварке, так и для самой сварки. Обычно такие вопросы касают¬
ся скорее изготовителя, а не ученого, но если пытаться самостоятельно раз¬
рабатывать специальную вакуумную камеру или усовершенствовать уже суще¬
ствующую, то об этом нельзя забывать.Другая важная часть разработки вакуумной системы, которая имеет от¬
ношение не только к СВВ-камере, - это обеспечение соответствующей ско¬
рости откачки во всех точках откачиваемого объема. Даже в приборах извест¬
ных фирм, которым следовало бы это учитывать, к сожалению, этот недоста¬
ток еще не редкость. Проектируется идеальная вакуумная система для спект¬
рометра, которая хорошо работает, пока камера пуста и к ней ничего не при¬
соединено, а затем помещаются внутрь или присоединяются к камере устрой¬
ства таких размеров и форм, что сечения, через которые осуществляется от¬
качка, уменьшаются до величин, при которых скорость откачки этих устройств
сильно ограничивается. Это основная причина того, что для получения СВВ с
помощью отжига в некоторых спектрометрах требуется много времени. Взаимо¬
расположение составных частей в камере должно рассматриваться еще на ста¬
дии проектирования не только с точки зрения требований электронной спектро¬
скопии, но и с точки зрения возможности получения необходимого вакуума.Это в равной мере относится к устройствам, присоединяющимся к фланцам яа
стенках камеры. В последнем случае, когда это необходимо для увеличения
скорости откачки, следует предусмотреть байпасные линии откачки, связан¬
ные с основным насосом или даже со вспомогательными насосами. Оценка
пропускной способности и, следовательно, локальных скоростей откачки с нуж¬
ной точностью может быть проведена при помощи различных известных фор¬
мул, основанных на кинетической теории газов, в интервале давлений, соответ¬
ствующем режиму молекулярного течения. Их можно найти в любом хорошем
учебнике [2] по физике вакуума, и мы не будем их здесь повторять.При производстве камер и других деталей из нержавеющей стали приме¬
няют широко распространенную сейчас дуговую сварку в атмосфере инерт¬
ного газа. Для СВВ-систем, однако, существуют некоторые практические ре¬
комендации по проведению сварки, которым необходимо следовать, в против¬
ном случае получение СВВ осложнится или станет невозможным. Эти практи¬
ческие рекомендации помогают избежать образования полых объемов-ловушек,
соединенных с объемом камеры каналами с малой пропускной способностью;
ясно, что такие объемы могут содержать грязь или газы, которые станут по¬
стоянным источником загрязнений, т. е. фактически будут вести себя как
"виртуальные течи". Во избежание таких образований все соединительные
швы должны выполняться изнутри камеры и быть непрерывными. Это означа¬
ет, что необходимо иметь расходуемый электрод достаточной длины с самого
Дж. К. Ривьер35начала сварки, чтобы, например, в случае кругового сварочного шва обеспе¬
чить непрерывную сварку вплоть до возвращения в исходную точку. Только
таким образом можно предотвратить образование объемов-ловушек. В идеаль¬
ном случае такая сварка должна производиться электронным пучком в ваку¬
уме, но это слишком долго и неудобно из-за сложной геометрии, которой часто
обладают внутренние поверхности сверхвысоковакуумных камер.Последняя операция, выполняемая при изготовлении камер, - это финиш¬
ная обработка (доводка) и очистка внутренних поверхностей, и от ее эффектив¬
ности в дальнейшем будет зависеть вакуум, достигаемый в камере или в ва¬
куумных компонентах. Сейчас считается, что для доводки внутренних поверх¬
ностей лучше всего применить обработку маленькими стеклянными шариками,
которые не только удаляют шлаки, загрязнения и т. п. со сварочных швов и
окружающих их поверхностей, но и пассивируют поверхность благодаря накле¬
пу приповерхностных слоев. Кроме того, микротопография поверхности сглаг
живается, следовательно, удаляются потенциальные ловушки для загрязнений.
В любом случае следует избегать доводки электрополировкой, которая еще не¬
давно широко применялась, так как полировка достигается физическим удале¬
нием поверхностных слоев, а сварные швы особенно чувствительны к такого
рода воздействию и в них вскрываются трещины и маленькие отверстия. Пос¬
ле обдува стеклянными шариками важно убедиться, что все осколки удалены,
так как если какой-нибудь из них останется, то он, случайно попав в движу¬
щиеся части, может вызвать разрушительные последствия.Удаление загрязнений химически чистым растворителем после доводки
создает для поверхности условия, позволяющие получать высокий вакуум, но
для достижения СВВ необходима еще одна операция - предварительный отжиг.
Под этим подразумевается отжиг отдельных деталей во вспомогательной ва¬
куумной системе; в идеальном случае этот отжиг проводится при температу¬
ре выше температуры отжига всего спектрометра в целом, чтобы удалить с
внутренней поверхности следы растворителя, использовавшегося для очистки.
После этого компоненты должны быть завернуты в пластиковые листы или
уложены в пластиковые коробки, чтобы они оставались чистыми до момента
сборки камеры. Нет нужды говорить, что после доводки ни одну деталь нель¬
зя трогать голыми руками; необходимо использовать перчатки. Желательно
также, чтобы предварительный отжиг, хранение перед использованием, и, ко¬
нечно, сборка происходили в очищенных от пыли помещениях, т. е. в помеще¬
ниях с профильтрованным воздухом.Последующие стадии разработки и конструирования обычно неважны для
отдельных исследователей, так как сейчас лишь немногие лаборатории сами
изготавливают свое вакуумное оборудование. Мы также не имеем возможно¬
сти в отпущенном на изложение объеме останавливаться на различных част-
362. Оборудованиеностях проектирования оборудования, которые необходимо учитывать. Однако
при сравнительной оценке стандартов, используемых возможными поставщи¬
ками при изготовлении приборов, практические рекомендации, данные выше,
помогут найти общий подход, обеспечивающий выбор оборудования, обладаю¬
щего требуемыми надежностью и качеством.2.2.1.3. Вакуумные засосыВ этом разделе будут рассмотрены только СВВ-насосы, а затем кратко
изложены лишь их достоинства и недостатки, поскольку описание режимов их
работы можно найти во многих других источниках.В коммерческих спектрометрах применяются четыре типа насосов в раз¬
личных комбинациях: диффузионные1J, ионные, турбомолекулярные и титано¬
вые сублимационные, и очевидно, что каждая фирма предпочитает свой тип
насоса. По-видимому, единственное, в чем фирмы-производители соглашают*
ся между собой, - это использование титановых сублимационных насосов в
качестве дополнительных, а не основных для получения СВВ.Диффузионные насосы можно использовать в широком диапазоне требуе¬
мых уровней предельного вакуума в зависимости от используемого масла;
для СВВ требуется масло с давлением паров менее 10“9 мм рт. ст. с большой
устойчивостью против разложения, ведущего к появлению легколетучих прог
дуктов, и, кроме того, оно не должно обладать склонностью к поверхностной
диффузии. Полифениловые эфиры успешно использовались в течение несколь¬
ких лет, но теперь появились другие масла с лучшими характеристиками.Хотя сами диффузионные насосы относительно дешевы, они нуждаются в эф¬
фективных ловушках с жидким азотом, расположенных между насосом и СВВ,
а такие ловушки практически стоят дороже, чем насосы. Если учесть также
стоимость жидкого азота, водяного охлаждения и затрат мощности за дли¬
тельный период времени, то станет ясно, что низкая стоимость является толь¬
ко кажущейся. Противовесом этому выступает основное преимущество диффу¬
зионного насоса по сравнению с описываемыми ниже - они надежны и не ка¬
призны. Они способны откачивать почти любой газ, который не реагирует с
горячим маслом, и при хорошем уходе могут работать в течение очень дли¬
тельного времени, не требуя особого внимания.^ Существенным недостатком применения диффузионных насосов, как это сле¬
дует из имеющегося опыта эксплуатации установок для исследования поверхности
фирмы Vacuum Generators Ltd. (/^глия), является то, что при длительной работе мае*,
ло разлагается и скорость откачки падает. Кроме того* масло хотя и медленно, но по¬
падает в камеру и, озедая на изоляторах, подверженных электронной бомбардировке,
разлагается, образует пленки и вызывает утечки. — Прим. ред.
Дж. К. Ривьер37Ионные насосы обладают многими привлекательными чертами. Они не ис¬
пользуют жидкостей, не нуждаются в охлаждении водой или жидким азотом,
не требуют большой мощности, могут непосредственно соединяться с откачи¬
ваемым объемом без переходных ловушек. Их можно приводить в действие
щелчком выключателя, им не нужен долгий прогрев и столь же просто они
могут быть выключены. Хотя эти насосы довольно дороги, поскольку к ним
требуется блок управления, они не нуждаются ни в каких дополнительных час¬
тях и затратах на охлаждение. Вероятно, при длительном использовании они
дешевле, чем диффузионные насосы. Однако при использовании их в качест¬
ве основных СВВ-насосов оказывается, что у них больше недостатков,,чем
достоинств. При этом существенно, что именно они будут откачивать и в ка¬
ком количестве. Пока идет откачка обычных составляющих остаточного газа,
таких, как азот, кислород и углекислый газ, - проблем не возникает. В совре¬
менном улучшенном исполнении они даже откачивают инертные газы с прием¬
лемой скоростью при условии, что давление инертных газов остается ниже
10“6 мм рт. ст. и насос не откачивает их непрерывно в течение длительного
времени. Однако следует избегать откачки ионным насосом гелия, так как
гелий диффундирует в титановый катод и может вызвать образование трещин
в катоде, если объем продиффундировавшего гелия будет достаточно велик.То же относится к водороду, но тут возникают дополнительные сложности из-
за того, что титан образует ряд твердых растворов и соединений с водородом,
так что в результате длительной откачки водорода может произойти разложе¬
ние катодов. Более того, поскольку пары воды откачиваются в результате дис¬
социации молекул воды на водород и ион гидроксила в разрядном промежутке
насоса с последующим взаимодействием продуктов реакции с титаном, то дли¬
тельная откачка водяного пара при высоком давлении последнего даст тот же
эффект, что и откачка водорода. Другой недостаток ионных насосов - это эф¬
фект памяти. Например, так как гелий и водород откачиваются посредством
растворения в титане, то последующее распыление заполненного гелием или
водородом титана вызовет восстановление этих газов. Проблему до некоторой
степени можно решить с помощью высокотемпературного (~ 500 °С) отжига на¬
соса, но эта операция по меньшей мере неудобна. Кроме того, хотя ионные на¬
сосы эффективно откачивают гидрокарбонаты, углерод образует соединения
с титаном и может рекомбинировать с другими газами, вызывая появление за¬
грязнений. Таким образом, в системе, откачиваемой ионным насосом, в спект¬
ре остаточного газа всегда будут присутствовать молекулы, содержащие один,
два, часто три атома углерода; особенно часто встречается метан, который
вместе с водородом может являться основной составляющей при рабочем дав¬
лении ниже 10~9 мм рт. ст. По этой же причине появление в ионном насосе
чистого кислорода вызовет увеличение парциального давления моноокиси угле-
382. Оборудованиерода до такой степени, что это существенно повлияет на ход эксперимента.
Итак, ионно-разрядные насосы пригодны и очень удобны для откачки атмосфер¬
ных газов, но в случаях, отличных от этого, их применение связано с возник¬
новением многочисленных проблем.Турбомолекулярные насосы1* также привлекательны с точки зрения удоб¬
ства. Турбомолекулярный насос - единственный вид насосов, который в прин¬
ципе может откачать систему от атмосферного давления до СВВ (разумеется,
с отжигом), хотя на практике он никогда не используется таким образом, так
как подобное регулярное использование связано со слишком большой нагруз¬
кой на насос. Он откачивает любой газ, но эффективность откачки уменьшает¬
ся прямо пропорционально молекулярному весу газа, таким образом, эффек¬
тивность его при откачке гелия и водорода мала; следовательно, остаточный
газ будет в основном состоять из гелия и водорода. Единственными ограниче¬
ниями на природу откачиваемого газа будут либо возможная реакция со сма¬
зочным маслом с низковакуумной стороны насоса, либо возможная коррозия
вращающихся лопастей. В обычной практике ни одна из этих возможностей не
вызывает никаких осложнений. Турбомолекулярные насосы не нуждаются в ло¬
вушках и экранах, как диффузионные насосы, и, так же как ионные насосы, ве¬
роятно, дешевле при длительном использовании, чем диффузионные. Существу¬
ет одно соображение, ограничивающее их применение: позволяют ли они сами
достигать давления ниже 10”10 мм рт. ст., так как обычно их стремятся ис¬
пользовать в сочетании с титановыми сублимационными насосами, но то же са¬
мое можно сказать про диффузионные насосы. На самом деле большинство
фирм не гарантирует предельный вакуум Ю~10 мм рт. ст. и выше без помощи
сублимационного насоса, какой бы тип насоса ни использовался в качестве ос¬
новного. Сейчас турбомолекулярные насосы работают очень тихо, одним из
первых возражений против их применения был шум, и, по-видимому, они не ис¬
пользуются в спектрометрах чаще только из-за возможности вибрации и при¬
вычного недоверия к их надежности, основанного на прежнем опыте.Титановые сублимационные насосы наиболее просты и дешевы среди СВВ-
насосов и они действительно очень широко используются, но, как бь*ло отмече¬
но раньше, никогда не применяются в качестве основного насоса спектромет¬
ра. Они могут работать при давлении порядка 10~6 мм рт. ст., но в электрон¬
ных спектрометрах и других СВВ-системах их основной вклад заключается в
достижении предельного вакуума или в уменьшении давления, необходимом1>Опыт использования турбомолекулярных насосов показывает, что пары смаз¬
ки подшипников рано или поздно попадают в камеру. Применение турбомолекулярных
насосов с подвеской ротора на воздушной подушке пока, к сожалению, невозможно из-
за их низкой надежности и малого срока службы. - Прим. ред.
Дж. К. Ривьер39для получения вакуума 10~10 мм рт, ст. и меньше. Для максимальной скоро-
сти откачки титан должен быть напылен на поверхность, охлажденную жидким
азотом, но ни один стандартный спектрометр не дает такой возможности, и
преимущества простоты использования в основном исчезают, если надо обес¬
печивать питание жидким азотом. По крайней мере следовало бы использовать
водяное охлаждение, но, к сожалению, даже это не столь обычно, как должно
было бы быть. Первое распыление с титанового катода следует производить
осторожно, так как при первоначальном разогреве освобождается значитель¬
ное количество газов, но если обезгаживание проведено, то при последующей
работе ток накала можно выводить сразу на максимальное значение. Обычно
время работы титанового насоса регулируется блоком управления, так.что
титан не расходуется напрасно; по грубой оценке насос должен работать каж¬
дые несколько часов только по две минуты при давлении порядка 5 • 10“10 мм
рт. ст. Поскольку сублимационные насосы работают как геттеры, т. е. откач¬
ка происходит благодаря реакции распыленного титана с активными газами,
то они великолепно откачивают основные атмосферные составляющие, водо¬
род и гидрокарбонаты, но совсем не откачивают инертные газы. Более того,
так как титановая пленка впоследствии не разрушается, как катод в ионном
насосе, то эффекты памяти очень малы.Исходя из высказанных замечаний, можно прийти к выводу, что оптималь¬
ными с точки зрения отсутствия осложнений при откачке были бы системы,
состоящие из диффузионного или турбомолекулярного насоса, работающего
параллельно с титановым сублимационным насосом. По указанным выше при¬
чинам не рекомендуется использовать ионные насосы в качестве основных,
хотя они могут применяться как дополнительные насосы для откачки вспомо¬
гательных объемов, куда не поступают инертные газы, или как поддерживаю¬
щие насосы на случай поломки или остановки работы основного.2.3. Подготовка и установка образцаВ этом разделе будут рассмотрены все этапы подготовки образца для
последующего анализа с помощью энергоанализатора, начиная от обработки
образца вне спектрометра до его ввода и размещения внутри камеры, кончая
дальнейшей обработкой в вакууме, если она окажется необходимой.2.3.1. Подготовка образцаБольшая часть работы по подготовке образца вне спектрометра заключа¬
ется в укреплении образца на зонде, или на манипуляторе, или на стержне та¬
ким образом, чтобы изучаемая поверхность была минимально загрязнена. Сле¬
довательно, во избежание прямого контакта кожи с образцом или любой другой
деталью, которая будет помещена в СВВ, необходимо использовать перчатки.
402. ОборудованиеВсе инструменты, используемые при подготовке образца, необходимо очистить,
держать их только руками в перчатках и хранить в чистом месте. В идеальном
случае это должна быть отдельная комната или небольшое отгороженное поме¬
щение, где очищен воздух, но не многие лаборатории имеют свободное помеще¬
ние, чтобы выделить его для этой цели, и обычно достаточно использовать за¬
крытое место или препарационный шкаф с перчатками, в котором создается
избыточное давление проходящим через него потоком воздуха.Способы установки образцов изменяются от образца к образцу и от лабо¬
ратории к лаборатории, часто при этом проявляется большая изобретатель¬
ность. В том случае, когда образцы анализируются в обычных условиях при
комнатной температуре, т. е. когда не требуется специальных устройств для
их нагрева и охлаждения, в современных спектрометрах используются стан¬
дартные держатели образцов, конструкции которых отличаются у различных
фирм-изготовителей, но все они обладают двумя общими конструктивными
особенностями. Во-первых, у них есть углубления, пазы или пружинные зажи¬
мы, которые позволяют перемещать образец из системы ввода в манипулятор,
и, во-вторых, они имеют плоскую площадку, на которой располагается обра¬
зец. Изобретательность проявляется в том, каким образом закрепить на этой
площадке образец неудобной формы так, чтобы материал, используемый для
крепления, не повлиял на результаты анализа исследуемой поверхности об¬
разца. Плоские образцы всегда являются самыми простыми; если обратная
сторона не представляет интереса, то образец можно приклеить к держателю
каплей метаноловой суспензии графита (а не аквадагом), которая быстро вы¬
сыхает и крепко удерживает образец после высыхания. В другом случае об¬
разцы могут быть прижаты зажимами из бериллиевой бронзы или из какого-
либо другого упругого металла, прикрепленного к держателю.Крепление образца неправильной формы определяется его конкретной
геометрией. В некоторых случаях образец достаточно хорошо можно прикре¬
пить к держателю с помощью тонкой полоски какого-нибудь пластичного
металла типа никеля, который может принять форму образца; концы полоски
можно приварить контактной сваркой к держателю или прижать винтами. В
других случаях может понадобиться завернуть образец в металлическую
фольгу, часто в платиновую, оставив исследуемую поверхность открытой;
обертка из фольги служит чашкой, в которой лежит образец, концы фольги
либо приварены контактной сваркой, либо закреплены головками винтов, что¬
бы зафиксировать ее положение. Каким бы образом ни осуществлялось креп¬
ление образца, всегда следует принимать меры, чтобы материал крепления не
перекрывал хотя бы частично исследуемую поверхность; в случае когда это
неизбежно, необходимо, чтобы фотоэлектронный спектр материала, исполь¬
зуемого при монтаже образца, не интерферировал с ожидаемым спектром об¬
разца.
Дж. К. Ривьер41Введение порошковых образцов составляет особую проблему. Если их
нельзя предварительно спрессовать или обжать, то их можно разместить в
нише, укрепленной на держателе или являющейся частью модифицированного
держателя, но при этом имеется очевидный риск. Откачка, начинающаяся при
атмосферном давлении, если она происходит слишком быстро, распылит боль¬
шую часть порошка по всей вакуумной системе; порошок почти наверное рас¬
сыпется при движении держателя или при перемещении его из одной части
спектрометра в другую, а в некоторых спектрометрах анализируемая поверх¬
ность должна быть наклонена к горизонтали под углом по крайней мере 30°,
что также может привести к рассыпанию порошка. Рекомендуется следующий
метод фиксации порошкового образца для анализа: следует насыпать порошок
равномерным слоем на индиевую фольгу, а затем осторожно надавить на слой
порошка куском чистого твердого металла типа молибдена или вольфрама.
Достаточное количество порошка вдавится в поверхность индия, что обеспе¬
чит непрерывность анализируемой поверхности, избыток порошка можно стрях¬
нуть. Этот метод имеет дополнительное преимущество: близость индия умень¬
шит эффект поверхностной зарядки, который может быть значителен в случае
свободно насыпанного порошка. Другой распространенный метод фиксации по¬
рошка - использование двусторонней липкой ленты, но его не следует приме¬
нять в случае ЭОС.В более сложной ситуации, когда образец необходимо нагреть или охла¬
дить либо до, либо во время исследования, закрепление образцов* должно про¬
изводиться с большей точностью. Обычно программы экспериментов, включаю¬
щие такой анализ, более длительны, чем те, в которых образцы перемещают¬
ся на держателе, и, следовательно, в этом случае образцы закрепляются ин¬
дивидуально и непосредственно на манипуляторе, а не передаются на него.
Большинство фирм-производителей продают либо специальные зонды, либо
специальные приспособления к манипулятору для использования в спектромет¬
ре, когда требуются нагрзв и охлаждение. В качестве примера устройство та¬
кого комбинированного приспособления приведено на рис. 2.2. Косвенный на¬
грев обеспечивается изолированным горячим катодом, расположенным за под¬
ложкой, на которой крепится образец, охлаждение - пропусканием жидкого
азота через резервуар, который соединен с подложкой многожильным медным
проводом большого сечения. В общем случае рекомендуется использовать зонд
или приспособление, предназначенное либо для нагревания, либо для охлаждения, так
как устройство, служащее для выполнения обеих задач, обязательно будет иметь ог¬
раничения по достигаемой температуре. Так как фактическая температура образца
при нагреве и охлаждении существенно зависит от теплового контакта между
образцом и подложкой, всегда желательно, если это возможно без разруше¬
ния образца, крепить термопару непосредственно к образцу, а не полагаться
на показания термопары, закрепленной на подложке. Если это не получается,
Рис. 2.2. Фотография комбинированного устройства для нагрева и охлаждения об¬
разцов. Нагрев осуществляется с помощью изолированного горячего като¬
да, расположенного под подложкой, на которой укреплен образец. Охлаж¬
дение производится передачей тепла от подложки за счет теплопроводнос¬
ти к хладопроводу — многожильному медному проводу, прикрепленному к
резервуару, через который протекает жидкий азот. Установка монтируется
на оси универсального манипулятора (см. рис. 2.9). (С разрешения фирмы
Vacuum Generators Ltd., Hastings.)1 - трубки, по которым подводится жидкий азот; 2 - электрический
ввод; 3 - хладопровод (многожильный медный провод); 4 — азимутальный
держатель образца; 5 - радиационные экраны; 6 - резервуар с жидким
азотом.
Дж. К. Ривьер43то необходимо сначала, используя пробный образец, измерить зависимость
температуры подложки от действительной температуры образца.Одним из недостатков изложенного выше метода, т. е. разогрева образца
за счет теплопроводности от нагреваемой подложки, является то, что присут¬
ствующие внутри или на поверхности подложки примеси при высокой темпера¬
туре могут обрести подвижность и загрязнить образец. Это, конечно, приведет
к искажению экспериментальных результатов. Поэтому необходимо, чтобы в
начале блок нагрева в течение длительного времени работал при предполагае¬
мых температурах без образца, при этом следует контролировать чистоту по¬
верхности подложки. Если какие-либо загрязнения появляются и остаются, их
требуется удалить ионной бомбардировкой. Циклы нагрева и ионной бомбарди¬
ровки следует повторять до тех пор, пока примеси не перестанут появляться
при нагреве до заданной температуры. Другими словами, перед началом рабо¬
ты подложка рассматривается как образец и очищается таким же образом.В основополагающих экспериментах, где исследуются очень чистые мате¬
риалы, часто монокристаллы, указанный способ нагрева образца не может счи¬
таться удовлетворительным. В таких экспериментах недопустима возможность
загрязнения образца материалами несущих конструкций, и нагрев образца дол¬
жен быть однородным. В зависимости от формы, толщины и природы образца
нагрев может производиться прямым пропусканием тока через образец, путем
теплопередачи от нагретой подложки, излучением от близко расположенной
нити накала или электронной бомбардировкой с термокатода. В общем
случае держатель должен быть изготовлен либо из того же материа¬
ла, что и образец, и обладающего такой же чистотой, либо из тугоплавкого ме¬
талла типа вольфрама или молибдена, который необходимо очистить прогре¬
вом при высокой температуре, прежде чем использовать в качестве держате¬
ля. Для измерения температур до ~ 850 необходимо использовать термо¬
пару, аккуратно приваренную контактной сваркой к краю образца или, если
это невозможно, прикрепленную к подложке возможно ближе к образцу. Термо¬
пары из благородных металлов, например Pt - Pt /10%Rh, следует предпо¬
честь наиболее распространенным термопарам NiCr - NiAl, так как риск вне¬
сения загрязнений при использовании последних намного больше. Для измере¬
ния температур выше 850 °С используется оптический пирометр, так как при
высоких температурах увеличивается вероятность реакции между образцом
и материалом термопары; конечно, следует помнить, что данные, получаемые
с помощью пирометра, всегда нуждаются в корректировке за счет излучения
образца и поглощения излучения в стекле Окна, через которое ведется наблю¬
дение. В случае некоторых конструкций для крепления возможно измерение
температуры образца по температурной зависимости сопротивления проволо¬
чек, на которых закреплен образец.
442. Оборудование2.3.2. Введение образца в спектрометрВ случае когда образец должен быть непосредственно установлен на ма¬
нипуляторе, как это обсуждалось выше, ввод образца в спектрометр, очевид¬
но, будет связан с заполнением камеры, в которой проводится анализ, возду¬
хом и присоединением манипулятора с помощью болтового соединения непо¬
средственно к камере. Эта процедура занимает, безусловно, много времени,
так как камера затем должна быть откачена и весь спектрометр отожжен,
чтобы вновь получить СВВ. Однако если процедура исследования образца яв¬
ляется достаточно долгой, то время, затраченное на получение СВВ, будет
относительно невелико. До недавнего времени эта процедура применялась
всегда, так как не существовало никаких систем ввода образцов; это означа¬
ло, что при рутинном анализе в СВВ смена образцов требовала значительного
времени. Некоторого ускорения рутинного анализа можно достичь, устанав¬
ливая одновременно несколько образцов на карусели, которая может вращать¬
ся с помощью ввода вращения, находящегося на манипуляторе, но в общем
случае это незначительно расширяет возможности смены образцов при ана¬
лизе.Одним из основных направлений, благодаря которому в последние годы
было достигнуто существенное увеличение скорости смены образцов, явилось
развитие способа ввода образцов непосредственно в СВВ. В данный момент в
зависимости от концепции фирмы-изготовителя применяются две различные
модификации этого способа. В одном случае носитель (держатель с приспособ¬
лением, позволяющим осуществить передачу образца) с укрепленным на нем
образцом размещается на конце длинного стержня или штока, обладающего
очень хорошо отполированной цилиндрической поверхностью. Шток скользит
совершенно без смазки внутри ряда плотно прилегающих к стержню уплотняю¬
щих колец, которые различными фирмами изготавливаются из витона или теф¬
лона11, перенося образец из внешнего пространства через промежуточную ка¬
меру (шлюз) в СВВ-камеру. Шлюз откачивается по меньшей мере до 10~6 мм
рто ст., прежде чем открывается впускной клапан в СВВ. Внутри СВВ-камеры
шток движется контролируемым образом, пока держатель не достигнет кару¬
сели и образец не будет закреплен на одном из свободных мест. Затем шток
может быть отведен в исходную позицию. Удаление образца осуществляется
просто в обратном порядке.Преимущества способа введения образца на шгоке заключается в быстро¬
те, возможности выполнения операции ввода в полуавтоматическом режиме и
в том, что используется лишь осевое перемещение. Недостатком является то,
что часть поверхности штока вводится в СВВ-камеру непосредственно из ат-1) Используемое за рубежом название фторопласта. — Прим. ред.
Дж. К. Ривьер45мосферь: без дополнительного отжига и обезгаживания, кроме обусловленного
откачкой в шлюзе. То же следует сказать и об образце и его держателе, но их
общая поверхность значительно меньше, чем поверхность вводимой в СВВ-ка-
меру части шгока, кроме того, в любом случае держатель подвергается отжи¬
гу на какой-либо стадии эксперимента. Обычно при введении образца с помо¬
щью описанного выше способа давление в СВВ-камере повышается до такой
величины, что выходит из СВВ-диапазона, и требуется некоторое время, что¬
бы давление снова стало ниже 10“9 мм рт. ст. Практически после серии быст¬
ро чередующихся вводов и извлечений образцов получение вакуума 10 ~9 мм
рт. ст. без отжига может оказаться невозможным. Таким образом, этот
способ ввода образцов пригоден для случая рутинного анализа образцов, обла¬
дающих сравнительно инертными поверхностями, и для случая, когда можно
пожертвовать поддержанием в камере, где проводится анализ, давления поряд¬
ка Ю~10 мм рт. ст. ради увеличения скорости смены образцов.Другой способ ввода образцов можно назвать способом передачи образ¬
цов. В этом случае устройство, в котором закреплен образец, размещается на
держателе в небольшой промежуточной камере (шлюзе), которая затем отка¬
чивается от атмосферного давления до 5 • Ю~3 мм рт. ст. форвакуумным на¬
сосом, имеющим ловушку. Затем открывается проходной вентиль между шлю¬
зом и второй препарационной камерой и устройство, в котором закреплен
образец (носитель), перемещается во вторую камеру на каретке, находящейся
в вакууме. После перенесения носителя на каретку, находящуюся в препара¬
ционной камере,с помощью управляемого вручную линейного ввода перемеще¬
ния с сильфонным уплотнением первая каретка возвращается в шлюз и про¬
ходной вентиль закрывается. Так как препарационная камера обладает суще¬
ственно большим объемом, чем шлюз, и откачивается большим диффузион¬
ным насосом, то давление в ней быстро восстанавливается до~10~9 ммрт. ст.
Затем открывается второй проходной вентиль между препарационной камерой
и аналитической камерой, носитель вводится в аналитическую камеру на вто¬
рой каретке и передается на манипулятор с помощью операций, осуществляе¬
мых вручную. По окончании передачи образца каретка возвращается обратно
и второй проходной вентиль закрывается. Так как при передаче образца давле¬
ние в препарационной камере меньше 10“9 мм рт. ст., СВВ в аналитической
камере едва ли ухудшится.Очевидно, что изложенный метод медленней, чем метод введения образ¬
ца с помощью штока, существует известный риск уронить носитель при пере¬
даче и трудно понять, каким образом этот способ ввода образца можно авто¬
матизировать. Значительное преимущество с точки зрения эксперимента за¬
ключается в том, что давление в камере для анализа сохраняется около
Ю~10 мм рт. ст,, т. е. таким же. какое было получено с помощью отжига и
462. Оборудованиет. д. Если будет обнаружено, что сам образец является по какой-либо причи¬
не источником газа, его можно перевести в препарационную камеру и выдер¬
жать там либо до тех пор, пока не закончится обезгаживание, либо пока не
будет решено, что данный образец не подходит для анализа.Не так уж редко необходимо исследовать образцы, которые нельзя выно¬
сить на воздух ни на одной из стадий, например, из-за того, что материал об¬
разца будет слишком сильно взаимодействовать с атмосферными газами, или
на поверхности образца присутствуют пленки, интересные для анализа, кото¬
рые могут разрушаться или заменяться при реакции с окружающей средой,
или даже из-за наличия в образце химических или радиоактивных загрязнений,
попадание которых в окружающую среду недопустимо. Применяются различные
методы для минимизации времени контакта образца с окружающей средой приРис. 2.3. Камера для введения в спектрометр образцов, чувствительных к воздейст¬
вию атмосферы. Камера проходит через стандартное отверстие препара-
ционного шкафа и должна быть сконструирована таким образом, чтобы ее
можно было подсоединить к входному окну ESCALAB. После помещения
образца в камеру инертного газа клапан закрывается и камера прикрепля¬
ется к входному окну шлюза. Затем шлюз заполняется инертным газом,
вентиль открывается и шлюз с камерой откачиваются вместе. После дости¬
жения требуемого вакуума образец передается в следующую камеру обыч¬
ным путем. (С разрешения фирмы Vacuum Generators Scientific Ltd., East
Grinstead.)1 - вентиль клапана в открытом положении; 2 - держатель образца;3 - ввод линейного перемещения механизма вентиля; 4 - транспортер в ра¬
бочем положении — осуществлен ввод образца в шлюз; 5 - вентиль для на¬
пуска воздуха; в - червячная пара; 7 - коническая передача; 8 - уплотня¬
ющее кольцо "С-образной формы; 9 - транспортер в конечном положении.
Дж. К. Ривьер47введении его в спектрометр. Самое простое - поставить большую пластмас¬
совую коробку, окружающую либо шток ввода, либо вход в шлюз, где поддержи¬
вается избыточное давление инертного газа типа аргона. Затем крепление об¬
разца осуществляется внутри коробки руками в перчатках, причем щели между
обшлагами перчаток и стенками коробки достаточно малы, чтобы воздух не
проникал в коробку. Более совершенная и эффективная модификация данного
метода - препарационный шкаф с перчатками, плотно присоединенный к тому
концу спектрометра, через который производится ввод образца. В препараци-
онном шкафу также поддерживается избыточное давление. Это устройство об¬
ладает тем преимуществом, что многие чувствительные к воздействию Окру¬
жающей атмосферы образцы могут быть сохранены в нем в качестве образ¬
цов для сопоставления в будущем и что в таком шкафу хорошо видны все мани¬
пуляции.Еще более совершенным способом защиты при вводе в спектрометр чув¬
ствительных к воздействию атмосферы образцов является применение специ¬
ально изготовленных камер, спроектированных таким образом, чтобы они
проходили через стандартное отверстие препарационного шкафа с перчатка¬
ми и были снабжены вентилем, имеющим откидной клапан. Пример такого
приспособления приведен на рис. 2.3. Внутри препарационного шкафа в атмос¬
фере инертного газа клапан открывается, образец вводится в камеру, после
чего клапан закрывается. Затем камера вместе с образцом, находящимся в
инертном газе, вынимается из препарационного шкафа и присоединяется к
шлюзу спектрометра. В шлюзе, также заполненном инертным газом, клапан
открывается, и камера вместе со шлюзом откачивается до обычного вакуума,
необходимого для ввода образца. Затем образец вводится согласно уже опи¬
санной процедуре; при этом чаще используется каретка, находящаяся в каме¬
ре, чем каретка шлюза.2.3.3. Подготовка образца в вакуумеСуществуют три основных типа подготовки образца после ввода его в
спектрометр: получение чистой поверхности, послойный анализ и проведение
химической обработки поверхности. Последняя включает нагрев для получе¬
ния сегрегации примесей на поверхности или для расплавления образца, если
исследуются жидкие металлы.2.3,3.1. Получение чистой поверхностиВ контексте этого раздела слово "чистый" будет означать такое состоя¬
ние поверхности, при котором РФЭС и ЭОС не могут на пределе чувствитель¬
ности обнаружения зарегистрировать характерные спектральные линии каких
бы то ни было элементов, кроме тех, из которых состоит сам образец.
482. ОборудованиеОчевидно, что если применить более чувствительный метод анализа к поверх¬
ности, которая в указанном выше смысле считается чистой, то загрязнения,
вероятно, будут обнаружены. Как всегда, чистота - понятие относительное.На первый взгляд простейшим методом очистки является прогрев. Неко¬
торые элементы, включая кремний и тугоплавкие металлы, могут быть очи¬
щены в соответствии с приведенным определением нагревом до достаточно
высоких температур, обычно на несколько сотен градусов Цельсия ниже их
температуры плавления; при этом их поверхность будет чистой, пока матери¬
ал находится при такой высокой температуре. Проблема заключается в под¬
держании этой степени чистоты при охлаждении до комнатной температуры,
так как при охлаждении образцы проходят через те диапазоны температур,
в которых примеси быстро сегрегируют на поверхности. Такая же проблема
возникает при работе с многокомпонентными материалами, такими, как спла¬
вы и соединения, которые нельзя нагреть до температуры, при которой поверх¬
ность станет чистой, но которые подвергаются термической обработке в ходе
эксперимента. В определенных температурных диапазонах загрязнения, при¬
сутствующие в объеме в небольшой концентрации, могут накапливаться на
поверхности. Наиболее часто сегрегируют сера, кислород и углерод, хотя в
некоторых случаях и другие примесные элементы, такие, как азот и фосфор,
скапливаются на поверхности в значительных количествах.Следовательно, нагрев образца сам по себе едва ли может быть эффек¬
тивным способом получения чистой поверхности при комнатной температуре.
Для удаления загрязнений с поверхности, как тех, которые образовали сегре¬
гации, так и присутствовавших на поверхности изначально, нужен другой ме¬
тод; чаще всего для этого используется ионная бомбардировка. В принципе
это очень просто. Пучок положительных ионов инертного газа с энергией
обычно от 500 эВ до 5 кэВ направляется на поверхность. В результате обме¬
на энергией на поверхности и в приповерхностных слоях некоторые атомы
или кластеры на поверхности получают достаточную кинетическую энергию
для того, чтобы покинуть поверхность, и удаляются с нее. Другими словами,
происходит эрозия поверхности. Обычно используется аргон, выбранный в ре¬
зультате компромисса между эффективностью удаления материала, которая
увеличивается с атомным весом, и величиной давления пара при температуре
ловушек, заполненных жидким азотом, если они используются; инертные га¬
зы с большим атомным весом имеют тенденцию к конденсации при низких тем¬
пературах, что затрудняет их откачку. Процесс эрозии поверхности называет¬
ся распылением, а источник ионов - ионной пушкой. Аргон для ионной пушки
может подаваться двумя способами: либо напуском аргона, так что давление
во всей системе достигает значения, при котором ионная пушка-может рабо¬
тать, либо напуском газа непосредственно туда, где образуются ионы, так
что только в этой области давление газа будет высоким. Первый метод - ста-
Дж. К. Ривьер49тический - используется в системах с ионной откачкой; при этом насос сле¬
дует отсекать с помощью вентиля, так как он не должен непрерывно отка¬
чивать аргон. Второй метод - динамический; так как выходные апертуры ион¬
ной пушки достаточно малы, что позволяет осуществлять дифференциальную
откачку, то давление в аналитической камере может поддерживаться доста¬
точно низким с помощью диффузионного или турбомолекулярного насоса.
Очистка образца происходит более успешно с динамически откачиваемой ион¬
ной пушкой, где примеси, покидающие поверхность при эрозии, сразу удаля¬
ются, чем в статической системе, где возможно повторное оседание приме¬
сей на поверхности. Описание различных типов ионных пушек и их режимов ра¬
боты будет дано в п. 2.4.3.Как указывалось выше, метод очистки ионной бомбардировкой в принципе
прост. Его можно непосредственно использовать на практике. Опубликовано
также множество примеров, в которых поверхность очищалась только ионной
бомбардировкой. Однако в большинстве случаев оказывается, что, хотя ион¬
ная бомбардировка позволяет удалить слой загрязнений, всегда остается не¬
большое количество атомов примеси порядка нескольких атомных процентов.
Обычно труднее всего полностью удалить кислород и углерод. Вэ многих при¬
ложениях при анализе допустим низкий уровень остаточных загрязнений, если
требовалось только удаление маскирующих примесей с помощью бомбардиров¬
ки, чтобы стало возможным проведение анализа образца. В случае других при¬
ложений существенным является получение чистой поверхности в соответст¬
вии с данным выше определением, тогда чаще всего применяется комбиниро¬
ванный способ очистки, т. е. чередование циклов нагрева и ионной бомбарди¬
ровки.Если используется тонкий образец, т. е. образец в виде фольги и уже до¬
статочно чистый, то можно соответствующим выбором температуры и дозы
ионного облучения полностью очистить образец от примесей, сегрегировавших
на поверхности при нагреве. При использовании такого образца легко при не¬
обходимости восстановить чистоту поверхности одним циклом бомбардировки
и/или нагрева, поскольку никаких новых примесей из объема не поступает.В случае толстого образца, очевидно, было бы нерациональным пытаться уда¬
лить сегрегирующие примеси из всего объема образца, но во многих случаях
правильный выбор температуры отжига и ионной дозы может привести к тому,
что прилегающий к поверхности слой толщиной до 1-2 мкм будет свободен от приме¬
сей. Очевидно, это состояние нестабильно относительно последующего нагрева
образца до температур, близких к используемой при очистке, но если не будет
последующей термической обработки или образец будет нагреваться до значи¬
тельно более низких температур, то такой способ очистки представляет со¬
бой полезный компромисс.4—249
502. ОборудованиеХотя ионная бомбардировка постоянно применяется для очистки поверх¬
ности либо самостоятельно, либо в сочетании с нагревом, потенциальный поль¬
зователь должен помнить о сопутствующих эффектах, с которыми связано ее
применение. Это могут быть химические [3] или топографические [4] измене¬
ния состояния на поверхности или те и другие вместе. Химические изменения
означают изменения элементного состава или химического состояния или и
того и другого вместе у основных компонентов, что может иметь место на
поверхности сплавов и соединений при ионной бомбардировке. Топографиче¬
ские изменения обычно связаны с увеличением шероховатости поверхности.Эти эффекты подробно обсуждаются в гл. 4, но полезно кратко рассмотреть
их здесь в связи с проблемой очистки поверхности.Поскольку коэффициент распыления, т. е. количество распыленных ато¬
мов поверхности на падающий ион аргона, существенно меняется в зависимо¬
сти от атомного номера, как показано на рис. 2.4, согласно работе Сиха [5],
отсюда следует, что в материале, состоящем из двух и более элементов, бу¬
дет происходить преимущественное распыление по крайней мере одного изРис. 2.4. Предсказанные величины коэффициентов распыления элементов в зависимос¬
ти от их номера в периодической таблице при бомбардировке ионами аргона
с энергией 500 и 1000 эВ. Значения нескольких элементов с очень высоким
коэффициентом распыления для простоты не показаны, но их можно найти в
оригинальной работе Сиха [б].о
Дж. К. Ривьер51них, так как он будет обладать высоким коэффициентом распыления. Анализ
поверхности после ионной бомбардировки обнаружит уменьшение концентра¬
ции этого элемента по сравнению с объемом или со стехиометрическим соста¬
вом. Уменьшение концентрации при ионной бомбардировке продолжается до
тех пор, пока не будет достигнуто равновесное состояние, при котором отно¬
сительная концентрация элементов на поверхности будет определяться отно¬
сительной скоростью распыления. Этот эффект часто наблюдался [6] в спла¬
вах, в которых компоненты имеют существенно различающиеся коэффициенты
распыления. Если нет матричных эффектов и если известны коэффициенты
распыления отдельных элементов, то можно определить состав поверхности
до распыления; это будет обсуждаться ниже в гл. 5.Другие химические изменения под действием бомбардировки также могут
быть достаточно серьезны. К ним относятся восстановление соединений [3] и
сохранение элементов в поверхностных слоях благодаря "вбиванию”. В первом
случае наиболее летучие элементы, такие, как кислород в оксидах, сера в суль¬
фидах ит. д., преимущественно теряются при бомбардировке, даже если их
коэффициент распыления такой же, как у остальных составляющих, или ниже.
Такая потеря неизбежно сопровождается уменьшением степени окисления, на¬
пример металла в металлическом оксиде. Эффект ’’вбивания", как и предпола¬
гает его название, заключается в том, что некоторые элементы, особенно лег¬
кие, могут быть "вбиты" в глубь материала либо прямым ударом первичного
иона, либо косвенно путем каскадной передачи кинетической энергии. Напри¬
мер, устойчивость углерода в некоторых материалах относительно продолжи¬
тельной бомбардировки обычно приписывается этому эффекту [7]. До сих пор
не удается учесть каким-либо систематическим образом роль восстановления
и "вбивания" при количественном анализе.Существует несколько путей увеличения степени шероховатости поверх¬
ности при ионной бомбардировке [8]. Если ионы падают на поверхность образ¬
ца только в одном направлении, т. е. из одного источника, что до сих пор яв¬
ляется наиболее часто встречающимся режимом работы, то в общем случае
шероховатость поверхности будет увеличиваться, так как эффективность рас¬
пыления выше при больших углах падения. В конце концов будет достигнута
равновесная конфигурация, при которой заселенность атомами каждого от¬
дельного топографического положения будет задаваться динамическим равно¬
весием, определяющим скорость распыления из данного положения. Более
сложная ситуация может возникнуть, если в материале присутствуют загряз¬
нения, скорость распыления которых значительно меньше скорости распыле¬
ния основной матрицы. В этом случае загрязнения "затеняют" материал от
падающих ионов и образуются конусообразные пилоны, высота которых быст¬
ро возрастает. Оба этих эффекта можно минимизировать, если не исключить
вовсе, при одновременном использовании двух и более источников ионов, бом-
522. Оборудованиебардирующих образец с разных сторон, но лучше непрерывно вращать образец
при бомбардировке, чтобы полностью сгладить угловые эффекты. К сожале¬
нию, лишь в очень немногих установках можно осуществить подобную процедуру.Итак, ионная бомбардировка, являясь достаточно эффективным методом
очистки поверхности, особенно в сочетании с прогревом, дает весьма нежела¬
тельные побочные эффекты, из которых одни поддаются учету, а другие - нет.
Тем не менее этот метод столь широко применяется, что очевидно, он будет
использоваться и в дальнейшем; и, следовательно, необходимо постоянно сле¬
дить за появлением артефактов при очистке любого образца.В особых случаях применяются другие способы получения чистых поверх¬
ностей. Например, мягкие металлы, такие, как свинец, индий и олово, успешно
очищались [9] посредством скобления (скрайбирования) их поверхности в ваку¬
уме инструментом типа лезвия бритвы, укрепленным на конце стержня, прохо¬
дящего через сильфон. Очевидно, очистка в этом случае равнозначна созда¬
нию новой поверхности за счет механического удаления первоначальной по¬
верхности и связанных с ней загрязнений. Другим специальным методом явля¬
ется вакуумный скол. Некоторые материалы типа щелочно-галоидных кристал¬
лов, кремния, германия и т. д. легко разламываются вдоль определенных пло-Рис. 2.5. Фрактографическая приставка для приготовления скола хрупких образцов в
вакууме. Вращающаяся турель снабжена отверстиями для пяти образцов, ко¬
торые закрепляются выступающими винтами. Вращение достигается с по¬
мощью вертикального нажимного стержня. Чтобы сколоть образец, молоток
поднимается, сжимая мощную пружину до тех пор, пока в определенной
точке спусковой механизм не освободит его, после чего молоток падает
вниз и бьет по свободному концу образца. Отломанный кусок затем по ска¬
ту попадает в держатель, с помощью которого его можно подвести к ана¬
лизатору. Образец перед сколом можно охладить пропусканием жидкого
азота через резервуар, на котором расположена турель. (По Коаду и др.[10].)1 - головка; 2 - сильфон; 3 - цанга и втулка спускового механизма;4 - опорная скоба; 5 - опорный подшипник; б - вращающаяся турель; 7 -
полость; 8 — образец, позиционированный для скола; 9- термопара; 10-
сколотый кусок образца; 11 — изолированный блок, обеспечивающий пово¬
рот сколотого куска образца вокруг оси, перпендикулярной плоскости ри¬
сунка; 12 - стержень привода; 13 - кожух пружины и оси; 14- фланец;15 - пружина; 16 - ударный стержень; 17 - молоток; 18 - направляющая
для скатывающегося образца; 19 - пружинящая пластина, которая прижи¬
мает образец при вращении блока; 20 - крепление к оси манипулятора с
двумя вращательными степенями свободы; 21 - регулируемый ограни¬
читель хода; 22 - ввод термопары; 23 - ввод жидкости или охлаждающего
газа; 24 - соединение; 25 - трубка для поступления охлаждающих газа
или жидкости в камеру; 26 - направляющая для скатывающегося образца;27 - сильфон; 28 - толкатель; 29- пружина; 30- предохранитель; 31 -
вращающаяся турель с пятью образцами; 32 - сколотый кусок образца; 33 -
изолятор блока-держателя образца.
Дж. К. Ривьер53скостей спайности; таким образом, для получения чистой поверхности с со¬
ставом, близким к объемному, необходимо лишь некоторое механическое уст¬
ройство, с помощью которого можно закрепить образец, сделать скол и пере¬
нести образец к энергетическому анализатору для исследования. Материалы
типа поликристаллических металлов и сплавов слишком пластичны для скалы¬
вания при комнатной температуре, однако их также можно сколоть, охлаждая
до температуры, при которой они становятся хрупкими. Плоскость скола мо¬
жет проходить через отдельные зерна (внутризеренная) или по границам зерен
(межзеренная); в любом случае получается поверхность, свободная от загряз-
542. Оборудованиенений, хотя межзеренная поверхность может содержать некоторые примеси,
сегрегировавшие из объема, как это описано в гл. 7. Пример устройства для
скалывания образцов в вакууме [10] приведен на рис. 2.5. Все подобные уст¬
ройства позволяют охлаждать скалываемый образец до температуры, близкой
к температуре жидкого азота.Методы получения атомно-чистых поверхностей на массивных образцах
были недавно описаны в полезном обзоре Маскета и др. [11]. Если не учиты¬
вать ионной бомбардировки и прогрева, то наиболее распространенным спосо¬
бом получения чистой поверхности является напыление чистого материала на
подложку в СВВ либо путем испарения с нагретой спирали или из тигля, либо
с помощью электронной бомбардировки. В принципе таким образом можно на¬
пылить любой элемент, находящийся в твердом состоянии в виде тонкой плен¬
ки, а также многие соединения, но на практике требование свободной от при¬
месей атомно-чистой поверхности налагает ограничения, которые не позволя¬
ют использовать некоторые элементы. Существуют два основополагающих
критерия: а) напыленная пленка должна иметь тот же состав, что и исходный
материал; б) при испарении не должны выделяться газы. Трудности с выпол¬
нением первого требования возникают, если испаряемый материал во время
испарения находится на каком-нибудь носителе, так как в этом случае воз¬
можна реакция с образованием сплава, или если испаряется многокомпонент¬
ный материал, поскольку из-за различных скоростей испарения отдельных
компонентов возможно образование пленки с другим составом. Второй крите¬
рий обычно выполняется при аккуратном проведении процесса напыления, т.е.
при обеспечении полного обезгаживания образца и держателя до нанесения
пленки.Наиболее простые испарители - это те, которые сделаны непосредствен¬
но из материала образца, при этом нет несущей конструкции. Они обычно пред¬
ставляют собой тугую спипаль, через которую пропускают ток, чтобы омиче¬
ским нагревом поднять температуру до точки, при которой происходит испаре¬
ние материала. Очевидно, что материал должен быть таким, чтобы спираль
не провисала при температуре испарения, и поэтому метод применим лишь
для нескольких тугоплавких металлов. Преимущества заключаются в том,
что отсутствует возможность реакции с материалом несущей конструкции и
что испаритель можно полностью обезгазить; к недостаткам относится то,
что скорость напыления мала и окружающие предметы сильно нагреваются,
несмотря на специальные экраны для защиты от излучения.Испаритель, состоящий из нагревательной спирали, на которой находит¬
ся материал образца, смачивающий материал спирали, но не образующий с
ним сплава, также конструкционно очень прост. В этом случае спираль изго¬
товляется всегда из пластичного (т. е. отожженного) вольфрама или молибде-
Дж. К. Ривьер55на. Среди металлов, легко испаряющихся с таких спиралей, следует назвать
медь, серебро и золото1). Можно испарять также барий и магний. При исполь¬
зовании благородных металлов желательно заранее расплавить образец на спи¬
рали во вспомогательной вакуумной системе или в потоке водорода, поскольку
сама спираль предварительно обезгаживается при температуре, значительно
более высокой, чем температура, используемая при последующем испарении.
Если процедура обезгаживания испарителя затем будет проведена до испа¬
рения и образец при этом распл авится еще один или два раза, то последующее
испарение не вызовет сколько-нибудь заметного повышения давления. В слу¬
чае многих легколетучих материалов, не взаимодействующих со спиралью,
испаритель трудно обезгазить в такой же степени. Всегда можно обезгазить
нагреватель без образца, но при попытке расплавить образец из легколетуче¬
го материала большая часть его может преждевременно испариться. Вместо
этого можно осуществлять прогрев легколетучего образца в течение длитель¬
ного времени при температуре, при которой давление паров еще мало. Некото¬
рые металлы типа кадмия, цинка, селена, теллура, щелочных металлов и рту¬
ти имеют при комнатной температуре или температуре отжига слишком высо¬
кое давление паров, чтобы эни могли находиться в виде массивного образца в
СВВ-системе.Большинство металлов, которые было бы желательно испарять для полу¬
чения чистой поверхности, обладают нежелательным свойством более или ме¬
нее быстро образовывать сплавы с материалом испарителя. Сюда относятся
все переходные металлы, алюминий, редкие земли, платина, цирконий, уран,
торий и некоторые другие реже встречающиеся материалы. Ясно, что если при
температуре ниже температуры плавления материала испарителя образуется
сплав материала образца и испарителя, находящийся в жидкой фазе, то это вы¬
зовет не только преждевременное разрушение спирали, но и напыление загряз¬
ненной пленки при условии, что образовавшийся сплав имеет заметное давле¬
ние пара. В этой ситуации необходимо разработать такую систему нагреватель -
образец, чтобы вероятность образования сплава, приводящего к разрушению,
была минимальной. На практике это означает, что материал образца должен
быть распределен вдоль нагревателя так, чтобы во всех точках локальное от¬
ношение объемов материалов образца и нагревателя было меньше чем 1:5.Если испаритель сконструировать таким образом, то можно будет испарить
большую часть образца в чистом виде, прежде чем растворение вызовет разру¬
шение катода или загрязнение пленки. Имеются различные пути, используемые1) На самом деле при длительных выдержках наблюдается разрушение вольфрама,
например при взаимодействии с жидкой медью или серебром [481 за счет межкрис-
таллитной коррозии поликристаллической вольфрамовой проволоки. — Прим. ред.
562. Оборудованиедля уменьшения локального отношения объемов материалов образца и нагреваг
теля, а изобретательность должна предложить новые. Один из наиболее оче¬
видных - это нанесение материала образца на катод как однородного покры¬
тия гальваническим методом или даже с помощью ионного покрытия. Толщину
покрытия и, следовательно, локальное отношение материалов можно точно конт¬
ролировать. К недостаткам метода относится возможность появления приме¬
сей, например воды в результате электролиза, которые в дальнейшем трудно
удалить и которые могут вызвать плохое покрытие спирали материалом образ¬
ца. Другой распространенный способ распределения материала образца вдоль
нагревателя - это использование нагревательной спирали с обмоткой из ма¬
териала образца. В этом случае применяется образец в форме тонкой прово¬
локи с диаметром, значительно меньшим диаметра нагревательной спирали;
до скручивания в спираль проволоки нагревателя образец наматывается на не¬
го с густотой витков, обеспечивающей нужное локальное отношение материа¬
лов. Можно также сделать катод из нескольких тонких проволочек, и тогда ма¬
териал образца, подвешенный на катод в виде маленьких петель, при расплав¬
лении попадает в щели между нитями под действием капиллярных сил и распре¬
деляется достаточно равномерно. Не следует подвешивать петли из материала
образца на нагреватель, сделанный из одной проволоки; при расплавлении на
изгибах нагревателя образуются капельки, приводящие впоследствии к быст¬
рому образованию сплава и разрушению нагревателя.Во всех описанных методах испарения металлов, образующих сплавы с
материалом нагревателя (который практически всегда изготовляется из воль¬
фрама), трудно обезгазить испаритель перед напылением пленки. Поскольку
температура интенсивного испарения всегда такова, что при ней может обра¬
зоваться сплав напыляемого материала с материалом катода, предваритель¬
ное обезгаживание должно проводиться при более низкой температуре и, сле¬
довательно, необходим длительный нагрев. Обычно требуемый рабочий режим
напыления находится методом проб и ошибок, так как в каждом случае
трудно предсказать, какая последовательность операций будет наиболее успеш¬
ной. Следует также просмотреть соответствующие публикации, чтобы выбрать
наиболее подходящий метод.С некоторыми металлами особенно трудно работать либо потому, что они
не смачивают тугоплавкие металлы, используемые для нагревателей, либо
потому, что они становятся достаточно летучими для испарения только при
температурах значительно выше их точек плавления. К этим металлам отно¬
сятся галлий, индий, олово, кремний, таллий, кальций, литий, мышьяк, сурь¬
ма и висмут. Поскольку их невозможно испарить с нагревательных спира¬
лей, их обычно испаряют из тиглей, изготовленных из алунда, окиси берил¬
лия, нитрида бора или графита, нагреваемых с помощью нагревательных эле-
Дж. К. Ривьер57ментов из тугоплавких металлов. Даже длительным прогревом эти испарите¬
ли трудно обезгазить до такой степени, чтобы использовать в СВВ, и поэто¬
му едва ли когда-либо предпринимались попытки получения чистых поверхно¬
стей таких металлов путем напыления.Другим распространенным методом является метод испарения электрон¬
ной бомбардировкой, и его часто можно с успехом применять к материалам,
которые по тем или иным причинам трудно испарить с помощью спирального
нагревателя. Электроны с термокатода (раскаленной нити) фокусируются на
испаряемом материале, нагревая его. При этом температура последнего оп¬
ределяется вкладываемой мощностью. Обычно эмиттер заземлен, а матери¬
ал находится под высоким положительным потенциалом, но при некоторых
конфигурациях полярность может и изменяться. В соответствии с условия¬
ми фокусировки и рассеиваемой мощностью можно нагревать до температу¬
ры испарения весь образец или какую-то его часть. Выпускаемые промыш¬
ленностью испарители с электронной бомбардировкой снабжены охлаждаемы¬
ми водой тиглями, в которых находится испаряемый материал; тем самым
решается указанная проблема образования сплава с материалом тигля. С
другой стороны, такие испарители не подходят для СВВ, так как их нельзя
отжечь и хорошо обезгазить перед использованием. Тем не менее испарение
электронной бомбардировкой успешно использовалось в СВВ [12] с помощью
так называемого метода подвешенной капли, в котором при бомбардировке
конца проволоки или стержня часть его плавится, силы поверхностного натя¬
жения стягивают расплавленный материал в каплю, свисающую с конца стерж¬
ня. С этой капли и происходит испарение. Такой стержень обезгаживается
расфокусированным электронным пучком, который нагревает образец по всей
длине, но не расплавляет его.Какой бы метод испарения ни применялся, всегда следует либо убирать
подложку с направления потока испаряемого вещества, либо ставить перед
подложкой экран на время предварительного обезгаживания испарителя во из¬
бежание осаждения на ней примесей и загрязнений. Это особенно важно, когда
значительная часть образца должна быть испарена во время предварительного
испарения при обезгаживании.2.3.3.2. Поспойный анализ1)Ионная бомбардировка уже упоминалась как возможный метод получения
чистой поверхности образца; кроме того, указывались некоторые недостатки1) В отечественной литературе обычно используется термин "снятие профилей
концентраций примесей по глубине образца”, или "послойный анализ”. — Прим. ред.
582. Оборудованиеэтого метода. Он также весьма часто используется в сочетании с анализом
поверхности как метод получения информации о составе образца в зависимо¬
сти от расстояния до поверхности. Если используется методика, для которой
скорость регистрации мала (в каждый момент), например РФЭС, то информа¬
цию получают ступенчато, т. е. чередуя циклы бомбардировки и анализа. При
использовании ЭОС которая представляет собой метод сравнительно быстро¬
го получения данных, и, если парциальное давление аргона (или другого бла¬
городного газа, используемого при бомбардировке) в окрестности энергети¬
ческого анализатора меньше 10“5 мм рт. ст., бомбардировка и анализ произ¬
водятся одновременно, что позволяет получать непрерывный профиль измене¬
ния концентрации исследуемого элемента в зависимости от глубины ионного
травления. Такой метод анализа называется методом послойного анализа.Все артефакты, которые могут быть внесены ионной бомбардировкой при очи¬
стке поверхности, конечно, присутствуют и также существенны при длитель¬
ной бомбардировке, используемой для измерения профиля концентрации. Эти
эффекты все сильнее будут проявляться с увеличением глубины травления.
При столь широко распространенном применении послойного анализа большое
внимание уделялось и уделяется всем этим эффектам, их влиянию на состав,
и на самом деле изучение профилей концентраций фактически само по себе
является предметом исследования. Поэтому мы не будем останавливаться на
нем здесь, а более полное обсуждение проведем в гл. 4.2.3.3.3. Химические реакции на поверхностиПолучение атомно-чистой поверхности необходимо в некоторых экспери¬
ментах по изучению физических свойств поверхности, но обычно желание при¬
готовить чистую поверхность (с учетом предельной чувствительности исполь¬
зуемого метода) связано с необходимостью достижения воспроизводимого,
хорошо известного начального состояния данной поверхности для изучения ее
склонности к различного рода взаимодействиям. Такая склонность к реакциям может
проявляться по отношению к взаимодействию с газами и парами по отдельности
или вместе, с жидкостями, другими твердыми телами, или с примесями, или
чужеродными атомами, диффундирующими из объема к поверхности. Так как
обычно в вакуумной системе легко контролировать концентрацию газов, то
неудивительно, что подавляющее большинство изученных реакций на поверх¬
ности - это реакции между газами и твердыми телами [13]. Мы будем здесь
рассматривать не сами реакции, а лишь средства их осуществления.Во всех коммерческих спектрометрах можно в качестве дополнительного
приспособления (опциона) установить систему для напуска и контроля давле¬
ния напускаемого газа. Обычно она отделена от системы подачи аргона для
ионной пушки. Система напуска газа должна обладать рядом особенностей.
Дж. К. Ривьер59а. Она должна содержать столько резервуаров со спектрально чистыми
газами, сколько различных газов требуется в эксперименте. Эти резервуары
могут быть как стеклянными баллонами, заполненными газом под атмосфер¬
ным давлением, так и металлическими сосудами с газом под давлением в не¬
сколько атмосфер. В обоих случаях газ напускается после вскрытия герметизи¬
рующей перемычки после того, как резервуар с газом уже присоединен к сис¬
теме. На рис. 2.6 показаны оба варианта исполнения системы напуска газа.б. Устройство для подсоединения газовых контейнеров должно состоять
из фланца и прикрепленного к нему контрольного вентиля. В случае стеклян¬
ного баллона на соединяющем фланце имеется спай металл - стекло, к кото¬
рому припаивается стеклянная трубка, находящаяся в верхней части контей¬
нера, когда контейнер подсоединен к системе. Металлический баллон с га¬
зом, находящимся при повышенном давлении, обычно снабжен собственным
вентилем, который является частью механизма для прокалывания герметизи¬
рующей перемычки в баллоне, но из-за высокого давления в баллоне необхо¬
дим второй вентиль в системе напуска газа, так как требуется дополнитель¬
ный контроль давления напускаемого газа. На рис. 2.6 показано взаимное
расположение компонентов обоих вариантов исполнения системы напуска газа.в. Система должна содержать трубопровод, к которому подсоединены
все баллоны и в котором имеется объем, достаточно большой для смешивания
в точной пропорции нескольких газов, если в эксперименте требуется газовая
смесь, но не настолько большой, чтобы газ расходовался напрасно. Его объ-
602. ОборудованиеРис. 2.6. а - установка для смешивания и напуска контролируемым образом чистых
газов в спектрометр, использующая металлические баллоны со сжатыми га¬
зами. Когда вентиль баллона впервые на него завинчивается, пустотелый
плунжер разрушает герметизирующую перемычку, освобождая газ. Однако,
поскольку давление в баллонах порядка 8 атм, требуются дополнительные
вентили между баллоном и системой как в качестве меры предосторож¬
ности, так и для обеспечения надлежащего точного контроля за процессом
напуска. (С разрешения фирмы Messer Griesheim Ltd.)б - стандартное устройство для напуска газа при использовании
стеклянных баллонов, содержащих по 1 л спектрально чистого газа при ат¬
мосферном давлении. Газ освобождается следующим образом: кусок мяг¬
кого железа поднимается с помощью магнита и затем падает на отпаянный
носик ("чертик"). При этом необходимо принять меры предосторожности,
чтобы измельченное стекло не попало в какой-нибудь вентиль. Для пре¬
дотвращения такой возможности обычно в горлышко баллона кладут кусо¬
чек чистой стекловаты.1 - распределительный вентиль; 2 - натекатель; 3 - к спектрометру;
4 - трубопровод; 5 - спай стекло — металл; 6 - шарик для разрушения
отпаянного носика; 7 - стеклянный отпаянный носик; 8 - спектрально
чистый газ при атмосферном давлении.
Дж. К. Ривьер61Рис. 2.7. Натекатель для точного контроля напускаемого потока газа. В целом кон¬
струкция натекателя показана на рис. 2.7, а, а детали механизма затвора —
на рис. 2.7, б. Затвор натекателя состоит из оптически гладкого сапфиро¬
вого диска, прижатого к металлическому кольцу. Отношение вращательного
перемещения механизма к поступательному движению затвора составляет
2 — 5«103, поэтому возможно очень точное перемещение сапфира. При усло¬
вии что клапан не закрывают слишком сильно, любая определенная скорость
напуска может быть воспроизведена. (С разрешения фирмы Varian Associ¬
ates Ltd.)1 - диафрагма; 2 - поршень; 3 - оптически плоский сапфир.ем также может определяться общим давлением газа или газов, необходимым
для эксперимента в камере, где происходят реакции, и объемом этой камеры.
Однако, вообще говоря, поскольку газ, если это необходимо, может хранить¬
ся в трубопроводе при сравнительно высоком давлении (вплоть до нескольких
миллиметров ртутного столба), и поскольку давления, используемые в экспе¬
рименте, редко бывают выше 10~4 мм рт. ст., то объем трубопровода может
быть небольшим. Очевидно, необходимо иметь возможность контроля давле¬
ния в трубопроводе в широких пределах от СВВ до нескольких миллиметров
ртутного столба. Это означает, что в трубопроводе должны быть установлены
два датчика давления с перекрывающимися диапазонами.г. Система должна содержать вентиль тонкого напуска (натекатель) вы¬
сокого качества для напуска газа из трубопровода в камеру, где происходят
реакции. Конструкция и точность изготовления натекателя должны быть тако¬
вы, чтобы скорость напуска газа контролировалась точно и плавно и чтобы
поток любого выбранного газа можно было воспроизвести. Пример конструк¬
ции такого натекателя приведен на рис. 2.7. Естественно, что натекатель в
622. Оборудованиезакрытом состоянии должен полностью изолировать камеру от попадания га¬
за, находящегося под повышенным давлением в трубопроводе.Чтобы избежать загрязнения газа или газов, напущенных в трубопровод,
необходимо изготавливать его и все подводящие трубки, используя те же
СВВ-стандарты, что и при изготовлении спектрометра, т. е. так, чтобы все
его магистрали можно было отжигать. С другой стороны, стеклянные и метал¬
лические баллоны нельзя отжигать, а это означает, что между трубопроводом
и баллонами будет иметься область, в которой во врёмя отжига возникнет
градиент температур. Поскольку баллоны расположены вне зоны отжига, то
к области, находящейся между баллонами и трубопроводом, будет иметься до¬
ступ, и ее следует осторожно прогреть с помощью ручной горелки, когда бу¬
дет достигнута температура отжига в остальных частях спектрометра, для
удаления со стенок конденсирующихся примесей.Хотя газ в трубопроводе можно считать чистым и приготовленная там
газовая смесь должна иметь хорошо известный состав, нет гарантии, что
при напуске газа его состав останется неизменным. Может произойти обмен
с адсорбированными на различных частях камеры газами, попадание газа на
горячий катод может вызвать диссоциацию, за которой последует рекомбина¬
ция с образованием новых молекул. Если эксперимент является динамическим,
т. е. если камера откачивается во время эксперимента, так что поток газа
проходит над поверхностью, то всегда существует риск, что и в самом насо¬
се будут происходить различные реакции. Это почти невероятно при диффузи¬
онной или турбомолекулярной системе откачки, но иногда наблюдается в слу¬
чае использования ионного и сублимационного насосов. Поэтому весьма же¬
лательно иметь в камере средства для проверки действительного по сравне¬
нию с предполагаемым состава газа, который будет взаимодействовать с
чистой поверхностью. Подобную проверку обычно осуществляют с помощью
небольшого квадрупольного масс-спектрометра. Поскольку такое устройство
также представляет ценность для диагностики, т. е. для контроля состояния
системы и обнаружения течей, то его скорее следует рассматривать как необ¬
ходимый инструмент, а не предмет роскоши.Едва ли когда-нибудь предпринимались попытки исследовать взаимодей¬
ствие жидкости с чистой поверхностью в СВВ; проблемы, связанные с конт¬
ролируемой подачей жидкости на поверхность в СВВ, представляются слиш¬
ком сложными, чтобы прилагать усилия для их решения. С другой стороны, в
последнее время началось активное изучение реакций между твердыми тела¬
ми. Эти исследования стимулированы потребностями микроэлектронной про¬
мышленности, связанными с производством поверхностных слоев с заданны¬
ми электронными свойствами. Эксперименты достаточно просты, они состоят
в напылении посредством испарения определенного количества одного мате-
Дж. К. Ривьер63риала, обычно металла, на поверхность другого, иногда это металл [14], но
чаще полупроводник [15]. Выше обсуждалось соблюдение требований вакуум¬
ной гигиены с целью обеспечения чистоты и отсутствия загрязнений при на¬
пылении. В данном случае это особенно важно, так как даже очень небольшие
количества нежелательных элементов в напыленных пленках или на границе
поверхность - пленка могут вызвать большие изменения электронных свойств
слоя. В некоторых случаях реакция происходит при комнатной температуре
спонтанно (например, при напылении палладия на кремний) [16], в других слу¬
чаях необходимо [17] программировать изменение температуры подложки во
время эксперимента либо для того, чтобы стимулировать протекание реакции
при напылении, либо для того, чтобы предотвратить ее до окончания напыле¬
ния. Поэтому в большинстве случаев для исследования реакций между твер¬
дыми телами необходимы тщательно разработанный испаритель и устройство
для нагрева и охлаждения подложки.Для изучения адсорбции сегрегировавших из объема образца загрязне¬
ний на поверхности для подготовки поверхности необходимо лишь устройство
для прогрева, способное разогреть материал до требуемой температуры. Ес¬
ли же исследуются сегрегации на границах зерен, то поверхность приготав¬
ливается с помощью вакуумного скола, осуществляемого с помощью одной
из фрактографических приставок, показанных на рис. 2.5. В этом случае вся
термообработка образца должна быть завершена до введения в СВВ, хотя в
большинстве случаев необходимо охладить образец перед сколом для увели¬
чения склонности к образованию хрупкого излома.2.3.4. Управление положением образцаСуществует несколько причин, по которым необходимо иметь возмож¬
ность управлять положением образца с большой точностью после введения в
спектрометр и установки его в непосредственной близости от энергетическо¬
го анализатора.Во-первых, формы и размеры образцов могут сильно различаться, поэто¬
му оптимальное положение одного образца для получения лучшей чувствитель¬
ности и разрешения по энергии при анализе поверхности необязательно будет
оптимальным для другого. Во-вторых, для анализа различных областей образ¬
ца может потребоваться воспроизводимое продольное перемещение; это, ко¬
нечно, в большей степени относится к РФЭС, чем к ЭОС, так как в последнем
случае электронный пучок можно отклонить для анализа других областей, при¬
чем в любом случае средняя анализируемая площадь для ЭОС меньше, чем
для РФЭС. В-третьих, желательно уметь перемещать образец вокруг оси, пер¬
пендикулярной его поверхности (это перемещение также должно быть воспро¬
изводимым), либо для изменения угла падения ионного пучка при бомбардиров¬
ке, либо для изменения угла выхода электронов, попадающих в анализатор,
642. Оборудованиечтобы провести послойный анализ, изменяя глубину выхода электронов (см.
гл. 3 и 4). В-четвертых, если в спектрометре можно использовать другие ме¬
тодики, например статический ВИМС, в котором частицы анализируются по
массам, а не по энергиям, то может понадобиться изменить положение об¬
разца по сравнению с тем, которое он занимает при исследовании с помощью
РФЭС и ЭОС. В-пятых, поскольку надежность и степень автоматизации меха¬
нических операций при использовании методов исследования поверхности воз¬
растают, то пользователи будут настаивать на автоматической смене образ¬
цов, обеспечивающей точное перемещение и установку положения.Требование точной установки образца не ново и даже не является специ¬
фическим для РФЭС и ЭОС. Точность всегда была необходима, например при
использовании метода ДМЭ (дифракции медленных электронов) на самых ран¬
них этапах исследования поверхности. По тем же причинам, по которым точ¬
ная установка требуется для РФЭС и ЭОС, она необходима и для целого ряда
других методик. Согласно изложенному выше, образец должен иметь как ми¬
нимум четыре степени свободы: линейные перемещения по осям X, Y и Z и
вращение вокруг оси. Для автоматической смены образцов и перемещения об¬
разца в положение, отличное от требуемого при РФЭС и ЭОС, желательно,
если не необходимо, иметь возможность азимутального вращения и одновре¬
менно вращения образца вокруг оси карусели. Перемещение образца для по¬
зиционирования, обычно предусматриваемое фирмами в их стандартном обо¬
рудовании для анализа поверхности, несколько варьируется и зависит от кон¬
структивных решений, используемых фирмой-изготовителем для ввода образ¬
ца. Когда образцы передаются на карусель с помощью длинного скользящего
штока, на котором находится держатель образца, то при использовании типо¬
вой конструкции держателя образца карусели очень трудно совместить враще¬
ние карусели вокруг своей оси со степенями свободы, необходимыми для по¬
зиционирования образца; таким образом, в подобной системе невозможно
изменить угол падения ионов и угол выхода электронов. С другой стороны,
азимутальное вращение карусели, конечно, дает возможность автоматиче¬
ски менять образцы и исследовать один образец с помощью различных мето¬
дик. Когда образец вводится методом передачи, его в итоге устанавливают
на конце стержня манипулятора, и он может перемещаться по трем взаимно
перпендикулярным направлениям и вращаться вокруг оси манипулятора. Если
манипулятор снабжен устройством для двойного вращения (часто встречаю¬
щимся в качестве опциона), то к возможным видам перемещений добавляется
азимутальное вращение с ограниченным радиусом, но такой механизм недо¬
статочно точен для использования с системой автоматической передачи об¬
разцов.Для экспериментов, носящих фундаментальный характер, когда образец
не вводится одним из двух описанных методов, а устанавливается индивиду-
Дж. К. Ривьер 65Рис. 2.6. Схема прецизионного манипулятора для обеспечения воспроизводимого пе¬
ремещения образцов в спектрометре. Возможные перемещения: перемеще¬
ние вдоль осей х, Y, Z, вращение вокруг центральной оси, вокруг осей,
расположенных под прямым углом к ней (азимутальное вращение), и изме¬
нение наклона.Электрические и другие вводы могут вводиться в вакуум
через ряд фланцев типа мини-конфлэт, расположенных на основ¬
ном фланце, и вся система может быть отожжвна при температуре < 250°С.
(С разрешения фирмы Vacuum Generators Ltd., Hastings.)1 - азимутальное вращение; 2 - аксиальное вращение; 3 - перемеще¬
ние по оси Z; 4 - фланцы типа мини-конфлэт; 5 - полный стержень; 6 -
толкатель для вращения вокруг второй оси; 7 - перемещение по оси X;8 - перемещение по оси У; 9 - механизм изменения наклона.ально, все фирмы выпускают манипуляторы, обеспечивающие точно воспроиз¬
водимые перемещения. Пример такого ман/шулятора приведен на рис. 2.8. Ли¬
нейные перемещения осуществляются с помощью очень гибкого сварного силь-
фона, длина которого может существенно увеличиваться в стандартном прибо¬
ре до 50 мм, если требуется дополнительное перемещение по оси Z. Вращение
вокруг оси обеспечивается движением бокового шарнира, который также при¬
водится в действие с помощью сильфона. Если необходимо азимутальное вра¬
щение, то добавляется механизм, управляющий положением троса или стерж¬
ня, проходящих внутри трубки, с помощью толкающих и тянущих усилий; трос
прикреплен к блоку, приводящему во вращение держатель образца. Применяв¬
шиеся ранее конструкции универсальных манипуляторов такого рода приходи¬
лось частично разбирать для проведения отжига, но все модели, выпускаемые
в настоящее время, не нуждаются в разборке, а точность и воспроизводимость
обеспечиваемых ими перемещений не зависят от повторяющихся отжигов при
662. Оборудованиетемпературах до 250 °С. Большинство современных манипуляторов позволяет
использовать различные приспособления для нагрева, охлаждения, азимуталь¬
ного вращения, вращения вокруг оси, смещенной относительно оси манипуля¬
тора, почти в любой комбинации, так что возможности использования их для
самых различных приложений достаточно велики.2.4. Источники2.4.1. Источники рентгеновского излученияУстройства для генерации мощных потоков рентгеновских фотонов, обла¬
дающих характеристическими энергиями, применялись задолго до появления
РФЭС в основном в приборах, использующих дифракцию рентгеновских лучей
(РД). Однако для РД обычно требуются характеристические энергии порядка
многих десятков килоэлектронвольт, а, как видно из описания РФЭС в гл. 3,
исследование поверхности с помощью РФЭС основано на использовании мягко¬
го рентгеновского излучения с характеристической энергией порядка несколь¬
ких килоэлектронвольт. Кроме того, источники, используемые для РД, обыч¬
но устроены следующим образом: электроны, вылетающие с термоэлектрон¬
ного катода, бомбардируют анод; при этом катод находится под высоким от¬
рицательным потенциалом, а анод и другие окружающие поверхности заземле¬
ны. Таким образом, электроны их также бомбардируют. Так как в случае
РФЭС фотоны должны перейти из вакуумированного источника в аналитиче¬
скую камеру через тонкостенное окно, преграждающее путь электронам и
возможным загрязнениям, то при применении рентгеновского источника обыч¬
ной конструкции окно также будет облучаться электронами. Поэтому в источ¬
никах, применяемых в РФЭС, катод заземлен, а анод находится под высоким
положительным потенциалом. На рис. 2.9 показаны две наиболее часто ис¬
пользуемые конструкции.При выборе материала для анода источника необходимо в первую очередь
рассмотреть вопрос об энергетическом разрешении. Соотношение Эйнштейна,
описывающее взаимодействие фотона с электроном, находящимся на внутрен¬
нем атомном уровне,(2.1)где Е - кинетическая энергия испущенного фотоэлектрона; hv - характе-
ристическая энергия падающего фотона; Ев - энергия связи электрона, на¬
ходящегося на внутреннем атомном уровне; <р - работа выхода, показывает,
что ширина линии Еке зависит, в частности, от ширины линии hv. (Ширина
линии в данном случае означает полную ширину контура возбужденной рент¬
геновской линии или излученной фотоэлектронной линии, измеренную на поло-
Рис. 2.9. a - источник мягкого рентгеновского излучения с одним катодом из маг¬
ния или алюминия, напыленным в виде толстой пленки на плоскую охлаж¬
даемую водой медную пластину. Анод окружен цилиндрическим фокусирую¬
щим экраном, находящимся под потенциалом катода. Корпус служит ради¬
ационным экраном, и в нем имеется тонкое алюминиевое окно, которое долж¬
но находиться между анодом и образцом. ( С разрешения фирмы РегИп-Е1-
mer, Physical Electronics Division.)б - источник мягкого рентгеновского излучения с двойным анодом, по¬
зволяющим использовать К а -излучение магния или алюминия; переход от
одного анода к другому осуществляется с помощью внешнего переключателя.
Анод имеет острый конец, образованный двумя наклонными плоскостями, на
одну из которых напылена пленка магния, а на другую — алюминия. Перед
каждой плоскостью анода расположен полукруглый катод. Фокусирующие ус¬
тройства такие же, как и в случае источника с одним анодом на рис. а. (По
Барри И Стриту [ 18] С разрешения Institute of Physics.)1 - алюминиевое окно; 2 - внешний экран; 3 - магниевый или алюминие¬
вый анод; 4 - катод; 5 - фокусирующий и супрессорный экраны; 6,7 - во¬
да для охлаждения; 8 - фокусирующие экраны; 9 - второй катод; 10— вто¬
рая анодная плоскость; 11 — алюминиевое окно; 12 - первая анодная плос¬
кость; 13 — первый катод.
682. Оборудованиевине высоты контура.) Так как в РФЭС мы непрерывно получаем химическую
информацию при детальном анализе индивидуальных спектров элементов, то
наиболее достоверную информацию можно получить, работая при лучшем раз¬
решении по энергии, обеспечивающем достижение необходимой чувствитель¬
ности, т. е. определенное отношение сигнала к шуму. На практике это озна¬
чает, что во время записи спектра разрешение по энергии должно быть поряд¬
ка 1,0 - 2,0 эВ, в основном ближе к нижнему, чем к верхнему пределу. Ясно,
что во избежание ограничения достижимой разрешающей способности шири¬
ной линии рентгеновского источника необходимо изготавливать анод из ма¬
териала, у которого ширина линии меньше 1 эВ. В табл. 2.1 приведены энер¬
гии и ширины различных характеристических линий элементов, которые мож¬
но было бы использовать в качестве анодов рентгеновских источников по дру¬
гим параметрам, например по их устойчивости к продолжительной электрон¬
ной бомбардировке. Видно, что на самом деле лишь немногие элементы об¬
ладают достаточно узкими характеристическими линиями. М£- линии иттрия
и циркония узки и успешно используются в некоторых случаях, но их энергии
слишком малы, для того чтобы их можно было применять в общем случае,
так как число фотоэлектронных линий, которые их кванты могут возбудить,
недостаточно для надежного анализа. Среди других материалов только Ка-ли¬
нии магния, алюминия и кремния удовлетворяют приведенным требованиям.
Хотя кремний иногда применяется [19], но, поскольку он является неметал¬
лом, его теплопроводность мала и его трудно использовать в качестве ано-Таблица 2.1. Энергии и ширины некоторых
линий мягкого характеристичес¬
кого рентгеновского излученияЛинияЭнергия ли¬Ширина линии,нии, эВэВYMC132,30,47Zr Л!?151,40,77NbMC171,41,21Mo MC192,31,53Ti La395,33,0Cr La572,83,0Ni La851,52,5Cu La929,73,8MgKa1253,60,7Al Ka1486,60,85Si Ka1739,51,0У La1922,61,5Zr La2042,41,7T\Ka4510,02,0Cr Ka5417,02,1Cu Ka8048,02,6
Дж. К. Ривьер69да.. Таким образом, остаются Ка-линии алюминия и магния, и на самом деле
именно эти два элемента повсеместно используются в РФЭС.Хотя энергии характеристических Кос-линий магния и алюминия порядка
1250-1500 эВ, для эффективной генерации рентгеновского излучения не¬
обходимо использовать электроны с энергией на порядок выше в соответст¬
вии с зависимостью эмитируемого потока рентгеновских квантов от энер¬
гии бомбардирующих электронов [20], приведенной на рис. 2.10. Во всех ком¬
мерческих спектрометрах максимальный ускоряющий потенциал равен 15 кВ,
что вполне достаточно. Для улучшения чувствительности при данном разре¬
шении по энергии необходимо также использовать такой большой ток бомбар¬
дирующих электронов, какой только сможет выдержать источник, так как по¬
ток фотонов будет прямо пропорционален этому току. Однако этому препят¬
ствует требование, предъявляемое к источнику рентгеновских фотонов, обу¬
словленное тем, что размеры источника должны быть как можно меньше, по¬
скольку он должен быть расположен как можно ближе к образцу. Поток, об¬
лучающий образец, конечно, изменяется обратно пропорционально квадратуРис. 2.10. Зависимость эффективности возбуждения Ха-линий характеристического
излучения А1 и С от энергии бомбардирующих электронов. (По Долби
[ 20] С разрешения Institute of Physics.)1 — углерод; 2 — алюминий.
702. Оборудованиерасстояния между анодом и поверхностью образца. Все эти требования неиз¬
бежно приводят конструкторов к конфигурации типа показанной на рис. 2.9.
Необходимость создания компактного источника приводит к тому, что макси¬
мальная рассеиваемая мощность достигает 1 кВт при условии, что обеспечи¬
вается соответствующее водяное охлаждение. Интенсивное водяное охлажде¬
ние анода необходимо, чтобы он не расплавился, поэтому анод должен обла¬
дать хорошей теплопроводностью, т. е. он сам и подводящие водяные трубы
должны быть выполнены из меди. Таким образом, анод, т. е. сама эмитирую¬
щая поверхность, обычно представляет собой пленку толщиной порядка 10 мкм,
напыленную на медную пластину. Такая толщина пленки является компромис¬
сной: она достаточно велика, чтобы исключить излучение La-линии меди, и
достаточно мала для обеспечения нужного теплоотвода. В большинстве бло¬
ков питания таких источников электронный ток задается в виде набора фикси¬
рованных значений, например 5, 10, 20 мА и т. д.; конкретные значения зави¬
сят от фирмы-изготовителя. С другой стороны, ускоряющее напряжение мо¬
жет быть любым от 0 до 15 кВ, хотя обычно источник становится нестабиль¬
ным при напряжении ниже 2 кВ.Наиболее доступны из выпускаемых источники типа изображенного на
рис. 2.9,6. В нем имеются два катода для электронной бомбардировки и две
анодные поверхности, одна - магниевая, а другая - алюминиевая. Следова¬
тельно, с помощью простого переключения извне можно перейти от излучения
MgKa к излучению МКа за несколько секунд. Желательно иметь два ано¬
да по следующим причинам. Во-первых, эти две Ka-линии имеют различ¬
ную ширину, что видно из табл. 2.1, и, хотя А1£а используется несколь¬
ко чаще, чем MgKa, бывает, что необходимо лучшее энергетическое разреше¬
ние, которое можно получить с помощью линии магния. Во-вторых, в любом
спектре, возбуждаемом рентгеновскими лучами, помимо фотоэлектронных пи¬
ков, появляются оже-пики и возможна интерференция. Однако, поскольку энер¬
гии фотоэлектронов прямо связаны с энергиями падающих фотонов, в то вре¬
мя как энергии оже-электронов фиксированы, изменение энергии рентгенов¬
ского источника даст возможность разделить интерферирующие пики. В осо¬
бых случаях приготавливаются другие комбинации анодных материалов, часто
используются Mg/Zr и Al/Zr; La-линия циркония полезна [21], когда требует¬
ся увеличение чувствительности для обнаружения алюминия и кремния.Как уже упоминалось, необходимо использовать тонкостенное окно меж¬
ду анодом и образцом, чтобы экранировать последний от случайных электро¬
нов, от нагрева и от всех возможных загрязнений, возникающих при работе
источника. Для окна следует выбирать материал, который хорошо пропускает
используемое рентгеновское излучение, так, для MgKa и А\Ка удобно ис¬
пользовать алюминиевую фольгу толщиной приблизительно 2 мкм. При такой
Дж. К. Ривьер71толщине излучение MgKa ослабляется на 24%, а излучение А1£ос - на 15%.
При использовании других возбуждающих излучений следует заменить алюми¬
ниевое окно каким-либо другим. Например, для излучения М£-линии циркония с
энергией 151,4 эВ алюминий непрозрачен и вместо него следует использовать
бериллий или углеродную пленку.До сих пор при описании источников рентгеновского излучения для РФЭС
речь в основном шла о "естественном” или, точнее, немонохроматическом
излучении. Следует помнить, что спектр излучения любого материала очень
сложен и содержит широкий непрерывный фон (называемый тормозным излу¬
чением), на который накладываются более или менее узкие характеристиче¬
ские линии. В качестве примера на рис. 2.11 приведен спектр алюминия. Ин¬
тенсивность тормозного излучения является функцией энергии бомбардирую¬
щих электронов и после прохождения через окно ее максимум будет находить¬
ся между 20 и 40% значения этой энергии в зависимости от толщины окна;
непрерывный спектр простирается в сторону больших и меньших энергий от
основной характеристической линии и, следовательно, будет полезен для воз¬
буждения оже-переходов с атомных уровней, расположенных слишком глубоко,
чтобы их можно было ионизировать непосредственно характеристическим из¬
лучением. Сложность спектра обусловлена эмиссией сателлитов, связанных с
основными пиками, а также мультиплетностью линий при увеличении атомно¬
го номера, что обсуждается в гл. 3.2.4.1.1. Монохроматизация рентгеновского излученияУдаление сателлитов, улучшение отношения сигнала к шуму исключени¬
ем тормозного излучения и выделение одной линии из неразрешенного дубле¬
та могут быть достигнуты путем монохроматизации рентгеновского излуче¬
ния. Для этого имеется несколько методов, но все они зависят от дисперсии
рентгеновских лучей при дифракции на кристалле, которая описывается из¬
вестным соотношением Брэгга:7iA = 2dsin0, (2.2)где п - порядок дифракции; Л - длина волны рентгеновского излучения; d -
постоянная кристаллической решетки; 0 — брэгговский угол.Для первого порядка дифракции fCa-линии А1, Л = 0,83 нм, очень удобен
кристалл кварца, так как его постоянная решетки в плоскости 1010 равна
0,425 нм и, следовательно брэгговский угол равен 78,5°. Кварц обладает мно¬
гими достоинствами: можно получать идеальные монокристаллы кварца очень
больших размеров, которые легко изгибать и шлифовать и которые можно от¬
жигать при высоких температурах, не вызывая их разрушения или искажения
структуры.
722. ОборудованиеРис. 2.11. Спектр эмиссии рентгеновского излучения, эмитируемого алюминиевой
мишенью при бомбардировке электронами с энергией 15 кэВ, записанный
литиевым детектором через бериллиевое окно толщиной 7,5 мкм. На кри-
вой а с линейной шкалой интенсивности практически ничего не видно» кро¬
ме интенсивной характеристической Ка-линии. Заметим, что за счет энер¬
гетического уширения линии твердотельным детектором пик уменьшает¬
ся приблизительно в 100 раз. Кривая б - та же, что и а, нов логарифми¬
ческом масштабе, благодаря которому более ясно виден широкий фон тор¬
мозного излучения, простирающийся до значительно более высоких энер¬
гий, чем характеристическая линия. Интенсивность фона при низких энер¬
гиях ослабляется вследствие поглощения в бериллиевом окне. (Фирма
Materials Development Division, Harwell.)Принцип, лежащий в основе метода монохроматизации, широко используе¬
мого в коммерческом оборудовании, и носящий название тонкой фокусировки
[22], иллюстрируется на рис. 2.12. Кристалл кварца помещают на поверхность
сферы Роуланда,или фокусирующей сферы, при этом его аккуратно изгибают
по форме сферы, т. е. искривляют по двум направлениям. При этом анод, об¬
лучаемый сфокусированным пучком электронов, также лежит на сфере; Ка-
излучение диспергируется благодаря дифракции на кристалле и фокусируется
Дж. К. Ривьер73Рис. 2.12. Принципиальная схема монохроматизации рентгеновского излучения. Вы¬
деление одной длины волны достигается благодаря дифракции на кристал¬
ле с подходящей постоянной решетки. Для Ха-линии А1 обычно использу¬
ют кристалл кварца. Кристалл кварца помещают на поверхность сферы
Роуланда, предварительно придав ему соответствующую кривизну, источ¬
ник рентгеновских лучей находится в другой точке той же сферы. Фотоны
исслеадемой длины волны будут фокусироваться в третьей точке этой сферы,
где расположен образец. Для Ка * -линии А1 угол между падающим и ди¬
фрагированным пучками равен 23 . Обычно диаметр сферы равен 0,5 м.(По Келли и Тайлеру [22J.)1 ~ дисплей; 2 - многоканальный анализатор; 3 - многоканальный
детектор; 4 - электронный спектрометр; 5 - линза; 6 - сфера Роуланда;7 - диспергирующий кристалл; 8 - фотоэлектроны; 9 - образец; 10 - ис¬
точник рентгеновского излучения (анод).в другом месте сферы Роуланда, где расположен образец. Если необходимо
выделить Ка 1 -компоненту Ка-линии А1, то угол между падающим и дифраги¬
рованным пучками должен быть равен 23°. В зависимости от выбранного диа¬
метра сферы Роуланда дисперсия на окружности будет изменяться* Для обыч¬
ного монохроматора диаметром 0,5 м дисперсия составляет 1,6 мм/эВ, для
сферы диаметром 1 м - 3,2 мм/эВ. Следовательно, для получения линии с
шириной меньше естественной ширины Ка-излучения, например ~ 0,4 эВ, не¬
обходима такая фокусировка электронного пучка на аноде, чтобы размер об¬
лучаемой площади образца в плоскости дисперсии был меньше ~ 0,6 мм, ес¬
ли используется полуметровый монохроматор.Так как монохроматор обеспечивает попадание на образец лишь выделен¬
ной малой доли полного Ка-излучения, получаемый поток рентгеновских фо¬
тонов при одинаковой диссипируемой в аноде мощности будет значительно
меньше, чем поток от немонохроматизованного источника * Например, для
алюминиевого анода при использовании метрового монохроматора поток умень¬
шится приблизительно в 40 раз по сравнению со стандартным источником.
Таким образом, скорость счета сильно уменьшится, но при этом возрастет
742. Оборудованиедетектирующая способность, т. е. отношение сигнала к шуму, так как фон в
значительной мере удален, сателлиты отсутствуют, и мы получаем ’’чистый"
спектр и великолепное разрешение. Многие фирмы производят монохромато¬
ры в качестве дополнительных устройств, но они всегда громоздки и дороги.
Совсем недавно одна фирма описала монохроматизацию Ка-излучения Ag
[23].2.4.1.2. Синхротронное излучениеВсе описанные до сих пор источники мягкого рентгеновского излучения
генерировали дискретные линии. Эти источники, конечно, полезны, но имеют
принципиальные недостатки при использовании их для РФЭС. Один из самых
очевидных недостатков заключается в том, что сечение фотоионизации внут¬
реннего атомного уровня при данной энергии излучения источника может быть
близко к максимуму для одной группы линий и близко к минимуму для другой.
Чтобы изменить сечение при обычном оборудовании, необходимо взять источ¬
ник с другой фиксированной энергией, однако вовсе не обязательно, что при
этом удастся подобрать именно необходимую энергию, и, во всяком случае,
число имеющихся источников линейчатого излучения будет сильно ограничено.
В идеальном случае желательно иметь перестраиваемый источник с непрерыв¬
ным спектром и высокой интенсивностью при любой требуемой энергии. Син¬
хротронное излучение является хорошим приближением к такому идеалу.Известно, что заряженное тело излучает, двигаясь с ускорением; такой
процесс особенно эффективен в случае электрона из-за его малой массы.Этот эффект используется в синхротроне, где электроны ускоряются, двига¬
ясь практически по окружностям в круглых торах (или "дуантах") под воз¬
действием пульсирующих магнитных полей. Так как электроны ускоряются,
двигаясь по круговым траекториям, они излучают непрерывный спектр, мак¬
симум интенсивности которого прямо пропорционален радиусу кривизны и об¬
ратно пропорционален кубу энергии электронов. Электроны разгоняются до
релятивистских скоростей и тогда излучение концентрируется в очень узком
конусе, направленном по касательной к электронным орбитам, с угловым
расхождением порядка 0,1 мрад (0,006°). Это очень удобно для выделения нуж¬
ной энергии при монохроматизации. В общем случае форма спектра, излучае¬
мого электронами, движущимися по круговым орбитам [24], приведена на
рис. 2.13. Типичная критическая длина волны Л , соответствующая максиму¬
му интенсивности, порядка 0,1 - 0,4 нм, и энергия максимума находится меж¬
ду 1 и 10 кэВ. Из рис. 2.13 видно, что с увеличением длины волны, т. е. с
уменьшением энергии интенсивность падает довольно медленно, так что при
энергии порядка нескольких десятков электронвольт интенсивность спектра
достаточно велика, чтобы излучение можно было использовать.
Дж. К. Ривьер75Рис. 2.13. Форма спектра излучения электрона, движущегося по криволинейной орби¬
те. Поток фотонов, представленных по оси ординат, зависит только от
энергии электронов и их тока, в то время как ось абсцисс определяется
так называемой критической длиной волны А . Если требуется, чтобы мак¬
симум интенсивности лежалопри энергии порядка 1000 эВ, то величина Ас
должна составлять около 1 А. Для получения таких значений электроны,
движущиеся по орбитам, должны разгоняться до нескольких гигаэлектрон-
вольт, а радиусы их орбит должны составлять несколько метров. (По Фар-
жу и Дьюку [ 24].)Типичный источник синхротронного излучения обладает следующими па¬
раметрами: его радиус составляет несколько метров, энергия электронного
пучка порядка гигаватт, а ток достигает 1 А, поэтому такие источники невоз¬
можно создать и использовать в одной лаборатории. Они так велики, что мо¬
гут принадлежать только государству, поэтому пользователям приходится
либо привозить свои спектрометры, либо использовать те, которые имеются
при синхротроне.Несмотря на то что излучаемый спектр по своей природе непрерывен,
синхротрон еще недостаточно непрерывно перестраивается, так как выделе¬
ние нужной энергии или интервала энергий производится монохроматорами.
Разработанные до сих пор монохроматоры работают при непрерывной пере¬
стройке в интервале энергий от 20 до 500 эВ, но еще остается большой энер¬
гетический промежуток до высоких энергий, которые дают источники линей¬
чатого спектра. Ожидается, что этот промежуток будет заполнен, но при раз¬
работке следует учесть особенности работы в СВВ и необходимость исполь¬
зования отжига для уменьшения загрязнений отражающих поверхностей мо¬
нохроматора. Необходимость увеличить интервал энергий до 1000 эВ и больше
связана с тем фактом, что поток рентгеновских фотонов от обычного синхро¬
трона в этой части спектра по крайней мере на два порядка больше, чем по¬
ток в источнике Ка-излучения А1. Даже при потерях на монохроматоре поток
будет интенсивнее того, который мы имеем сейчас.
762. Оборудование2.4.2. Источники электроновОбычно в ЭОС единственным назначением электронов первичного пучка
является ионизация внутренних уровней, чтобы инициировать оже-переход.Для этой цели энергия пучка несущественна1*. Если же необходимо исследо¬
вать спектр электронных потерь, то ширина линии по аналогии с рентгенов¬
скими источниками должна быть минимизирована. Если не выделять опреде¬
ленный интервал энергий, то ширина будет определяться тепловым разбро¬
сом в соответствии с распределением Больцмана и равна ~0,3 - 0,5 эВ2>.
Такая ширина неприемлема в случае спектроскопии характеристических по¬
терь энергии электронов (СХПЭЭ), в которой изучаются колебательные спект¬
ры, но обсуждение СХПЭЭ выходит за рамки нашего рассмотрения. В случае
ЭОС нет необходимости предпринимать какие-либо усилия для уменьшения
ширины энергетического распределения первичного пучка,В ЭОС используются два типа электронных источников: термоэмиссион¬
ные и автоэмиссионные, причем до сих пор чаще встречаются термоэмиссион¬
ные. Все материалы характеризуются работой выхода, которая является прос¬
то энергией, необходимой для перемещения электрона из материала на беско¬
нечность. Работа выхода обычно [25, 26] равна нескольким электронвольтам
и, таким образом, представляет собой барьер на поверхности, препятствую¬
щий выходу электронов; при комнатной температуре и в отсутствие электри¬
ческого поля электроны не могут покинуть металл и эмиссии нет. Два типа
источников используют различные физические методы для создания ситуации,
когда значительное число электронов может преодолеть барьер и таким обра¬
зом создать эмиссию электронов из материала. В случае термоэмиссии ма¬
териал просто нагревается до температуры, достаточно высокой для того,
чтобы сообщить части электронов дополнительную энергию для прохождения
над барьером в вакуум, т. е. чем выше температура, тем больше термоэмис¬
сионный ток. Эмиссия описывается уравнением Ричардсона(2.3)где ] - плотность тока; А ~ постоянная Ричардсона, в идеальном случае
равная 120 А/(см2 • град2), но изменяющаяся в зависимости от рода матери-Точнее важна монокинетичность электронного пучка, тогда как энергия долж¬
на быть порядка энергии, соответствующей максимуму сечения ионизации данного
внутреннего уровня, чтобы обеспечить достаточно интенсивную оже-эмиссию. -
Прим. ред.^ На самом деле полный разброс будет определяться конфигурацией полей в
системе ускорения электронов, а не только тепловым разбросом электронов, эми¬
тируемых термоэлектронным катодом. — Прим. ред.
Дж. К. Ривьер77ала; Т - коэффициент отражения падающих электронов в отсутствие поля от
потенциального барьера на границе материал - вакуум; Т - абсолютная тем¬
пература; q> - работа выхода.Автоэмиссия имеет совершенно иной механизм. По существу электронам
в материале не сообщается дополнительная энергия, но высота барьера, соз¬
даваемого наличием работы выхода, уменьшается, так что электроны могут
выйти из металла при комнатной температуре. Поскольку для большинства
металлов работа выхода лежит между 4 и 5 эВ [25, 26] и протяженность барь¬
ера порядка нескольких нанометров, можно рассчитать, что поле на поверх¬
ности очень велико. Попытка исключить его полностью путем создания рав¬
ного по величине и обратного по направлению практически неосуществима, но
этого и не требуется, так как даже создание небольшого по величине поля
существенно уменьшит барьер и создаст возможность эмиссии. Это происхо¬
дит потому, что электроны имеют конечную вероятность ’’туннелировать"
сквозь барьер; вероятность слишком мала для измерения при комнатной тем¬
пературе и в отсутствие поля. Но, когда высота и форма барьера заметно
изменяются при приложении сильного электрического поля, эта вероятность
становится заметной. Для получения требуемого поля из соответствующего
материала изготавливается острие с радиусом закругления на конце ~50 нм
и между ним и вытягивающим электродомк прикладывается положительное
электростатическое поле порядка многих киловольт11. Процесс называется
автоэмиссией и описывается уравнением Фаулера - НордгеймаА/см2, (2.4)где / - плотность тока; Е - напряженность поля; е(х) - эллиптическая функ¬
ция Нордгейма; х = 3,62 • ТО"*4#1 /2/<р. Из выражения (2.4) легко понять, что
плотность тока автоэмиссии сильно зависит от значения работы выхода эми¬
тирующей поверхности, так что обычно в качестве эмиттера используют игло¬
образный монокристалл, ориентированный таким образом, чтобы плоскость с
малой работой выхода находилась на вершине иглы. Поскольку адсорбция ос¬
таточных газов на чистой металлической поверхности почти всегда увеличи¬
вает работу выхода, необходимо, если плотность тока в результате не умень¬
шится, поддерживать очень низкое давление вблизи эмиттера. Перед включе¬
нием автоэмиссионного источника адсорбированные газы удаляются ’’вспыш¬
кой”, т. е. быстрым увеличением температуры эмиттера до очень высоких1) В настоящее время создан автоэмиссионный источник электронов, позволяю¬щий получать достаточно интенсивные высокостабильные пучки автоэмиссионныхэлектронов при приложении напряжения, не превышающего 100- 300 В,и работающе¬го в относительно низком вакууме (10~6-10""7 мм рт. ст.) [49]. - Прим. ред.
782. Оборудованиезначений. Из-за необходимости вспышки, а также из-за того, что материал
эмиттера должен выдерживать сильное электростатическое поле, не разруша¬
ясь, всегда используется вольфрам, ориентированный так, чтобы плоскость
с малой работой выхода (310) была параллельна вершине.И термоэмиссионные, и автоэмиссионные источники обладают существен¬
ными достоинствами и недостатками. Благодаря тому, что в автоэмиссион-
ном источнике излучающая поверхность мала и эмитируемые электроны
сконцентрированы в малом угле, плотность тока на единицу телесного угла
значительно выше, чем в случае термоэмиссионного источника, и по этой при¬
чине автоэмиссионные источники известны как источники с большой яркостью.
Достигаемые значения [27] превышают 107 А/(см2 • ср), в то время как тер¬
моэмиссионный вольфрамовый катод обычно дает значения порядка
Ю4 А/(см2 • ср). При использовании материала с меньшей работой выхода
можно получить большую яркость [27] при термоэмиссии; например, дляLa В плотность тока достигает Ю6 А/(см2 • ср), однако следует учесть,6что изготовление как автоэмиссионных источников, так и источников из
La В6 сопряжено с большими трудностями и, кроме того, они не надежны в
работе1 ]. Хотя обычный вольфрамовый термоэмиссионный катод имеет мень¬
шую яркость, его относительно легко изготовить и заменить, его эмиссион¬
ные свойства полностью воспроизводимы, его можно использовать в широ¬
ком диапазоне значений тока эмиссии и он прочен. С развитием технологии
предпочтение будет все более отдаваться источникам с высокой яркостью,
но в настоящее время в подавляющем большинстве электронных источников
используются вольфрамовые катоды, работающие в режиме термоэлектрон¬
ной эмиссии.Основным направлением развития электронных источников для ЭОС всег¬
да было и остается обеспечение наименьшей облучаемой электронной пушкой
поверхности образца (или получение пятна минимальных размеров); при
этом электронная пушка должна соответствовать требованиям, предъявляе¬
мым СВВ-системами. До недавнего времени последнее требование означало,
что электронная оптика пушки должна быть электростатической, поскольку
сложно разработать и изготовить такие полюсные наконечники и кольца для
электромагнитной оптики, чтобы они выдерживали отжиг СВВ-системы. Одна¬
ко эти трудности были преодолены, а в последние несколько лет появилось
оборудование для ЭОС, обеспечивающее высокое пространственное разреше¬
ние, как будет видно ниже.1) Монокристаллические острийные термоэмиттеры из LaB6 работают вполне на¬дежно и в течение ряда лет используются в оже-спектрометрии фирмой Perkin Elmer. -Прим. ред.
Дж. К. Ривьер79В пушках с электростатической фокусировкой, использующих вольфрамо¬
вый катод, размер пятна уменьшался от 400 мкм при ^ 20 мкА и 2,5 кэВ, по¬
лучаемого в случае несколько переделанной осциллографической электрон¬
ной пушки, до 40 мкм при ~ 5 мкА и 5 кэВ в пушках, использовавшихся в сере¬
дине 70*х годов, затем до 5 мкм при ~!0~7 А и 5 кэВ в конце 70-х и 0,5 мкм
при ~10~8А и 10 кэВ в начале 80-х годов. При очень тщательной юстировке
катода и оптики, обеспечив изготовление апертур с необходимой точностью и
исключив влияние рассеянных магнитных полей, в современных электростати¬
ческих пушках можно достигнуть размера пятна ^ 0,2 мкм. Однако для боль¬
шинства приводимых исследований чаще всего используется пятно диаметром
0,5 мкм. По-видимому, в случае простых электростатических систем, работаю¬
щих при напряжениях, при которых не требуются громоздкие изоляторы или
дорогостоящие высоковольтные источники питания, существующие в настоя¬
щее время конструкции электронных пушек являются практически оптималь¬
ными.Один из коммерческих вариантов такой пушки схематически изображен
на рис. 2.14. Конструкция, на которой смонтированы вольфрамовый V-образ-
ный катод и первый электрод, цилиндр Венельта, укреплена на гибком силь-
фоне и снабжена винтами для юстировки катода и апертуры цилиндра Венель¬
та относительно остальной оптики. Юстировка самого катода по отношению
к цилиндру Венельта осуществляется с помощью подвески. Однако обеспечить
полную воспроизводимость установки такого устройства трудно, и достиже¬
ние оптимальной юстировки требует опыта.Фокусировка и форма электронного пучка определяются двумя конденсор-
ными и одной объективной линзами. Астигматизм (несовпадение фокуса в на¬
правлениях X и У) можно корректировать набором стигматоров, устанавливае¬
мых перед объективной линзой. Наконец, сканирование пучка осуществляется
X- и У-отклоняющими пластинами на выходе из пушки. Пушка откачивается че¬
рез выходную апертуру, но так как она мала и, следовательно, скорость от¬
качки пушки, и особенно области вблизи катода, низка, то необходимо позабо¬
титься об обезгаживании нового катода, чтобы вблизи него не возникла об¬
ласть повышенного давления.Как отмечалось выше, для достижения минимальных размеров пятна сле¬
дует перейти от электростатической к электромагнитной оптике. Необходимо
также работать при более высоких энергиях падающего пучка порядка
10-30 кэВ, так как из электронной оптики известно, что размеры пятна
уменьшаются, если увеличивается энергия электронного пучка. Поэтому даль¬
нейшее уменьшение размеров пятна от ~ 0,5 мкм связано с усложнением кон¬
струкции и увеличением стоимости электронной пушки. Нужно быть очень
уверенным, что дополнительная информация, которая может быть получена,
стоит затрачиваемых усилий!
802. ОборудованиеРис. 2.14. Зйектронная пушка с электростатической фокусировкой, создающая пучки
электронов с энергией до 10 кэВ. При токе пучка 10“® - 10""8 а оптималь¬
ный размер пучка на образце составляет приблизительно 0,5 мкм. Катод,
представляющий собой вольфрамовую шпильку, можно юстировать с цилин¬
дром Венельта при помощи приспособления для юстировки, в то время как
юстировка относительно оптической оси пушки достигается установкой ка¬
тодного узла с помощью котировочных винтов и сильфон а. (С разрешения
фирмы Vacuum Generators Scientific Ltd., East Grinstead.)1 - вводы катода и цилиндра Венельта; 2 - крепежные винты; 3- ка¬
тод; 4 - конденсорные линзы; 5 - полюсные наконечники стигматоров;6 - линза объектива; 7 - отклоняющие пластины; 8 - вводы линз; 9 -
цилиндр Венельта; 10- котировочные винты.Как и в случае электростатических пушек, появились различные вариан¬
ты конструкций электромагнитных пушек, которые, конечно, в основном схо¬
жи. На рис. 2.15 приведен пример конструкции электромагнитной пушки. В
пушке имеются две электромагнитные конденсорные линзы для "сжатия"
электронного пучка до малого диаметра и электромагнитная объективная лин¬
за для фокусировки. Обмотки линз полностью закрыты; таким образом, об¬
легчаются проблемы, связанные с отжигом, хотя наличие линз ограничивает
температуру отжига до 160 ЧГ. Крепление катода такое же, как в электроста-
Дж. К. Ривьер 81Рис. 2.15. Электронная пушка с электромагнитной фокусировкой, создающая пучок
электронов с энергией до 30 кэВ. При токе пучка порядка нескольких на¬
ноампер оптимальный размер пятна на образце составит приблизительно
50 нм. Так же,как в пушке, показанной на рис. 2.14, катодный узел юсти¬
руется относительно оси пушки с помощью котировочных винтов и силь¬
фон а. Поскольку объем этой пушки значительно больше, чем пушки на
рис. 2.14, требуется ввод дифференциальной откачки. Полюсные нако*
нечники и обмотки можно отжигать до температуры 160°С. (С разреше¬
ния фирмы Vacuum Generators Scientific Ltd., East Grinstead.)1 - апертуры; 2 - железные полюсные наконечники; 3 - устройство
для юстировки конденсорных линз; 4 — апертура экстрактора; 5- крепеж¬
ные винты; 6 - образец; 7 - катушки сканирования и стигматоры; 8 -
объективная линза; 9 - конденсорная линза; Ю - ответвление для диффе¬
ренциальной откачки пушки; 11 - изолятор на 30 кВ; 12— котировочные
винты; 13 - вводы катода и цилиндра Венельта.тической пушке, и систему можно юстировать с помощью юстирующих вин¬
тов, смещающих сильфон. Поскольку в настоящее время максимальное рабо¬
чее напряжение составляет 30 кВ, вся система довольно громоздка из-за не¬
обходимости использования дополнительной изоляции. Дальнейшая юстировка
электронного пучка вдоль оптической оси пушки может быть произведена ре¬
гулировкой положения самих конденсорных линз с помощью юстирующих вин¬
тов. Корректировка астигматизма и сканирование электронного пучка осу¬
ществляются до прохождения электронов через выходную апертуру, и в этом
822. Оборудованиеслучае стигматор и катушки сканирования встраиваются в объективную лин¬
зу, которая имеет такую форму, чтобы обеспечивать доступ к образцу. Вме¬
сто фиксированной выходной диафрагмы здесь используется скользящая пла¬
стина с тремя высверленными диафрагмами различных размеров, любую из
которых можно установить перемещением пластины в* соответствующее по¬
ложение. Объем пушки необходимо откачивать отдельно через боковое отвер¬
стие ввиду очень низкой скорости откачки через любую из выходных диафрагм.При длительном использовании возможность получения и поддержания в
течение нужного времени пятна размером порядка нескольких десятков нано¬
метров зависит не столько от базовой конструкции (она практически не меня¬
ется), сколько от точности изготовления линз и особенно от механической
обработки и юстировки железных полюсных наконечников. Важно также, что¬
бы конструкция пушки обладала высокой механической стабильностью и что¬
бы пушка и образец были экранированы от рассеянных переменных магнитных
полей. Различия в исполнении между пушками различных фирм будут в основ¬
ном зависеть от этих факторов, а не от возможности создания совершенно
новой конструкции.В автоэмиссионных источниках также используют электромагнитную
электронную оптику типа изображенной на рис. 2.15, однако редко. Сочетание
автоэмиссии при высоких напряжениях (20-60 кэВ) и электромагнитной оп¬
тики приводит к созданию минимального на сегодняшний день размера пятна
порядка 20 нм, но пока в ЭОС используется мало подобных спектрометров.
Чаще в автоэмиссионных источниках применяется электростатическая оптика;
при этом получаемые размеры пятна сравнимы с размерами пятна термоэмис¬
сионного источника с электромагнитной оптикой. Схема электронной пушкиРис. 2.16. Схема электронной пушки с автоэмиссионным источником электронов. Эми¬
тирующий наконечник Т выполнен из монокристаллической
вольфрамовой проволочки и ориентирован вдоль оси 1310]. F — вспомога¬
тельный катод, испопьзуемый для обезгаживания анода А1, расположенно¬
го вблизи F; А2 — второй анод, SD — восьмиполюсный стигматор-дефпектор.
При энергии пучка 21 кэВ, токе 10“® А и расстоянии от SD 55 мм пушка обео*
печивает пятно размером 50 нми (По Праттону и др. [28]) 1 - образец.
Дж. К. Ривьер83с автоэмиссионным катодом представлена на рис. 2.16 [28]; на этом же ри¬
сунке приведены размеры, показывающие, что габариты пушки невелики. Та¬
кая пушка при токе на образец 10“9 А, энергии пучка 21 кэВ и рабочем отрез¬
ке 55 мм обеспечивает размер пятна на образце ~50 нм. Отличительной чер¬
той является расположение катода F близко к первому аноду Л1, используе¬
мому для обезгаживания катода. Одним из ограничений при работе является
шум, возникающий при бомбардировке вершины автоэмиттера ионами, посту¬
пающими с поверхности анода А\ вследствие электронной бомбардировки
анода.Непосредственное сравнение трех эмиттеров, а именно вольфрамового
термоэмиттера, LaBg и вольфрамового автоэмиттера, показывает [27], что
при токах пучка порядка 10~8 А лучше использовать катод из LaBgc точки зре¬
ния получаемых размеров пятна и отношения сигнала к шуму. Однако при то¬
ках меньше 5 • 10“9 А автоэмиссионный источник превосходит два других
по обоим параметрам*).2.4.3. Ионные источникиХотя оже-эмиссию можно возбудить первичным ионным пучком и сущест¬
вует несколько исследовательских групп, применяющих метод ионной ЭОС, в
этом разделе будут обсуждаться устройства, используемые для ионного трав¬
ления поверхностей, как это было описано в п. 2.3.3. Для очистки поверхно¬
сти и определения профилей концентрации примесей при использовании ион¬
ного травления в настоящее время разработано несколько типов источников
ионов с определенными характеристиками и режимами работы, и выбор того
или иного источника зависит частично от личных склонностей эксперимента¬
тора, частично от того, чем снабжен используемый им спектрометр.Простейший тип, очень редко используемый в настоящее время, по кон¬
струкции и режиму работы похож на обычный ионизационный манометр, схе¬
ма его приведена на рис. 2.17. Как и в манометре, горячий катод эмитиру¬
ет электроны к находящейся под положительным потенциалом сетке, где
электроны совершают несколько колебаний и затем попадают на коллектор.В систему напускается аргон до давления 10”5 - 10~4 мм рт. ст., который
ионизуется в околосеточном пространстве. В отличие от ионизационного ма-1) Применение автоэмиссионных источников позволяет создавать при использо-ованных (до 60 кВ) напряжениях пятно диаметром менее 50 А. Однако их использование длянужд оже-спектрометрии осложняется тем, что, во-первых, ряд образцов при плотностях
тока на образцах, обеспечиваемых автоэмиссионным источником, разрушается и,
во-вторых, уменьшение тока пучка, неизбежное при уменьшении размеров пятна, силь¬
но увеличивает продолжительность анализа, что в ряде случаев неприемлемо. — Прим.
ред.
842. ОборудованиеРис. 2.17. Конструкция ионной пушки, использующей геометрию ионизационного
манометра. Горячий катод F эмитирует электроны, движущиеся к
сетке G, находящейся под положительным потенциалом, где электроны
совершают несколько колебаний, прежде чем попадают на коллектор. Ато¬
мы аргона, напущенного в пушку при давлении порядка 10-5 - КГ4 мм
рт.ст., ионизуются внутри сетки. Затем положительные ионы ускоряют¬
ся к цилиндрическим электродам л, окружающим катод и сетки и обеспе¬
чивающим градиент потенциала 200 В. Ионы попадают в систему через
сетки в Л и 5, внешний экран заземлен. Апертуры сеток смещены, чтобы
избежать попадания загрязнений с катода на образец. Изменяя потенциал
катода, сетки и ускоряющего электрода относительно земли, можно полу¬
чать ионы с энергией от 200 до 500 эВ. При давлении аргона Ю*4 мм рт.ст.
плотность ионного тока на образец равна 1 мкА/см2.нометра в ионной пушке нет коллектора положительных ионов, вместо этого
положительные ионы аргона ускоряются по направлению к цилиндрическому
электроду, окружающему катод и решетку, отрицательным потенциалом по¬
рядка 200 В. Они проходят через сетчатые диафрагмы в этом электроде и во
внешнем заземленном корпусе; диафрагмы смещены относительно оси пушки
во избежание попадания возможных загрязнений с горячего катода. Катод,
сетка и ускоряющий электрод могут находиться под плавающим положитель¬
ным потенциалом относительно экрана, так что энергии ионов в пучке могут
изменяться от 200 до 500 эВ.Источник ионов типа ионизационного манометра не обладает большой
мощностью при максимальном рабочем давлении 10~4 мм рт. ст., максималь¬
ная создаваемая им плотность тока составляет 1 мкА/см2. Он обладает сле¬
дующими достоинствами: дешев, надежен и создает относительно чистый пу¬
чок ионов, так как в ионизационном промежутке образуется мало высоко-
энергетичных нейтральных атомов.Вероятно, сейчас наиболее широко распространены источники, использую¬
щие газовый разряд для ионизации газа в пушке. Разряд может быть возбуж¬
ден в виде либо пеннинговского, либо высоковольтного разряда. В первом
случае под объединенным воздействием магнитного и электростатического
полей в ограниченном пространстве, заполненном аргоном при высоком дав¬
лении, возникает разряд с холодным катодом, из которого будут вытягивать¬
ся положительные ионы. Обычная конфигурация такого источника приведена
на рис. 2J 8. Аргон вводится непосредственно в заднюю часть пушки и отту¬
да попадает в разрядную камеру, вместо того чтобы заполнять весь объем
Дж. К. Ривьер85Рис. 2.18. Конструкция ионной пушки, в которой для создания положительных ионовиспользу ется разряд Пеннинга. В этом случае аргон напускается непосред¬
ственно в пушку, а не заполняет всю систему, и так как скорость его ухо¬
да мала, то давление внутри пушки значительно выше, чем давление арго¬
на в системе. Во время работы ионной пушки давление в системе состав¬
ляет приблизительно 3-10""7 _ з*1 (Г6 мм рт.ст. К ионизационной камере,
в которой происходит разряд, приложены высокое напряжение и осевое
магнитное попе. Положительные ионы вытягиваются из разряда в область
фокусировки. Энергия ионов изменяется от 500 эВ до 10 кэВ, и пушка мо¬
жет создать ток от 10 до 100 мкА на площади диаметром 5 мм в зависи¬
мости от давления аргона в ионном источнике. (С разрешения фирмы
Vacuum Generators Ltd., East Grinstead.)1 - дополнительная труба для ограничения размера пучка диамет¬
ром 5 мм; 2 - фланец из бескислородной меди для напуска газа; 3 - эк¬
ран вводов; 4 - вводы ионного источника; 5 - ионизационная камера; 6 -
выходная апертура источника; 7 - область вытягивания ионов; 8 - элек¬
тромагнит; 9 - блок фокусировки 0 — 10 кВ; 10- фокусирующий элек¬
трод; 11 - фланец из бескислородной меди.аналитической камеры, как это делается в случае источника типа ионизацион¬
ного манометра. После вытягивания положительных ионов пучок фокусирует¬
ся в той или иной степени в зависимости от сложности используемой системы
линз. Более простые пушки не могут создать ионный пучок диаметром мень¬
ше 3 мм, более сложные могут сфокусировать пучок до 0,1 мм, если образец
находится на нужном расстоянии от выходной диафрагмы. В случае хорошо
сфокусированных пучков удобно использовать сканирование пучком по образ¬
цу, поэтому после фокусирующей системы линз ставятся X - У-отклоняю-
щие пластины. Такие пушки работают при ускоряющем напряжении от 2 до
862. Оборудование10 кВ и при давлениях в аналитической камере от 3 • 10-7 до 3 .10”6 мм рт. ст., пос¬
кольку скорость откачки разрядного громежутка через пушку мала. Ионный ток на
образец зависит и от напряжения, и от давления; например, пушка будет соз¬
давать ток 20 мкА в пятне диаметром 5 мм при ускоряющем потенциале 5 кВ
и давлении в камере 4 • 10“7 мм рт. ст. Фокусирующее напряжение около
3,6 кВ. При предельных значениях напряжения 10 кВ и давления 3 • 10“6 мм
рт. ст. ток достигает 200 мкА.Заметное увеличение плотности тока ионной пушки такого типа по срав¬
нению с пушкой типа "ионизационного манометра" сопровождается некоторы¬
ми нежелательными явлениями. Ионы, образовавшиеся внутри пушки, не толь¬
ко поступают в пучок, но и могут бомбардировать внутренние поверхности и
тогда в пушке будет постепенно накапливаться распыленный материал. Когда
количество распыленного материала, осевшего на изоляторах, достаточно
для возникновения вакуумного пробоя или дуги, работа пушки становится ненаг
дежной и невоспроизводимой. В такой ситуации не остается ничего другого,
кроме демонтирования пушки и очистки ее в соответствии с инструкцией фир¬
мы; вообще желательно иметь запасные части для замены изоляторов. Дру¬
гим недостатком является то, что пучок, выходящий из пушки, недостаточно
чист; в нем наряду с положительными ионами присутствует заметная доля
нейтральных атомов. Очевидно, что можно измерить лишь ток положитель¬
ных ионов, бомбардирующих образец, поэтому о дополнительном разрушении
поверхности нейтралами известно мало. Однако все разрядные ионные пушки
страдают этим недостатком в той или иной степени.Ионные источники, использующие высоковольтный разряд для полученил
пучка ионов, должны иметь такую геометрию, чтобы увеличивать длину про¬
бега осциллирующих электронов и, следовательно, вероятность ионизации в
пересчете на один электрон. Возможно использование различных геометрий,
но цель остается одной и той же: сформировать такое электрическое поле,
чтобы электроны находились в течение длительного времени в одной или не¬
скольких потенциальных ямах, образуемых полем. Такие источники получили
название источников с "седлообразным полем" благодаря одной из наиболее
часто используемых конфигураций поля. Принципиальная схема подобного
ионного источника представлена на рис. 2.19. Две вольфрамовые проволочки,
окруженные экранами на большей части своей длины, введены в корпус пуш¬
ки, который в данном случае имеет сферическую форму. К проволочкам при¬
ложен высокий положительный потенциал от 5 до 10 кВ, корпус пушки заземлен. Ар¬
гон напускается непосредственно в пушку до давления порядка 10-4 мм рт. ст., после
чего возникает разряд между анодом и корпусом, выполняющим роль катода.Под действием такого поля электроны движутся по траекториям, имеющим
форму восьмерки, как это показано на рис. 2.19» Ионизация аргона в основ-
Дж. К. Ривьер87Рис. 2.19. Конструкция ионной пушки, использующей высоковольтный разряд. Высо¬
кое напряжение приложено к паре вольфрамовых проволок, введенных
внутрь корпуса пушки, который в этом случае имеет сферическую форму.
Аргон напускается непосредственно в пушку через трубку, и, как было
показано при разработке, скорость откачки аргона в пушке мала, поэтому
давпение внутри корпуса пушки значительно выше, чем снаружи. Разряд
происходит между проволоками и корпусом, электронные траектории ог¬
раничены обозначенными "восьмерками", причем ионизация в основном
происходит в петлях на концах траекторий. Положительные ионы вылета¬
ют через прорезь в корпусе, лежащую в плоскости поля. Из-за конфигу¬
рации поля этот тип источника также известен как источник с "седловым
полем". При энергиях ионов порядка 5-7 кэВ такой источник способен
создать ионный пучок с током 100 — 200 мкА и размером пятна около 1 см2.
(С разрешения фирмы Ion Tech Ltd., Teddington.)1 - анод; 2 - высоковольтный ввод; 3 - катод; 4 - ионы; 5 - аргон;6 - экран; 7 - траектория электронов; 8 - пучок ионов аргона и высоко-
энергетичных нейтралов.ном происходит в петлях на краях поля. Положительные ионы вытягиваются
через прорезь в корпусе, лежащую в плоскости поля, ширина ионного пучка
равна ширине щели в плоскости рисунка; ниже и выше данной плоскости пу¬
чок существенно расширяется. Регулируя расстояние от образца до щели, мож¬
но по желанию изменять форму бомбардируемой поверхности от прямоуголь¬
ной до квадратной. Так как откачка может производиться только через про¬
резь, в системе с нужной скоростью откачки устанавливается давление по¬
рядка 10“6 - 10~5 мм рт. ст.Источники с седлообразным полем способны давать очень высокие плот¬
ности ионного тока. Даже такой небольшой источник, как показанный на
882. Оборудованиерис. 2.19, может дать пучок с плотностью тока 200 мкА/см2 при напряжении
6 — 7 кВ, в то время как большие источники обладают такой мощностью, что
их обычно используют не для ионной бомбардировки в СВВ, а для так называ¬
емого "ионного дробления". При этом, как и предполагает название, удаляет¬
ся большое количество материала., Однако, как и все другие источники, они
имеют свои достоинства и недостатки. В отличие от источников, использую¬
щих пеннинговский разряд, они нестабильны при напряжениях ниже 5 кВ и пло¬
хо управляемы, а поэтому обычно их применение ограничено областью высо¬
ких рабочих напряжений. Кроме того, как видно из рис. 2.19, положительные
ионы не только вытягиваются через щель, но и бомбардируют внутренние по¬
верхности; таким образом, распыленный материал может, выйдя через щель,
напыляться на образец. По этой причине необходимо либо изготовлять корпус
пушки из труднораспыляемого материала, например из алюминия или титана,
либо вводить внутренний лайнер из такого материала. Наконец, такие источ¬
ники создают довольно большое количество высокоэнергетичных нейтральных
атомов в пучке порядка 50% при максимальном рабочем напряжении, т0 е0 до¬
ля их оказывается выше, чем в случае использования пеннинговского разряда.Новейшим из разработанных ионных источников [29] является автоэмис-
сионный жидкометаллический источник ионов, принципиальная схема которого
изображена на рис. 2.20. Игла с радиусом закругления ~1 -10 мкм, проходя¬
щая через плотно прилегающий капилляр, другим концом погружена в резерву¬
ар с жидким металлом. Материал иглы выбран таким образом, чтобы жидкий
металл смачивал его, но не вступал в химическую реакцию. При смачивании
жидкий металл благодаря воздействию капиллярных сил движется вверх по вы¬
ступающему концу иглы к ее вершине, а любая потеря металла возмещается
за счет металла, поступающего из резервуара. Между иглой и вытягивающим
электродом, расположенным на небольшом расстоянии от конца иглы, прикла¬
дывается высокое напряжение от 4 до 10 кВ; при этом пленка жидкого метал¬
ла на конце иглы образует выступ в форме острия копья. Этот выступ обра¬
зуется благодаря равенству сил электростатического притяжения и поверх¬
ностного натяжения1). Вблизи конца острия возникает сильное электростати¬
ческое поле, которое обеспечивает формирование пучка положительно заря¬
женных ионов металла, которые вытягиваются через круглое отверстие в
экстракторе. Несмотря на то что источник называется автоэмиссионным, на
самом деле до сих пор неясно, происходит ли ионизация благодаря десорбции1) Вопросы, связанные с неустойчивостью жидкости в сильном электростатиче¬
ском поле, приводящей к образованию микровыступов, были впервые рассмотрены в
работах Тонкса и Френкеля ГбО, 511, а вопросы отрыва ионов от острий в сильном
электростатическом поле — в работе Мюллера [52]. - Прим. ред.
Дж. К. Ривьер89Рис. 2.20. Схема принципа действия автоэмиссионного жидкометаллического источ¬
ника ионов. Игла с наконечником 1 — 10 мкм погружена в резервуар с жид¬
ким металлом и окружена плотно прилегающей капиллярной трубкой. Жид¬
кий металл поднимается по игле и ее наконечнику благодаря капиллярным
силам при условии, что жидкость смачивает материал иглы. Затем к вытя¬
гивающему электроду, расположенному перед самым острием иглы, при¬
кладывается напряжение от 4 до 10 кэВ, после чего жидкий металл вытя¬
гивается в острие, из которого под влиянием приложенного поля экстраги¬
руются попожительно заряженные ионы жидкого металла. Поскольку при
этилл получают очень большие токи порядка 100 мкА с очень небольшого
объема острия, источник характеризуется большой яркостью и пучок фо¬
кусируется в маленькое пятно. (Из работы Преветта и Джеффриса [29]С разрешения Institute of Physics.)1 - жидкий металл; 2 - игла; 3 - ионы металла; 4 — вытягивающий
электрод; 5 - пленка жидкого металла; б - капилляр.полем или ионизации полем или тому и другому вместе, или же здесь имеют
место какие-либо другие процессы. В настоящее время в таких источниках
используются цезий, индий, галлий, олово, висмут, медь и золото; наиболь¬
шее внимание привлек цезий благодаря возможному использованию в ’’ионном
дроблении" и в ВИМС.Жидкометаллический ионный источник, так же как и автоэмиссионный
электронный источник, обладает большой яркостью. Получаемые высокие зна¬
чения тока вплоть до 100 мкА в сочетании с очень малым эмитирующим объ¬
емом предполагают наличие очень большой яркости. К сожалению, трудно
902. Оборудованиеуказать точное ее значение, так как размеры эмитирующего объема неиз¬
вестны. Однако большая яркость, как и в случае электронного источника, по¬
зволяет получать нужный ток в хорошо сфокусированном пучке, что в принци¬
пе дает возможность производить ионное травление с бЪлее высоким простран¬
ственным разрешением, чем достигаемое с источником, использующим газо¬
вый разряд, Предварительные эксперименты [30] показали, что размер ионно¬
го пятна меньше .0,5 мкм можно получить без больших усилий, и вероятно,
скоро будет получено разрешение 0,1 - 0,2 мкм.В настоящее время неизвестны значения некоторых параметров жидкоме¬
таллических источников ионов, хотя выполнен значительный объем исследова¬
ний и уже недалеко то время, когда мы будем знать о них значительно больше.
По-бидимому, в некоторых диапазонах изменения рабочих параметров они не¬
стабильны, и, возможно, это связано с механизмом образования жидкого ост¬
рия. Состав ионного пучка в смысле отношения однократно и двукратно заря¬
женных ионов и т. д. зависит от используемого металла и до некоторой степе¬
ни от приложенного напряжения. Возможно также образование молекулярных
кластеров. Ионный пучок может иметь значительную расходимость по сравне¬
нию с выходной диафрагмой, поэтому для оптимального использования его яр¬
кости необходима хорошо спроектированная система линз. Однако эти недос¬
татки преодолимы, и ожидается, что жидкометаллические ионные источники
будут играть все возрастающую роль при анализе поверхности.2.5. Электронные энергоанализаторыУстройство, измеряющее энергии (или, точнее, скорости) электронов,
эмитированных или рассеянных с поверхности, является центральной частью
любого спектрометра. В этом разделе такое устройство мы будем называть
"анализатором”, чтобы отличать от ’’спектрометра" - термина, которым в
данной главе обозначается все оборудование, как указывалось в разд. 2.1.
Однако следует отметить, что многие авторы, употребляя термин "спектро¬
метр", имеют в виду только анализатор. Ни один из вариантов не является
совершенно правильным или совершенно неправильным, и это зависит от соб¬
ственного желания и принятого определения.За время развития РФЭС и ЭОС предлагались различные типы анализато¬
ров, будто бы наилучшим образом соответствовавшие этим методикам, но
сейчас, кажется, выработалось единое мнение относительно наиболее подхо¬
дящих типов анализаторов. Из-за низких энергий анализируемых электронов,
а также из-за того, что в СВВ трудно создавать магнитные поля, во всех ана¬
лизаторах используется электростатическое поле. Существуют два основных
типа анализаторов: анализатор типа цилиндрическое зеркало (АЦЗ) и концент¬
рический полусферический анализатор (ПСА); оба они являются дисперсион-
Дж. К. Ривьер91ными анализаторами, т. е. в них электроны диспергируют по энергиям под
действием отклоняющего электростатического поля таким образом, что при
заданном значении поля измеряются лишь энергии, лежащие в узком интер¬
вале. Используются также и бывают весьма полезны анализаторы с задержи¬
вающим полем (АЗП); в них используется сферическая электронная оптика,
применяемая в дифракции медленных электронов (ДМЭ), где к сетке перед
коллектором прилагается задерживающий потенциал и только электроны с
энергией выше этого потенциального барьера могут достичь коллектора. Опи¬
сание этих трех анализаторов будет дано в приблизительно хронологическом
порядке, т. е. по мере того, как они начинали использоваться для анализа
поверхности. Но прежде необходимо установить требования, предъявляемые
РФЭС и ЭОС с точки зрения энергетического разрешения.2.5.1. Требования к энергетическому разрешениюСуществуют два определения энергетических разрешений и необходимо
ясно понимать разницу между ними. Первое - абсолютное разрешение Д£,
обычно измеряемое как полная ширина на половине высоты наблюдаемого пи¬
ка (ПШПВ). Вместо этого иногда используют ширину пика у его основания
ЛЕв, и очевидно, что в идеальном случае Д£в = 2Д£. Второе — относитель¬
ное разрешение, определяемое какгде Eq - кинетическая энергия электронов при данном положении пика. Раз¬
решение R часто выражают в процентах, т. е. (ДE/Eq) . 100. Относитель¬
ное разрешение также выражают через разрешающую способность, которая
просто является величиной, обратной R:Таким образом, аЬсолютное разрешение может быть получено независимо
от положения пика в спектре, а относительное - только по отношению к оп¬
ределенной кинетической энергии.Сейчас в РФЭС для определения возможных различий в химическом со¬
стоянии элементов необходимо иметь одно и то же абсолютное разрешение
для любого фотоэлектронного пика в спектре, т. е. для любой кинетической
энергии. Как было показано, ширины естественных (т. е. немонохроматизо-
ванных) линий обычно используемых источников мягкого рентгеновского из¬
лучения равны 0,70 эВ для MgKa и 0,85 эВ для А1 Ка. Чтобы абсолютное
разрешение соответствовало таким ширинам линий на максимально возмож¬
ных энергиях фотоэлектронов (т. е. 1253,6 эВ в случае Mg, 1486,6 эВ в слу¬
чае А1), необходимо относительное разрешение ~6 • 10“4 (или разрешающая
способность ~1700). Такое относительное разрешение несложно получить,(2.5)(2.6)
922. Оборудованиено это будет сопровождаться неизбежными потерями в интенсивности. Кроме
того, если требуется использовать монохроматическое излучение, то необхо¬
димому абсолютному разрешению ~ 0,2 эВ будет соответствовать относитель¬
ное разрешение ~10~4 (разрешающая способность 10 ООО); такое относитель¬
ное разрешение очень трудно получить, не используя очень большой и доро¬
гой анализатор.По указанным причинам обычно фотоэлектроны замедляют либо до выб¬
ранной энергии анализатора, называемой энергией пропускания, либо в опре¬
деленное число раз. В обоих случаях энергия пропускания или соотношение
сохраняется постоянным в процессе записи одного спектра. Таким образом,
если фотоэлектроны замздляют до энергии пропускания 50 эВ, то при абсо¬
лютном разрешении 0,7 эВ требуется относительное разрешение всего ~10”2.
Следовательно, торможение дает возможность получать такое же абсолютное
разрешение при более низком относительном разрешении, но, как будет вид¬
но ниже, это имеет и свои недостатки.В ЭОС естественная ширина линии (монокинетичность) возбуждающих па¬
дающих электронов не существенна, так как ширина оже-линии не зависит от
ширины линии первичного возбуждающего излучения (если применяется и
спектроскопия электронных потерь, то ситуация, конечно, меняется). Разре¬
шение по энергии определяется собственной шириной оже-линий. Так как боль¬
шая часть оже-пиков имеет ширину порядка нескольких электронвольт, то
здесь не требуется такое же абсолютное разрешение, как в РФЭС, и достаточ¬
но среднего абсолютного разрешения 2-3 эВ. На практике желательно рабо¬
тать с высокой чувствительностью при высоких энергиях оже-электронов
(> 1000 эВ) и с высоким абсолютным разрешением при низких энергиях
(< 150 эВ); это легче осуществить, работая при постоянном относительном
разрешении, чем при постоянном абсолютном.Обычное относительное разрешение оже-анализатора составляет 4« 10~3
(разрешающая способность 250). При таких условиях абсолютное разрешение
при энергии оже-электронов 50 эВ будет составлять 0,2 эВ, что более чем
достаточно, и при 1000 эВ оно составит 4 эВ. Хотя при высоких энергиях
абсолютное разрешение ухудшается, чувствительность при 1000 эВ значитель¬
но больше чем при 50 эВ.Если же необходимо исследовать оже-спектр с высоким разрешением для
материалов с узкими оже-линиями [31], то спектр записывается в условиях
возбуждения рентгеновским излучением и работы анализатора в режиме РФЭС,
т. е. с предварительным торможением. В этом случае интерес представляют
только отдельные участки спектра и любые изменения в чувствительности
или абсолютном разрешении в больших диапазонах изменения энергии несу¬
щественны. Этот вариант ЭОС иногда называют РЭОС (рентгеновская элект-
Дж. К. Ривьер93ронная оже-спектроскопия) или ЭОСВР (электронная оже-спектроскопия вы¬
сокого разрешения).2.5.2. Анализатор с задерживающим попем (АЗП)Задолго до появления РФЭС и ЭОС в ДМЭ использовалась сферически-сим-
метричная электронная оптика для получения дифракционных картин при по¬
падании ускоренных пучков дифрагировавших электронов на покрытый фосфо¬
ром коллектор или экран. Первоначальная конструкция состояла из двух кон¬
центрических сеток и экрана; внутренняя сетка была заземлена, вторая сет¬
ка находилась под отрицательным потенциалом, близким к энергии первичного
пучка, экран находился под высоким положительным потенциалом. После того
как примерно в 1967 га стало ясно [32], что ДМЭ-оптику можно использовать
в качестве энергетического анализатора, количество сеток возросло до трех,
а затем до четырех; окончательная используемая и в настоящее время кон¬
струкция анализатора с задерживающим полем приведена на рис. 2.21.В четырехсеточном АЗП, показанном на рис. 2.21, электроны вылетают
либо из пушки, используемой для получения картин в ДМЭ и проходящей сквозь
электронно-оптическую систему анализатора, и падают на образец нормально
к его поверхности, либо из боковой пушки, находящейся вне электронно-опти-
ческой системы, и возбуждают вторичную электронную эмиссию при наклон¬
ном падении. Внутренняя и внешняя сетки заземлены, чтобы создать однород¬
ное поле вокруг образца и экранировать коллектор (т. е. экран ДМЭ) от наво¬
док при модуляции напряжения, приложенного к другим сеткам, которые мог¬
ли бы иметь место за счет емкостной связи. Две средние сетки соединены
вместе, и к ним приложен задерживающий потенциал; две сетки используют¬
ся для уменьшения существенных линзовых эффектов, возникающих от про¬
хождения электронов через ячейки сетки. На задерживающий потенциал на¬
кладывается небольшое модулирующее напряжение от генератора. Обычно мо¬
дуляция синусоидальная, но это необязательно.Промодулированный ток коллектора при помощи синхронного детектора
сравнивается с опорным сигналом от генератора Первая гармоника тока кол¬
лектора на основной частоте со дает распределение по энергии N(E), а вторая
гармоника на частоте 2со дает производную распределения N'(E). Эти соотно¬
шения можно легко получить посредством разложения сигнала в ряд Тейлора
и алгебраических преобразований. Поскольку общий ток коллектора / являет¬
ся функцией задерживающего потенциала V, то если модуляция имеет вид
A sin cot, ток / можно записать как I(V + A sin сot). Разложение в ряд Тейлора
имеет вид(2.7)
942. ОборудованиеРис. 2.21. Схема конструкции анализатора тормозящего поля (АТП) для ЭОС, осно¬
ванного на использовании четырехсеточной ДМЭюптики. Электронная пуш¬
ка ДМЭ должна была бы выступать из коннектора (экрана) и проходить че¬
рез сетки, но она здесь отсутствует, так как не подходит дпя анализа, по¬
скольку ее диапазон изменения энергии слишком ограничен, а размеры пят¬
на слишком веники. Задерживающий потенциан, приложенный к двум внут¬
ренним сеткам, модунируется небольшим потенциалом, поступающим с ге¬
нератора сигналов через разделитеньный трансформатор, и усиленный сиг¬
нал коллектора сравнивается в синхронном детекторе нибо в основной, пи-
бо с первой гармоникой частоты модупяции. В первом спучае, согпасно вы¬
ражению (2.8), получаем распределение по энергии N (£), во втором - про¬
изводную энергетического распределения dN{E)/dE согпасно выражению
(2.9).1 - эпектронная пушка; 2 - образец; 3 - предусилитепь;4 - мапошу-
мящий усипитепь; 5 - фазовращатепь; 6 - синхронный детектор; 7 - ось У;
8 - двукоординатный самописец; 9 - ось X; 10 - программируемый источ¬
ник питания; 11 - блок задерживающего напряжения; 12 - удвоитель час¬
тоты; 13 — генератор; 14 - разделительный трансформатор.Произведя алгебраические преобразования, получим, что коэффициент при
sin со Г равен
Дж. К. Ривьер95коэффициент при cos 2 со* есть(2.9)Так как dl/dV - распределение по энергии N(E) и d2I/dV2 - производная
распределения N'{E), то амплитуда первой гармоники пропорциональна рас¬
пределению по энергии и амплитуда второй - производной распределения по
энергии при условии, что производными более высоких порядков можно пре¬
небречь. Это выполняется, только если величина k достаточно мала. На прак¬
тике этот критерий удовлетворяется, если амплитуда модуляции меньше по¬
ловины ширины оже-пика; поскольку ширина оже-пиков не постоянна, модуля¬
цию следует выбирать так, чтобы воспроизводить без искажений самый узкий
пик спектра. Используя большое напряжение модуляции, можно повышать чув¬
ствительность, однако при этом спектр будет искажен.Обратив особое внимание на конструкцию и внеся некоторые изменения
в пространственное расположение используемых в ДМЭ сеток, можно полу¬
чить относительное разрешение ~0,3%. Для большинства АЗП обычное разре¬
шение составляет ~ 0,5 —1,0%, что приблизительно соответствует значениям,
требуемым в ЭОС. В области очень низких кинетических энергий (ниже 100 эВ)
АЗП превосходит дисперсионные анализаторы; так, при относительном разре¬
шении 0,5% абсолютное разрешение будет равно 0,5 эВ, т. е. можно будет за¬
писать спектр с высоким разрешением.Несмотря на простоту, АЗП неудобен для применения в оже-спектроскопии
из-за одного очень серьезного недостатка: задерживающий потенциал сеток
пропускает на коллектор все электроны с энергией больше этого потенциала.
Следовательно, всеми электронами, а не только электронами анализируемой
энергии генерируется дробовой шум, поэтому для АЗП характерно низкое от¬
ношение сигнала к шуму. Кроме того, существуют проблемы, связанные с од¬
нородностью структуры сеток и их параллельностью, поскольку даже неболь¬
шие отклонения от требуемой геометрии могут заметно ухудшить разрешение.
Однако для тех, кто в основном изучает структуру поверхности методом ДМЭ,
АЗП представляет собой удобное средство для проверки чистоты поверхности
на любой стадии эксперимента.2.5.3. Анализатор типа цилиндрическое зеркало (АЦЗ)Недостатки, присущие АЗП, побудили первых исследователей, использо¬
вавших ЭОС, искать другие типы анализаторов, более подходящие для этой
методики, и вскоре стало ясно [33], что АЦЗ обладает многими достоинствами,
делающими его практически идеальным для данной цели. За прошедшее время
предлагались различные варианты АЦЗ, как коммерческие, так и самодель-
962. Оборудованиеные, но в общем случае различия между ними заключались в отдельных осо¬
бенностях конструкции, а основная форма и принцип действия оставались не¬
изменными.Схема АЦЗ изображена на рис. 2.22. Два цилиндра с радиусами г (внут¬
ренний) и г2 (внешний) расположены строго коаксиально. Внутренний цилиндр
заземлен, а внешний находится под потенциалом - V. Электроны, эмитируе¬
мые источником, расположенным на оси, под углом а проходят в апертуру
внутреннего цилиндра и те из них, энергия которых равна Е отклоняются
потенциалом внешнего цилиндра так, что, пройдя через другую апертуру, фо¬
кусируются на оси. В общем виде соотношение, связывающее V и Е описы¬
вается следующим образом:АЦЗ может иметь различные входные углы а, но в случае, когда а = 42° 18',
АЦЗ становится прибором с фокусировкой второго порядка [34]. В этом слу¬
чае К = 1,31 и приведенное соотношение для £q и V принимает вид(2.11)На самом деле апертуры обладают конечной шириной и в любом случае
желательно получить максимальную чувствительность в сочетании с соответ¬
ствующим разрешением при использовании АЦЗ с апертурами, допускающимиРис. 2.22. Схема конструкции анализатора типа цилиндрическое зеркало (АЦЗ). Ради¬
ус внутреннего цилиндра r1t радиус внешнего т2 . Внутренний цилиндр за¬
землен, к внешнему приложен потенциал - V. Электроны, испускаемые ис¬
точником S, расположенным на оси, с кинетической энергией EQf вновь
фокусируются в точке F согласно выражению (2.10). Входной угол выбран
равным 42° 18', поскольку при этом угле АЦЗ становится устройством с
фокусировкой второго порядка. Обычная угловая апертура Да равна 6°.L. — расстояние между S и F, гс — положение минимальной ширины
траектории, гm _ максимальное расстояние от оси электронов, влетаю¬
щих в анализатор под углом 42° 18'. (По Бишопу, Коаду и Ривьеру [37].)
Дж. К. Ривьер97разброс по углам обычно Да =6°. При таких условиях электронные траекто¬
рии в анализаторе, изображенные на рис. 2.22, имеют некоторую ширину, и
можно показать [34, 35], что наименьший размер, характеризующий сечение
траектории со^ для Да < 6°, определяется уравнением(2.12)Важной характеристикой минимального сечения траектории является его
положение; оно располагается вне оси и немного впереди фокальной плоско¬
сти. Расстояние до него от оси гс определяется следующим образом:(2.13)Важность этого факта заключается в том, что если в том месте, где достига¬
ется наименьшее сечение, поставить дополнительную апертуру [36] такого же
размера, то можно улучшить разрешение, лишь немного потеряв в чувствитель¬
ности. В некоторых разработанных конструкциях [37] можно изменять ширину
дополнительной апертуры, но эту операцию трудно провести извне, т. е. не
вскрывая вакуумную систему, и поэтому в настоящее время наметилась тен¬
денция фиксации размера этой апертуры при некотором среднем значении и
предоставления возможности изменения размеров основных апертур.Для единственного угла 42°18' входа электронов расстояние на оси меж¬
ду источником и фокусом равно 6,1 Гг Другой важный параметр конструкции -
это максимальное расстояние от оси, при котором электроны еще смогут за¬
тем пройти в выходную апертуру. Это расстояние задается выражением [29](2-14)Таким образом, все параметры АЦЗ определяются радиусом внутреннего
цилиндра г и, если он выбран, получаются более или менее автоматически.На рис. 2„22, чтобы не загромождать схему, не показаны некоторые дру¬
гие важные рабочие детали, а именно пластины для снятия краевых эффек¬
тов,, Из практических соображений, например из-за необходимости доступа к
образцу, источник, испускающий электроны, должен находиться вне АЦЗ, и
сразу видно, что такое расположение вызывает ограничение размеров анали¬
затора,, Со стороны образца на самом деле все фирменные АЦЗ срезаны, так
что их концы имеют коническую форму„ Но электроны, проходящие через ана¬
лизатор, должны находиться в однородном радиальном поле, в противном
случае будут иметь место сильные аберрации, поэтому поле на концах анали¬
затора должно обрываться так, чтобы не искажать поле в области апертур.В некоторых АЦЗ это достигается [ 36] применением ряда круговых электро-
982. Оборудованиедов, потенциалы которых устанавливаются с помощью разделяющих их сопро¬
тивлений, так чтобы обеспечить плавный переход от отклоняющего потенциа¬
ла V к потенциалу земли» В других анализаторах этот же эффект достигает¬
ся с помощью керамических дисков, покрытых пленкой, радиальное сопротив¬
ление которых осуществляет ту же функцию, что и разделяющие сопротивле¬
ния, но изменение потенциала в этом случае происходит более плавно.Как и во всех дисперсионных анализаторах, изменяя потенциал -V, при¬
ложенный к внешнему цилиндру АЦЗ, можно получить энергетическое распре¬
деление электронов, прошедших через анализатор, но важно отметить, что,
поскольку функция пропускания АЦЗ изменяется как £q, получаемое распре¬
деление есть не N(E), a EN(E). Таким образом, модуляция V небольшим сину¬
соидальным сигналом, как было показано в случае АЗП, может быть исполь¬
зована для выделения производной энергетического распределения, но в
этом случае настройкой на основную частоту со, а не на вторую гармонику;
полученное при этом распределение будет иметь вид EN'(E), а не N'(E).Разрешение по энергии для АЦЗ и ПСА можно записать [37, 38], исполь¬
зуя разрешение по основанию Д£^, в виде(2.15)где w- ширина щели (одинаковая для входа и выхода), А, В и п - постоян¬
ные. Лучшее приближение дает выражение, использующее разрешение по по¬
луширине Д£:(2.16)которое справедливо только тогда, когда В(Да)п « Аю/2. Для АЦЗ и ПСА
значения постоянных приведены в табл. 2.2.Таблица 2.2. Значения коэффициентов для опреде¬
ления энергетического разрешения по основанию и
энергетического разрешения по полуширине АЦЗ и ПСААВпАЦЗ (42,3°)0,36/г,5,553ПСА (180°)1/*о12Подставив значения для АЦЗ в два приведенных выше выражения, полу¬
чим для разрешения по основанию пика(2.17)
Дж. К. Ривьер99и для разрешения по полуширине пика(2.18)В случае АЦЗ относительно широкие угловые апертуры могут ограничи¬
вать достижимое разрешение; в действительности в выражениях (2.15) - (2.18)
ширина щели должна быть заменена характерным размером источника. На¬
пример, АЦЗ с внутренним радиусом г = 3 см и полушириной угловой аперту¬
ры Да = 6° будет иметь теоретическое разрешение по полуширине ДЕ/Е0 = 0,15%
для источника размером 5 мкм. Однако для Ьольших размеров источника тео¬
ретическое разрешение ухудшается следующим образом: 10 мкм, 0,16%;50 мкм, 0,18%; 100 мкм, 0,21%; 500 мкм, 0,48%; 1 мм, 0,80%. Разрешение,
получаемое на практике, будет, конечно, зависеть от точности изготовления
и влияния рассеянных магнитных полей, но в достаточно хорошо сконструи¬
рованном анализаторе эти эффекты могут вызвать ухудшение разрешения по
полуширине от 0,16 до 0,25% для пятна размером 10 мкм.Простой однокаскадный АЦЗ, описанный выше, был наиболее эффективен
в ЭОС благодаря очень высокому пропусканию. Это наиболее важное свойст¬
во для оже-анализатора, так как площадь источника в ЭОС в случае хорошо
сфокусированного электронного пучка всегда значительно меньше, чем пло¬
щадь сбора анализатора. Другими словами, светосила, определяемая произ¬
ведением площади сбора на функцию пропускания, не является решающей в
ЭОС. С другой стороны, в РФЭС светосила является самым важным парамет¬
ром, поскольку, как отмечалось, пучок из стандартного немонохроматизиро-
ванного источника рентгеновского излучения не фокусируется, а покрывает
большую площадь на поверхности образца. Кроме того, в РФЭС обычно требу¬
ется лучшее абсолютное разрешение, чем в ЭОС. По этим причинам однокас¬
кадный АЦЗ не подходит для РФЭС. Другая причина заключается в том, что
точное положение фотоэлектронного или оже-пика настолько зависит от по¬
ложения образца перед однопролетным анализатором, что не будет обеспечи¬
ваться воспроизводимость, необходимая для РФЭС (например, Сикафус и Хол¬
лоуэй [40] показали, что при 1500 эВ осевой сдвиг образца в 1 мм вызывает
сдвиг пика на 17,5 эВ).Большое пропускание, характерное для АЦЗ, привлекательно для
РФЭС, но более высокую светосилу хотелось бы получить, сохраняя
нужное разрешение. Используя два АЦЗ, расположенные последовательно,
т. е. двукаскадный АЦЗ, с предварительно тормозящими сетками перед вход¬
ной щелью, удалось создать анализатор, приемлемый для РФЭС [41]. Схема
его приведена на рис. 2.23. Две сетки, осуществляющие предварительное тор-
1002. ОборудованиеРис. 2.23. Схема конструкции двухпролетного АЦЗ, используемого как в ЭОС, так и в
РФЭС. Выходная апертура первой ступени является входной апертурой вто¬
рой. Перед входом в анализатор расположены две сферические сетки с цент¬
ром в пятне источника на образце, они замедляют фотоэлектроны до постоян¬
ной энергии пропускания в случае РФЭС, в случае ЭОС сетки заземлены так
же, как внутренний цилиндр. Управляемый извне ввод вращения позволяет из¬
менить входную и выходную апертуры второй ступени от больших размеров
для РФЭС до малых для ЭОС. Электронная пушка расположена внутри анали¬
затора на его оси, в то время как рентгеновский источник типа изображен¬
ного на рис. 2.9, а находится снаружи и настолько близко к образцу, насколь¬
ко ЭТО ВОЗМОЖНО. (По Палмбергу [41] С разрешения фирмы Elsevier Scienti¬
fic Publishing Company.)1 - образец; 2 - рентгеновский источник; 3 - внутренний и внешний
магнитные экраны; 4 — внешний цилиндр; 5 - внутренний цилиндр; 6 — ввод
вращения для изменения апертуры; 7 - электронный умножитель; 8 - вто¬
рая ступень; 9- первая ступень; 10- электронная пушка; 11 - сферичес¬
кие замедляющие сетки.можение, имеют сферическую форму и их центр совпадает с положением пят¬
на источника на образце.Действие предварительного торможения заключается в том, что при пере¬
ходе от кинетической энергии Eq к фиксированной полосе энергий анализатора
разрешение по энергии улучшается (т. е. Д£/£0 уменьшается) в (£^/£0) раз.
Но в то же время эффективный объем источника сокращается в (Ep/E0)Vi
раз. Таким образом, для поддержания общей светосилы на уровне, необходи¬
мом в РФЭС, входную и выходную апертуры второй ступени следует увели¬
чить, насколько это возможно, что на практике означает увеличение до тако¬
го размера, пока не начнет ухудшаться общее разрешение по энергии. Для
этого апертуры необходимо изменять извне, выбирая маленькую для ЭОС и
Дж. К. Ривьер101большую для РФЭС. Следовательно, в двухпролетном АЦЗ эффективное разре¬
шение по энергии определяется первой ступенью, а чувствительность - вто¬
рой, хотя ясно, что эти две функции неразделимы.В случае ЭОС двухпролетный АЦЗ работает в ’'нормальном1' режиме, т. е.
тормозящие сетки заземлены. Если попытаться использовать торможение в
ЭОС, рассеяние на сетках сильно уменьшит функцию пропускания. Однако в
РФЭС, несмотря на то что рассеяние на сетках уменьшает пропускание при
использовании торможения, эффективная площадь сбора пропорционально уве¬
личивается, поэтому светосила остается прежней.Противоположные и связанные между собой эффекты сжатия площади
источника при замедлении и увеличения эффективной площади источника бла¬
годаря рассеянию на сетках являются функциями и кинетической энергии, и
энергии пропускания и с трудом поддаются оценке. Общий эффект для
любой выбранной энергии пропускания необязательно равен нулю и даже не¬
обязательно линейно изменяется в зависимости от кинетической энергии.
Обычно абсолютное разрешение ДЕ хуже, чем 1,4 эВ, высокая светосила в
случае двухпролетного АЦЗ означает, что интенсивность сигнала для идентич¬
ных образца и рабочих условий больше, чем в случае ПСА. Однако с уменьше¬
нием энергии пропускания для достижения абсолютного разрешения ^ 1,0 эВ
интенсивность сигнала падает быстрее, чеЫ для ПСА, и, вероятно, это вызва¬
но упоминающимися выше эффектами, обусловленными наличием замедляю¬
щих сеток. Практически фирмы непрерывно идут по пути простого увеличе¬
ния общих размеров АЦЗ, т. е. внутреннего радиуса, определяющего прочие
параметры анализатора, что приводит к уменьшению нежелательных эффек¬
тов при данной энергии пропускания.2.5.4. Полусферический концентрический анализатор (ПСА)Приблизительно в то же время (1969 г.), когда было установлено, что
АЦЗ удобно использовать в ЭОС, РФЭС получила признание как ценная мето¬
дика исследования поверхности, и анализатор, использовавшийся создателем
РФЭС Зигбаном [42], был приспособлен для работы в СВВ. Это был полусфери¬
ческий концентрический анализатор (ПСА), также называемый сферическим
отклоняющим анализатором; до сих пор он является наиболее широко распро¬
страненным в РФЭС анализатором, им снабжены спектрометры по крайней ме¬
ре четырех фирм, в то время как двухпролетный АЦЗ выпускается только
одной фирмой.ПСА схематически изображен на рис. 2.24. Две полусферические поверх¬
ности с внутренним радиусом Я1 и внешним радиусом R2 располагаются кон-
центрично. Потенциал AV приложен между поверхностями, так что внешняя
отрицательна, а внутренняя положительна. Посредине между ними существу-
1022. ОборудованиеРис. 2.24. Принцип работы концентрического полусферического анализатора. Анализа¬
тор состоит из двух концентрических полусферических поверхностей, ради¬
ус внутренней r1 , радиус внешней r2 . Между ними приложен потенциал AF,
так что внешняя поверхность отрицательна, а внутренняя положительна. RQ —
средняя эквипотенциальная поверхность между полусферами; входная и вы¬
ходная щели расположены так, что их центры лежат на rq. Если е - кине¬
тическая энергия электрона, движущегося по орбите с радиусом к0, то соот¬
ношение между Е и v дается выражением (2.21); фиг - соответствен¬
но угловая и радиальная координаты электрона с энергией Eq (= еД F0)f вле¬
тевшего в анализатор под углом а относительно нормали к плоскости щели.
Если этот электрон должен пройти через выходную щель , то его траектория
в анализаторе определяется выражением (2.22). (Из работы Роя и Каретта
[44] С разрешения фирмы National Research Council of Canada.)ет эквипотенциальная поверхность, имеющая радиус Rq, и центры входной и
выходной щелей расположены на расстоянии RQ от центра кривизны и лежат
на диаметре. В идеальном случае RQ = (R^ + R2)/2, и обычно это соотноше-
н Jfe довольно точно выполняется.Координаты электрона с начальной кинетической энергией Е движуще¬
гося между полусферами, определяются двумя условиями фокусировки. Во-
первых [43], если электрон влетает в анализатор под углом а к нормам че¬
рез центр щели, его положение определяется параметрами г, расстоянием
по радиусу от центра кривизны и <р, угловым отклонением:(2.19)где у = Rq/t; у q - начальная координата электрона;Здесь Е соответствует кинетической энергии, которой будет обладать элект¬
рон при движении по круговой орбите радиуса Rq; для ПСА с углом отклоне¬
ния 180° первое условие сводится к(2.20)
где у^ - конечная координата электрона. Отклоняющий потенциал и оптималь¬
ная энергия Е связаны соотношением(2.21)Второе условие фокусировки вытекает [44] из ограничений, налагае¬
мых на наибольший и наименьший радиусы электронных траекторий. Можно
выразить максимальное и минимальное отклонения от средней траектории
радиуса Rq в виде(2.22)Это второе условие просто выражает тот факт, что если электрон должен
пройти через анализатор, то его конечное положение должно находиться
внутри апертуры выходной щели, и что у+ должны проходить в пространстве,
ограниченном полусферическими поверхностями.В старых моделях спектрометров потенциал средней эквипотенциали
анализатора был равен потенциалу земли, а сканирование по энергии осуще¬
ствлялось с помощью изменения потенциала образца. Однако в настоящее вре¬
мя имеет место обратная ситуация в том смысле, что образец заземлен, а
весь анализатор изолирован от земли и сканирование по энергии осуществля¬
ется путем изменения потенциала (или пьедестала), на который поднят анали¬
затор.Разрешение по основанию и по полуширине можно получить из выраже¬
ний (2.15) и (2.16), а также из табл. 2.2:(2.23)(2.24)(Заметим, что входной угол а в ПСА здесь эквивалентен половине угловой
апертуры Да АЦЗ.) Обычно желательно работать с большим входным углом
для увеличения чувствительности, и, поскольку необходимо достижение ком¬
промисса между чувствительностью и разрешением, величину а выбирают
так, чтобы а2 » w/2Rq. Тогда выражения (2.23) и (2.24) принимают вид
1042. ОборудованиеТаким образом, если Rq и абсолютное разрешение фиксированы, то порого¬
вая энергия пропускания Е обратно пропорциональна ширине щели.В настоящее время используются два режима торможения в ПСА. В од¬
ном случае начальная кинетическая энергия электронов уменьшается в опре¬
деленное количество раз. Очевидно, что при этом сохраняется постоянное
относительное разрешение (ПОР); такой режим работы называется режи¬
мом ПОР. В другом случае электроны замедляются до определенной энергии
пропускания и при этом сохраняется постоянное абсолютное разрешение; этот
режим называется режимом постоянной функции пропускания (ПФП). Каждый
из способов имеет свои достоинства, недостатки и своих поклонников. При
использовании режима ПФП легче провести последующую количественную об¬
работку результатов, так как абсолютное разрешение будет одним и тем же
во всех частях спектра, но при этом ухудшается отношение сигнала к шуму
с уменьшением энергии. При использовании режима ПОР труднее провести ко¬
личественную обработку, зато небольшие пики при низких энергиях легко де¬
тектируются.Торможение в ПСА обычно осуществляется с помощью плоских сеток,
закрывающих входную щель, но оно может быть достигнуто и с помощью сис¬
темы-линз. Система линз также используется для увеличения эффективности
анализа. При удалении образца от входа анализатора и проецировании анали¬
зируемой площади образца на входную щель под линзами освобождается ра¬
бочее пространство, позволяющее приблизить рентгеновский источник, элект¬
ронную и ионную пушки. В случае использования пушек это необходимо, что¬
бы получить минимальный размер пятна. В зависимости от используемых ре¬
жимов замедления и применения режимэв ПОР или ПФП или и того, и другого
конструкция линз усложняется, так как если в режиме ПОР функция пропуска¬
ния линзы постоянна, то в режиме ПФП функция пропускания линзы меняется.
В первом случае можно использовать простую эквипотенциальную линзу, во
втором необходима более сложная система с тремя или более элементами.Для оценки и сравнения АЦЗ и ПСА используются различные критерии, в
частности пропускание [45], светимость [46] и полное пропускание (произве¬
дение площади входной щели на входной телесный угол). Согласно Рою и Ка¬
рету [47], которые рассмотрели все критерии для различных анализаторов,
включая и эти два, АЦЗ в режиме фокусировки второго порядка (т. е. а =42,3°)
является лучшим. Однако они также подчеркнули, что критерии не учитывают спо¬
собы применения конкретных анализаторов, и здесь следует обратить на это
особое внимание. Выбор типа анализатора должен зависеть от практических
требований. Именно по этой причине AU3 до сих пор наиболее часто исполь¬
зуются в ЭОС благодаря высокому пропусканию при приемлемом разрешении
по энергии, в то время как в РФЭС всегда предпочтительнее ПСА, поскольку
Дж. К. Ривьер105он обладает хорошей светосилой при исследовании в области высоких энергий,
особенно при использовании линз. Кроме того, ПСА благодаря небольшому
углу сбора удобен для измерений угловых зависимостей, в то время как АЦЗ
для этого непригоден, так как его угол сбора составляет 360° (хотя в новых
моделях имеется внутренний затвор, который может выделять различные
секторы; см. гл. 4). Развитие АЦЗ, вероятно, пойдет только по пути увели¬
чения размеров, так как трудно себе представить, как его использовать с
линзами, в то же время сомнительно, что конструкция входных линз в ПСА
достигла оптимума.Литература1)1. Seah М.Р., Dench W.A., Surf. Interface Anal., 1, 2 (1979).2. Pirani M•, Yarwood Principles of Vacuum Engineering, Chapman and
Hall, London, 1961.3. Kelly R., Surf. Sci., 100, 85 (1980).4. Lewis G.W*, Kiriakides G., Carter G., Nobes M.J., Surf. Interface Anal.,4, 141 (1982).5. Seah M.P., Thin Solid Films, 81, 279 (1981).6. Holloway P.H., Bhattacharya R.S., J. Vac. Sci. Techn., 20, 444 (1982).7. Hofer W.O., Littmark V., Phys. Lett., 71, 6 (1979).8. Kelly R., Auciello O., Surf. Sci., 100, 135 (1980).9. Parry-Jones A.C.9Weightman P., Andrews P.Т., J. Phys. C: Solid State
Phys., 12, 1587 (1979).10. Coad J.P., Riviere /.C., Guttmann М., Krahe P.R., Acta Met., 25, 161
(1977).11. Musket R.G., McLean W. et ah, Appl. Surf. Sci., 10, 143 (1982).12. Benndorf СSeidel H., Thieme FRev. Sci. Inst., 47, 778 (1976).13. Madden H.H., J. Vac. Sci. Techn., 18, 677 (1981).14. Barthes M.-G,, Pariset С., Thin Solid Films, 77, 305 (1981).15. Crider C.A., Poate J.M., Appl.. Phys. Lett., 36, 417 (1980).16. Okada SOura КHanawa Т., Satoh КSurf. Sci., 97, 88 (1980).17. Eizenberg М., Brener R., Thin Solid Films, 88, 41 (1982); 89, 355 (1982).18. YatesK., Barrie Л., Street F. J. Phys. E: Sci. Inst., 6, 130 (1973).19. Castle J.E., Hazell L.B., Whitehead R.D., J. Electron. Spec., 9, 247 (1976).20. Dolby R.M., Brit. J. Appl. Phys., 11, 64 (1960).21. Castle J.E., Hazell L.B., West R,H*f J. Electron. Spec., 16, 97 (1979).22. Kelly М.Л., Tyler C,E., Hewlett-Packard Journal, 24, 2 (1972).23. Yates K., West R.H., Surf. Interface Anal, (в печати).1) Литература, отмеченная звездочкой, добавлена редактором переводя
1062. Оборудование24. European Synchrotron Radiation Facility, Supplement I (eds. Y. Farge,P.J. Duke), European Science Foundation (1979).25.Riviere J.C., Solid State Surface Science, 1, 79 (1969).26.Michaelson H.B., J. Appl. Phys,., 48, 4729 (1977Ь27. Christou AJ. Appl. Phys., 47, 5464 (1976).28. Prutton М., Browning REl Gomati M.M., Peacock D., Vacuum, 32, 351 (1982).29. Prewett P.D., Jefferies D.K., J. Phys. D: Appl. Phys., 13, 1747 (1980).30. Prewett P.D., Jefferies D.K., Second International Conf. Low Energy Ion
Beams, Bath, 1980.31. Fuggle J.C., in: Electron Spectroscopy (eds. C.R. Brundle, A.J. Baker),
v. IV, Academic Press, London, 1982.32. Palmberg P.W., Rhodin Т./V., J. Appl. Phys., 39, 2425 (1968).33. Palmberg P.W., Bohn G.K., Tracy J.C., Appl. Phys., Lett., 15, 254 (1969).34. Зашквара B£., Корсунский М.И., Космачев O.C., ЖТФ, 36, 132 (1966).35. Sar-El H.Z., Rev. Sci. Inst., 41, 561 (1970).36. Hafner H., Simpson J.A., Kuyatt C.E., Rev. Sci. Inst., 39, 33 (1968).37. Bishop H.E., Coad J.P., Riviere J.CJ. Electron. Spec., 1, 389
(1972 - 1973).38. Simpson J.A., in: Methods of Experimental Physics (eds. V.W. Hughes,H.L. Schultz), v. 4A, pp. 124 — 135, Academic Press, New York, 1967.39. Rudd M.E., in: Low Energy Electron Spectrometry (ed. K.D. Sevier), pp.17 — 32, Wiley-Interscience, New York, 1972.40. Sickafus Holloway P.H., Surf. Sci., 51, 131 (1975).41. Palmberg P,W., J. Electron. Spec., 5, 691 (1974); J. Vac. Sci. Techn., 12,
379 (1975).42. Siegbahn КNordling СFahlman A., et ah, ESCA, Atomic, Molecular and
Solid State Structure Studied by Means of Electron Spectroscopy, Almqvist
and Wiksells Boktryckeri AB, Uppsala, 1967.43. Purcell EM., Phys. Rev., 54, 818 (1938).44. Roy D., Carette /.-D., Canadian J. Phys., 49, 2138 (1971).45. Seah M.P., Surf. Interface Anal., 2, 222 (1980).46. Heddle D.W.O., J. Phys. E: Sci. Inst., 4, 589 (1971).47. Roy D», Carette J.-D., Topics in Current Physics, v. 4, Electron
Spectroscopy for Chemical Analysis, pp. 13 — 58, Springer-Verlag, Berlin, 197248*. Раховский В.И., Любимов AM., ДАН, 135, № 4, 906 (I960).49*. Васин В.А., Запорожченко В.И., Раховский В.И., Авт. свид. N 594540,
Бюлл. изобрет., № 7, 25.02 (1978).50*. Tonks L., Phys. Rev., 48, 562 (1935).51*. Frenkel Phys. Zs., 9, 675 (1935).52*. Muller E.W., Naturwiss., 29, 533 (1941).
Г"лава. 3
ИНТЕРПРЕТАЦИЯ СПЕКТРОВД. Бриггс1 Дж.К. Ривьер13.1. ВведениеВ этой главе особенности, которые можно обнаружить в спектрах
вторичных электронов и в рентгеновских фотоэлектронных спектрах,
рассматриваются как с точки зрения их физической природы, так и в отно-
шении содержащейся в них информации. Поскольку оже-пики содержатся
в спектрах обоих типов, а спектры, возбуждаемые рентгеновским излу¬
чением, дополнительно содержат и фотоэлектронные пики, то логично
вначале рассмотреть спектры вторичных электронов. Однако прежде все¬
го необходимо обсудить терминологию, используемую для описания этих
двух основных типов пиков в спектрах.3.2. ТерминологияПрежде чем приступить к изложению обозначений, используемых в
двух методах, которые будут описаны в этой главе, необходимо вспомнить
теорию углового момента, связанного с орбитальным движением элект¬
ронов вокруг ядра атома. Поскольку электрон является заряженной части¬
цей, при его движении вокруг ядра возникает магнитное поле, величина и
направление которого зависят соответственно от скорости электрона и ра¬
диуса его орбиты. Очевидно, что две последние величины можно охарак¬
теризовать с помощью углового момента, называемого орбитальным мо¬
ментом, который, конечно, квантован, так как электрон движется только
по определенным дискретным орбитам., Соответствующим квантовым
числом является число I, которое может принимать значения 0, 1, 2, 3,4, . . о . Другим свойством электрона является его спин (т. е. положитель¬
ная или отрицательная величина), который также индуцирует собственное
магнитное поле; со спином в свою очередь связан спиновый момент
количества движения, характеризуемый спиновым квантовым числом 5,l^D. Briggs, ICI PLC,Petrochemicals and Plastics Division, Wilton, Middlesbrough,
Cleveland, UK.2)/.C. Riviere, UK Atomic Energy Research Establishment, Harwell, Didcot,
Oxfordshire.
1083. Интерпретация спектровкоторое может принимать значения +1/2 или - 1/2. Таким образом, пол¬
ный угловой момент электрона является комбинацией орбитального уг¬
лового и спинового моментов, и эта комбинация является просто вектор¬
ной суммой двух указанных моментов. Однако с точки зрения содержания
настоящей главы очень важно отметить, что векторное суммирование мож¬
но проводить двумя способами, которые соответствуют так называемым
/ / -связи и LS-связи (известной так же как связь Рассел - Саундерса).3.2.1. //-связьПри таком суммировании полный угловой момент изолированного элект¬
рона получают путем векторного суммирования его спинового и углового
моментов. Тогда для каждого отдельного электрона полный угловой мо¬
мент можно охарактеризовать квантовым числом /, где / = I + s. Оче-13 5видно, что / может принимать значения , -у- , -у- и т. д. Для получе¬
ния полного углового момента всего атома необходимо затем провести
суммирование по всем электронам; результатом такого суммирования
являются полный угловой момент атома и связанное с ним квантовое
число /, где J =2/. Такое правило суммирования известно как //-связь.Строго говоря, / / - связь является наилучшим способом описания
электронного взаимодействия в элементах с большим атомным номером,
т. е. при Z ^ 75. Однако основанная на этой связи терминология факти¬
чески использовалась для описания как оже-спектров, так и других пиков
для всех элементов периодической системы. Это не существенно в случае
спектров, возникающих при образовании фотоэлектронов, поскольку в этом
случае атом оказывается в однократно ионизованном конечном состоянии.
Однако для оже-процессов, при которых конечное состояние является дважды
ионизованным, взаимодействие между двумя дырками в конечном состоянии
может привести к ситуации, когда //-связь непригодна. Этот вопрос будет
обсуждаться ниже.В схеме ; / -связи обозначения основаны на использовании главного
квантового числа п и электронных квантовых чисел Z и / , упоминавшихся
ранее. В исторически сложившихся рентгеновских обозначениях состояния
с п = 1, 2, 3, 4, . . . обозначаются соответственно К, L, М, /V, . . . , в
то время как состояния с различными комбинациями / = 0, 1, 2, 3, ... ио с 7/ = _L -i-. —- , , . . . имеют условный индекс 1, 2, 3, 4,...1 2 2 2 2(табл. 3.1). Рентгеновские обозначения почти всегда используются для оже-пере-
ходов, так, например //-связь предсказывает шесть XLL-переходов, т. е. KL XLKLxLb, KL2L2, KL2L5 и KL3L3о Эти обозначения до сих пор наиболее
часто используются в ЭОС»Спектроскопические обозначения однозначно соответствуют рентге¬
новским обозначениям и в них непосредственно используются различные
Д. Бриггс, Дж. К. Ривьер
Таблица 3.1. Рентгеновские и спектроскопические обозначения109Квантовые числа
п 1 jРентгеновскийиндексРентгеновскийуровеньСпектроско¬пическийуровень10121К1*^ 1/220121Li2^1/2211222р 1/221323L32р3/230121Mi3s 1/231122м2Зр 1/2313.23мъЗРз/232324м43d3/232525М53^5/2и т.д.и т. д.и т.д.и т.д.квантовые числа. В этих обозначениях главное квантовое число стоит на
первом месте, далее состояния с / = 0, 1, 2, 3, . . . обозначаются соответ¬
ственно буквами s, р, d9 ft которые стоят после первого числа, и, нако¬
нец, значение / добавляется в виде индекса. Таким образом, записанное
в рентгеновских обозначениях состояние L3, для которого п = 2, / = 1 и/ = —, в спектроскопических обозначениях будет записываться какI 22^з/2 • В табл. 3.1 спектроскопические обозначения приведены после эк¬
вивалентных им рентгеновских обозначений. Принято связывать фотоэлек¬
тронные пики в спектрах со спектроскопическим названием атомного
уровня, с которого был эмитирован данный фотоэлектрон.3.2.2. LS-связьДругой способ векторного суммирования состоит в том, что в начале
суммируются угловые моменты всех электронов, а затем спиновые мо¬
менты всех электронов. Эти суммарные моменты описываются двумя
квантовыми числами: квантовым числом L полного орбитального углово¬
го момента атома, которое равно 1.1, и суммарным спиновым кванто¬
вым числом S атома, которое равно 3ls* Связь этих двух полных момен¬
тов дает в результате полный угловой момент атома и эту связь можно,
как и раньше, охарактеризовать с помощью квантового числа, которое
теперь, однако, равно | L ± S |. Так как L и S могут принимать значения
О, 1, 2, 3, . . . , то / может быть равно любому целому числу между
| L -5 | и | L + S |. Происхождение названия "LS-связь" теперь очевидно.Было обнаружено, что LS -связь пригодна для элементов с малым
атомным номером, т. е. с Z < ~ 20. Для описания распределения элект¬
ронов в конечном состоянии в этой схеме связи используются обозначения,
которые представляют собой символы термов вида (25 + Аналогично
1103. Интерпретация спектровспектроскопическим обозначениям в табл. 3.1 состояния с L = О, 1,2, 3,..
обозначаются заглавными буквами S, Р, D, F, „ . -, в то время как кван¬
товое число полного спина 5 указывается в виде индекса (2S + 1). (Сос¬
тояние S, соответствующее L = 0, не нужно путать с квантовым числом
полного спина S,) Как и в случае //-связи, LS-ёвязь предсказывает
шесть возможных компонент в KLL-серии, которые перечислены в
табл. 3.2, однако одна из них запрещена законом сохранения четности
[ 1]. В табл. 3.2 также представлен часто используемый способ представ¬
ления электронной конфигурации конечного состояния после оже-перехо-
да ABC, заключающийся в указании заселенностей уровней В и С, нал-
ример 2s‘2p5 для KLXL2 3,Таблица3.2. Обозначенияв LS■связиПереходКонфигурацияi LSТермKL,L,2s°2p6001SKL,L2 з2s12p5101P113 p001S^2,3 ^2,32s2 2p4П13pj1)201DЗапрещенКлассификация и обозначения в LS -связи используются в основном
теми, кто с целью получения данных для сравнения с теоретическими рас¬
четами регистрируют оже-спектры с высоким энергетическим разреше¬
нием, и они обычно не встречаются в каждодневной практике. То же са¬
мое справедливо и для смешанной (промежуточной) схемы связи, которую
необходимо использовать для тех элементов периодической системы, для
которых ни LS-, ни / /-связь не дают правильного описания конфигурации
конечного состояния. При промежуточной связи каждый LS-терм расщеп¬
ляется на мультиплеты с различными значениями J, так что обозначение
терма теперь имеет вид +^Lj. Как видно из табл. 3.3, для KLL-се-
рий предсказывается 10 возможных конечных состояний, однако одно из
них является запрещенным по тем же причинам, что и ранее. В ряде экс¬
периментов было показано существование девяти линий в случае KLL-спек¬
тра.Обычно используются смешанные обозначения, как это видно из
табл. 3.3, так что KLL-cepm в приблизительном порядке возрастания
энергии оже-электронов будет иметь обозначения
Д. Бриггс, Дж. К. Ривьер 111Таблица 3.3. Обозначения в промежуточной связиПереходКонфигурацияLS- термL5J1C- термKLXLX2s°2p6lS000%lP101%KLxL2j2sl2p51103PoЪР1113P11123P2\S000'So110*0KL2%3L2j2s22p43P[1113<P,P1123P,lD202•Z>21) Запрещен.Рис. 3.1. Переход при движении по периодической системе от чистой LS -связи через
промежуточную связь к чистой И -связи и влияние типа связи на относитель¬
ные энергии в KLL оже-сериях [2].
1123. Интерпретация спектров(запрещен), KL2lb(3P0) и KL3L3(3P2). На рис. 3.1 показан переход KLL-
серии от чистой LS-связи через промежуточную связь к чистой //-связи
при движении по периодической системе.3.3. Спектр вторичных электронов, возбуждаемыйэлектронным ударомПри анализе энергий вторичных электронов, возникающих вследствие
облучения поверхности твердого тела электронным пучком, типичное
энергетическое распределение испускаемых электронов выглядит анало¬
гично распределению, показанному в нижней части рис. 3.2. При энергии
первичных электронов наблюдается большой узкий пик, интенсивность
которого соответствует тем электронам, которые отразились от поверх¬
ности без потери энергии, т. е. упруго рассеянным электронам. Если по¬
верхность является поверхностью монокристалла, то эти электроны несутРис. 3.2. Нижняя кривая — энергетическое распределение вторичных электронов, ис¬
пускаемых поверхностью графита под действием электронного пучка с энер¬
гией 1000 эВ. Верхняя кривая - дифференциальное распределение в диапа¬
зоне энергий, содержащем KLL оже-пики углерода. В дифференциальном рас¬
пределении за "положение” пика обычно принимается положение высокоэнер-
гетичного минимума.
Д. Бриггс, Дж. К. РивьерИЗинформацию о структуре кристалла благодаря дифракции на упорядочен¬
ной атомной решетке, что фактически используется в методах ДМЭ и ДБЭ.
Вблизи упругого пика при более низких энергиях расположена серия мень¬
ших и более широких пиков, интенсивность которых уменьшается при уда¬
лении от упругого пика. Эти пики соответствуют плазмонным потерям.На другом конце энергетического распределения можно наблюдать второй
интенсивный очень широкий пик, максимум которого лежит при значении
энергии в несколько электронвольт и который распространяется на многие
десятки электронвольт вверх по шкале энергий. Этот пик создают так на¬
зываемые "истинные” вторичные электроны; конечно, все электроны,
эмитируемые поверхностью при облучении первичными электронами, явля¬
ются вторичными, однако общепринятая терминология выделяет ис¬
тинные вторичные электроны из всех других электронов главным обра¬
зом по историческим причинам. Многие исследования, опубликованные под
заголовком "вторичная электронная эмиссия" имели дело с низкоэнергети-
чными вторичными электронами, которые возникают благодаря каскадным
процессам.Между пиками упругих и истинных вторичных электронов и часто на
крыле последнего расположены другие особенности, одни из которых легко раз¬
личимы как пики, а другие - нет и которые связаны в основном с оже-эмио
сией. Те немногие особенности, которые не являются оже-пиками, относятся
к пикам плазмонных или ионизационных потерь. В традиционной ЭОС, когда
не пытаются достигнуть максимального пространственного разрешения и
поэтому можно поддерживать ток электронного пучка на уровне 10~б - 10~7А,
спектр обычно регистрируется в виде производной по энергии. Таким обра¬
зом, энергетическое распределение, показанное в нижней части рис. 3.2,
будет иметь вид дифференциального распределения, представленного в
верхней части этого же рисунка. Дифференцирование не только помогает
выделить слабые пики, но также в значительной степени уменьшает фон;
это особенно ценно при выявлении пиков, расположенных на крутом крыле
пика "истинных" вторичных электронов.В следующем разделе обсуждаются эти пики и их происхождение, свя¬
занное с оже-эмиссией и различными процессами, сопровождающимися по¬
терей энергии. Большая часть этого рассмотрения основывается на диф¬
ференциальных спектрах, так как именно эти спектры встречаются наибо¬
лее часто.3.3.1. Спектры оже-электронов
3.3.1.1. Оже*процессВ левой части рис. 3.3 показана схема энергетических уровней твер¬
дого тела, причем энергия отсчитывается вниз от уровня Ферми, принято¬
го за уровень нулевой энергии., Формально за нуль следует принимать
1143. Интерпретация спектровэнергию уровня вакуума на бесконечно большом удалении от поверхности
твердого тела, однако как в РФЭС, так и в ЭОС энергии связи измеряют¬
ся относительно уровня Ферми.В средней части рис. 3.3 показана последовательность событий после
ионизации остовного уровня. В данном примере рассматривается ионизация
К-Уровня падающим электроном, энергия Ер которого, очевидно, должна
превышать энергию связи Ек электрона на К-уровне. Как будет показано
в гл. 5, вследствие зависимости сечения ионизации от Ер на практике
для эффективной ионизации необходимо, чтобы величина Ер была больше,
чем примерно 5£к. После образования дырки на уровне К атом релаксиру-
ет, заполняя эту дырку вследствие перехода электрона с внешнего уровня,
который в данном примере обозначен Lt. В результате этого перехода
возникает избыточная энергия, равная разности энергий (Ек -£L ), и эта
избыточная энергия может быть использована атомом любым из следую¬
щих двух способов. Она может перейти в энергию фотона характеристи¬
ческого рентгеновского излучения или же она может быть передана дру-Рис. 3.3. Схема процесса оже-эмиссии в твердом теле, а - начальное состояние сис¬
темы; б — налетающий электрон с энергией е создает в результате иони¬
зации дырку на внутреннем уровне к; для эффективности этого процесса не¬
обходимо выполнение условия е^ %ЪЕК. Дырка в к-оболочке заполняется
электроном из Lr что приводит к выделению энергии (Ек - EL ), котораяможет быть унесена фотоном с энергией h\> = (Ек - EL ) или перейти к дру¬
гому электрону. В данном случае этот второй электрон находится на
L2 3-оболочке и может испускаться с энергией (EK-EL _ е! ); вели-1 2, з ’чина Е* помечена звездочкой, так как она является энергией связи L0 ,
2,3 '3не в основном состоянии, а при наличии дырки в ; в - дважды ионизо¬
ванное конечное состояние.
Д. Бриггс, Дж. К. Ривьер115Рис. 3.4. Относительные вероятности релаксации после образования дырки на К-обо-
лочке путем эмиссии оже-электрона и путем испускания рентгеновских
фотонов с характеристической энергиейегому электрону, находящемуся на том же уровне или на более высоком
уровне, вследствие чего этот второй электрон покидает атом. Первый
процесс является рентгеновской флюоресценцией, а второй - оже-эмис¬
сией. Ясно, что оба процесса не могут происходить с участием одной и
той же остовной дырки, так как они конкурируют между собой. Однако,
как это показано на рис* 3.4, при ионизации К-оболочки вероятность ре¬
лаксации в результате оже-эмиссии в огромной степени превышает вероят¬
ность рентгеновской флюоресценции для относительно неглубоких остов-
ных уровней, т. е. уровней с энергией ниже примерно 2 кэВ. Это же ут¬
верждение верно для атомных уровней L, М, N и т. д. Если бы это было
не так, то интенсивность сигналов в ЭОС оказалась бы значительно мень¬
ше и поэтому ЭОС не служила бы столь полезным методом, каким она яв¬
ляется.Оже-переход, изображенный на рис. 3.3, согласно обычно используе¬
мой jj-связи, обозначается как KLtL2 3«, Очевидно, эти же обозначения
можно использовать для других переходов в атоме, представленных на
том же рисунке, например KL1LU KL2t3^,з> Li^29zL293 и т* Д- Элект¬
роны, участвующие в оже-процессе, могут также находиться в валентной
зоне твердого тела, в этом случае общепринято обозначать переход, нап¬
ример, как KL2 3V, если участвует один электрон из валентной зоны, и,
например, как KVV, если оба электрона принадлежат валентной зоне.Энергия испускаемых оже-электронов в примере, представленном на
рис. 3.3, равна
1163. Интерпретация спектровгде £• - энергия связи i -го энергетического уровня атома. Величина
Е*L помечена звездочкой, так как она является энергией связи уровня213^2,з В присутствии дырки на уровне Lt и поэтому отличается от EL^ .
Различные причины отличия Е* и Ei будут рассмотрены с общих позиций
ниже. Здесь достаточно отметить, что оже-энергия, представленная урав¬
нением (3.1), зависит только от энергий атомных уровней и, таким обра¬
зом, для каждого элемента периодической системы имеется характерный
набор оже-энергий, причем невозможно указать два различных элемента
с одинаковым набором атомных энергий связи. Таким образом, анализ оже-
энергий сразу приводит к идентификации элементов* Даже если один или
оба электрона, участвовавшие в оже-процессе, находились в валентной зоне,
такой анализ позволяет идентифицировать элементы, так как наибольшим
членом в уравнении (3d) всегда является энергия связи ионизованного
остовного уровня.Для тяжелых элементов и уравнения (3.1) следует, что количество
энергетически возможных оже-переходов при данной энергии первичного
электрона может стать в действительности очень большим, поскольку
число атомных уровней увеличивается. К счастью, для ЭОС вероятность
переходов велика только для нескольких из очень большого числа возмож¬
ных переходов, так что даже для самых тяжелых элементов эта проблема
не является непреодолимойЗо&7.2. Энергии оже-переходовПриближенное значение энергии ЕАВС оже-перехода АБС в атоме с
атомным номером Z, которое оказалось достаточно точным для боль¬
шинства практических приложений* было получено эмпирически Чангом
и Дженкинсом [ 31:(3.2)Здесь Е{ (Z) - энергии связи i -х урозней в элементе с атомным номе¬
ром Z, a Ei (Z + 1) — энергии связи тех же уровней для следующего
элемента в периодической системе. Имеются различные таблицы ЕАВС
типа таблиц Коглана и Клаузинга [ 4], основанные на уравнении 3.2.Более приемлемым с физической точки зрения выражением для энер¬
гии оже-перехода является(3.3)где ?(ВС : х) - энергия взаимодействия между дырками В и С в конечном
атомном состоянии и Rx — энергии релаксации. Она возникает вследствие
Д. Бриггс, Дж. К. Ривьер1дополнительного экранирования атомного остова, которое необходимо учи¬
тывать при наличии дырки на остовном уровне, и обусловлена притяжени¬
ем или "релаксацией" внешних электронных орбиталей к этому остову.В случае релаксации увеличивается взаимодействие, описываемое с по¬
мощью кулоновского и обменного интегралов электронов внешних орбита-
лей с электронами внутренних орбиталей» Член Я|НУТР есть энергия
внутриатомной релаксации, т. е. энергия релаксации изолированного ато¬
ма. В молекуле или в твердом теле, когда в области ионизованного атома
имеются дополнительные экранирующие электроны, т. е. соответственно
от других атомов или из валентной зоны, появляется дополнительный
член называемый энергией внетомной релаксации. Члены 5 ийчасто имеют значительную величину. В табл. 3.4 приведены примеры рас¬
четов, выполненные Кимом, Гаренструмом и Виноградом [ 5], для
L3M4t5M4 6 : 3Р-переходов в чистых никеле, меди и цинке.Таблица 3.4. Расчетные значения энергий 5и R,
входящих в уравнение (3.3), для 1зМ45: ЗР-перехода в Ni,
Си и Zn (Ким, Гаренструм и Виноград [5]) Металл5(МА;М4,5: 3Р)яв‘5™Ni26.69,918,2Си26.310,611,0Zn29.412,39,6При расчетах энергий оже-переходов с использованием уравнения (3.3)
обычно привлекаются экспериментальные данные по энергиям связи Е-
и расчетные данные для других членов, так что такой подход является
полуэмпирическим. Различные авторы опубликовали таблицы таких полу-
эмпирических значений энергий оже-переходов, наиболее детальными
таблицами являются таблицы Ларкинса [ 6], который рассмотрел серии
оже-переходов KLL, KLM, LZMM, LBMN, MbNN, M5NO и N600; все эти
серии были рассчитаны в приближении промежуточной связи.Идентификацию пиков в спектре вторичных электронов, возникающих
вследствие оже-эмиссии, можно было бы в принципе осуществить путем
сравнения измеряемых кинетических энергий с энергиями, рассчитанны¬
ми по соотношениям (3.2) или (3.3); последнее соотношение является бо¬
лее точным и дает также энергии отдельных термов конечных состояний.С другой стороны, в настоящее время имеется несколько атласов экспе¬
риментальных оже-спектров, в которых отмечены основные оже-пики с
указанием их энергий, что позволяет в большинстве случаев легко осу¬
ществить отождествление элементов. Между этими атласами имеются
незначительные различия, связанные с разными условиями записи спект¬
ров и различием между анализаторами энергий, используемыми в каж¬
дом конкретном случае, однако эти расхождения обычно не столь вели-
1183. Интерпретация спектровки, чтобы их нужно было принимать во внимание. Следует напомнить, что
спектры во всех случаях представляются в дифференциальном виде
EdN(E)/dE, при котором за энергетическое положение пика условно при¬
нято считать положение минимума высокоэнергетичного отрицательного
отклонения. Ясно, что такая условная энергия не соответствует энергии
реального пика в недифференцированном или интегральном спектре и бу¬
дет отличаться от нее на величину, зависящую от ширины пика. Таким
образом, не следует ожидать точного согласия между энергиями оже-пе¬
реходов, полученными при дифференциальной регистрации и расчетным
путем, однако такого сравнения обычно никто не делает, На практике оже-
атласы ежедневно используются в прикладной ЭОС, в которой до сих пор
общепринятым все еще является дифференциальный режим регистрации,
в то время как данные точных энергетических расчетов используются те¬
ми, кто работает в недифференциальном режиме регистрации спектра и
обычно получает данные при рентгеновском, а не электронном возбуждении.3,3,1.3„ Интенсивности оже-пиковИ зучение любого конкретного спектра или сравнение спектров двух
соседних элементов в вышеупомянутых атласах сразу же показывает,
что относительные интенсивности оже-переходов в одном элементе или оди¬
наковые оже-переходы для разных элементов сильно различаются между
собой* Причины этих различий, как правило, хорошо известны, однако в
отличие от расчетов энергий оже-переходов расчеты интенсивностей оже-
пиков или вероятностей переходов не столь точны. Не существует прос¬
той полуэмпирической формулы, которая давала бы набор пригодных отно¬
сительных интенсивностей, и даже сложные формулы, которые вынужде¬
ны использовать теоретики, все еще бесполезны для практического пред¬
сказания интенсивностей, особенно в случае твердого тела. Связь между
интенсивностью оже-пика данного элемента и его концентрацией, когда
он находится на поверхности вместе с другими элементами, будет обсуж¬
даться в гло 5, здесь же достаточно отметить, что интенсивности невоз¬
можно предсказать с той же определенностью, с какой можно предсказать
энергии. Когда сравнение интенсивностей важно, необходимо еще раз при¬
бегнуть к помощи опубликованных оже-атласов или же к набору спектров
чистых элементов, полученных в вашей лаборатории.3.3.1.40 Характеристические серии оже-пиковПоскольку, как показано в гл. 5, зависимость сечения ионизации элект¬
ронным ударом от энергии первичного электрона проходит через максимум
при значении энергии, в 4 - 5 раз превышающем энергию связи, и посколь¬
ку используемые пучки имеют типичную энергию 10 кэВ, то для любой
Д. Бриггс, Дж. К. Ривьер119последовательности элементов периодической системы неизбежно возни¬
кают ограничения, налагаемые на совокупность возможных оже-переходов,
которые можно возбудить с оптимальной эффективностью* Так, практичео
кое использование оже-переходов при ионизации /С-оболочки возможно в
случае элементов периодической системы от лития (в твердом состоянии)
до кремния; при ионизации L3-оболочки - от магния до рубидия, при иони¬
зации М 5-оболочки - от галлия до осмия и т. д. Следствием этих ограни¬
чений является то, что в каждой части периодической системы имеется
характеристический набор или серии наиболее заметных при обычных
экспериментальных условиях оже-переходов. Несколько примеров таких
серий представлено на рис. 3.5- 3*9, которые заимствованы из книги
Дэвиса и др. [ 7] (см. также [ 8])<.Рис. 3.5. Дифференциальные оже-спектры самых лег¬
ких элементов. Основным пиком является
KL2 3L2 3. Относительные интенсивности
приведены не в масштабе [7].Рис. 3.6. Дифференциальные оже-спектры скандия, ти¬
тана и ванадия. Характеристическими линиями
являются LMM-триплет и низкоэнергетичный
^2t3VVunm W*На рис. 3.5 можно видеть, что для легких элементов в большей части
этого диапазона преобладают оже-переходы К£2,з^2,а (для бериллия
Lx). Для магния, алюминия и кремния, которые не представлены на
рисунках, интенсивным становится также переход L2 %VV, а для элемен¬
тов от фосфора до кальция этот переход столь интенсивен, что его мож-
1203. Интерпретация спектровРис. 3.7. Дифференциальные оже-спектры хрома, марганца и железа. LA/Af-триплет,
который был едва заметен в начале первых серий на рис, з.б, теперь хо.
рошо различим; более интенсивен также L у у.пик [7].2в 3но использовать в целях идентификации, причем, как упоминалось выше,
ионизация К-оболочки при используемых энергиях уже невозможна. В се¬
риях переходных 3^-металлов от скандия до меди характерным "отпечат¬
ком*1 является LMM-триплет, состоящий в порядке возрастания энергии
из переходов L3VV, L3M2iV и £,3М2>3М2>На рис. 3.6-3.8 представ-
лено изменение этого триплета в серии; легко видеть, как изменяются
относительные интенсивности каждой компоненты, особенно L3VV, по от¬
ношению к другим двум линиям (отметим в этой связи также рис, 5.7 в
гл. 5). Постепенное увеличение интенсивности этого перехода отражает,
Д. Бриггс, Дж. К. Ривьер121Рис. 3.8. Дифференциальные оже-спектры кобальта, никеля и меди, в LMM-триплете
теперь преобладает L3VV-пик, расположенный при максимальной представ-
ленной на графике энергии, что связано с заполнением в этих металлах d-зон;
для меди эта зона заполнена полностью [7].конечно, последовательное заполнение валентной зоны до ее полного за¬
полнения у меди. Еще одну замечательную характеристическую серию
оже-пиков можно найти среди переходных 4^-металлов, она приведена
на рис. 3.9. Основной особенностью в этом случае является близкое
энергетическое расположение M4 *N4e5/V4>5-дублета, причем расщепле¬
ние определяется расщеплением М4 и М5. Для других последователь¬
ностей элементов периодической системы имеются другие серии, харак¬
терные для элементов этих групп периодической системы, но ни одна из
них не является столь ярко выраженной, как описанные выше серии.
1223. Интерпретация спектровРис. 3.9. Дифференциальные оже-спектры второй переходной группы металлов. Заисключением родия и палладия, для которых расщепление мало, характерис¬
тические линии представляют собой узкие, близко расположенные M4^w4 5
N г «дублеты, расстояние между которыми определяется расщеплением
И/и ^5 [71Оже-спектры на рис. 3,5-3,9 имеют также многочисленные неболь¬
шие пики, в основном связанные с малоинтенсивными оже-переходами, а
в сплавах, смесях и других многокомпонентных материалах, очевидно,
всегда имеется вероятность перекрытия оже-линий одного или нескольких
элементов,, Если, как в случае переходных 3d!-мет ал лов, имеется несколь¬
ко заметных переходов, то всегда можно найти среди них один переход,
свободный от наложений, однако проблемы могут возникнуть в том слу¬
чае, когда происходит совпадение положений единственного оже-пика лег¬
кого элемента и интенсивного лика, принадлежащего тяжелому элементу.
Д. Бриггс, Дж. К. Ривьер123Примеры такого наложения дают хлор и аргон с молибденом, бор с ниоби¬
ем, сера с цирконием и углерод с рутением. Если возникают сомнения,
лучше всего использовать методы вычитания спектров, при которых
стандартный "чистый" спектр вычитается из сомнительного спектра.3.3.1.5. Тонкая структура ожв-пинийТонкую структуру оже-спектра часто можно наблюдать как для ме¬
таллов, так и неметаллов. Она может быть обусловлена либо химичес¬
кими эффектами, либо эффектами конечного состояния. Как правило,
структуру, обусловленную влиянием химических эффектов, можно наб¬
людать в обычной ЭОС с электронным возбуждением, однако структуру,
связанную с конечным состоянием или эффектами локализации дырки,
можно зарегистрировать при более высоком энергетическом разрешении
в интегральном или недифференциальном режиме записи спектра с ис¬
пользованием рентгеновского возбуждения.Химические эффекты могут возникать по тем же причинам, что и
так называемые химические сдвиги, которые являются сильным ору¬
жием РФЭС, т. е. при этом энергия связи электронов на остовном уров¬
не, который подвергается ионизации, может изменяться с изменением
химического окружения, приводя к аналогичному смещению последующего
оже-перехода. Если атомы данного элемента находятся ка поверхности в
более чем одном химическом состоянии, то возможно появление тонкой
структуры оже-линии благодаря этому механизму* В качестве примера
на рис. ЗЛО рассмотрен индий, у которого тонкая структура, отмеченная
стрелкой (2) в нижней части обычного дифференциального спектра, обуслов¬
лена смещенными оже-пиками, как это видно при численном суммировании
спектров, представленном в верхней части рисунка. В данном случае сме¬
щение при окислении остовных уровней М4 и Мб соответственно на 2,8 и
3,4 эВ приводит к сдвигу энергии оже-переходов M4#gAf4>5N4>5 и таким об¬
разом к появлению дополнительной структуры.Однако более часто наблюдаемая тонкая структура, обусловленная хи¬
мическими эффектами, возникает в оже-спектрах неметаллических элемен¬
тов, находящихся на поверхности в одном или многих возможных химичес¬
ких состояниях,, Когда, например, такой элемент является частью хемо-
сорбировэнного слоя на металлической подложке, то наиболее интенсивны¬
ми оже-переходами этого элемента будут те переходы, для которых после
ионизации остовного уровня последующая оже-релаксация осуществляется
переходом валентных электронов подложки. Другими словами, хотя ус¬
редненное энергетическое положение результирующего пика в оже-спект-
ре будет специфично для данного элемента, который таким образом можно
идентифицировать, структура пика, включая тонкую структуру, будет свя¬
зана с локальной плотностью состояний в валентной зоне вблизи участка
1243. Интерпретация спектровРис. 3.10. Пример тонкой структуры оже-спектров, обусловленной химическим сдви¬
гом энергии связи ионизованного остовного уровня. В нижней части рисун¬
ка показан полученный при электронном возбуждении дифференциальный
оже-слектр индия m4<5n4^ 5n4 ь в чистом [1) и частично окисленном (2)
состояниях, имеющий тонкую структуру после частичного окисления, кото¬
рая не наблюдается у чистого состояния. Анализ методом вычитания недиф»
ференцированных спектров, расположенных в верхней части рисунка, получен¬
ных при фотонном возбуждении,показывает, что тонкая структура определяется
сдвигами при окислении на 2,7 и 3,4 эВ уровней М4 и М5. Оже«спектр частично
окисленного состояния, таким образом, перекрывается с чистым и ’’окис¬
ленным" спектрами.хемосорбции. Так как локальная плотность состояний (ЛПС) изменяется
при переходе от одной подложки к другой и при переходе от одного типа
участков к другому типу на любой конкретной подложке, то будут наблю¬
даться изменения в форме и тонкой структуре оже-пика (пиков) этих
хемосорбированных соединений.
Д. Бриггс, Дж. К. Ривьер125Рис. 3.11. KLL оже-спектр углерода в различных химических ситуациях*, Значок* на
спектрограмме в верхней части рисунка указывает, что спектр записан
после ионной бомбардировки [9, 10].Самым ранним примером наблюдения этих изменении, который до сих
пор является самым впечатляющим, является KVV оже-спектр углерода
в различных химических ситуациях,Рис. 3.11, на котором приведены
данные разных авторов [9-12] показывает, что как общий вид оже-пика,
так и его тонкая структура заметно изменяются при переходе от графита
к различным карбидам и даже после ионной бомбардировки твердого угле¬
рода. Отметим особенно отчетливую тонкую структуру в спектре карбидов.
На рис. 3.12 приведен еще один пример L2 9VV оже-пиков серы трех пере¬
ходных металлов; различие не столь велико, как для углерода, однако
оно существенно. Аналогичные изменения были обнаружены для азота,
фосфора и даже кислорода [ 13], которые более электроотрицательны, чем
другие упоминавшиеся здесь элементы. Такие валентные спектры можно
использовать для получения методом обратной свертки информации о
ЛПС на ионизованных участках поверхности, или же, что проще и сразу да?*
ет полную информацию, использовать их как спектры и отпечатки" имею¬
щихся конкретных химических форм или соединений. В данном обсуждении
интерпретации спектров достаточно указать, что тонкая структура, обус¬
ловленная химическими эффектами, не является чем-то необычным.
1263. Интерпретация спектровРис. 3.12. L2 3vv оже-спектр серы на поверхности трех переходных металлов, де¬
монстрирующий химические эффекты, обусловленные различием в энергии
связи и как следствие в локальной плотности электронных состояний око¬
ло атомов серы, Приведены расчетные положения спектральных термов.
Пики, обозначенные X, и обусловлены так называемыми внутриатом-
н ыми п ереходами [ 12 J.Другой вид тонкой структуры, возникающий вследствие корреляции
между двумя дырками, которые остаются в атоме в конце оже-процесса,
является характерной чертой некоторых металлов. В этих металлах оже-
переходы типа CVVP где С - остовный уровень, приводят к форме линии,
ширина которой до проведения обратной свертки значительно меньше
ширины валентной зоны V и, таким образом, получение информации о плот¬
ности состояний валентной зоны этих металлов будет невозможно, Факти¬
чески эти ширины аналогичны ширинам линий, ожидаемым для тех же пе¬
реходов в свободном атоме с поправкой на уширение в твердом теле и
эти оже-спектры называют "квазиатомными". Физическое объяснение
состоит в том, что в таких металлах эффективная энергия взаимодействия
между двумя дырками в конечном состоянии значительно больше ширины
валентной зоны, и поэтому эти дырки не могут релаксировать быстро, что
было бы возможно при более низкой энергии их взаимодействия. В резуль¬
тате эти дырки локализуются на продолжительное время возле ионизован¬
ного атома, что приводит к ситуации, аналогичной случаю изолированного
Д. Бриггс, Дж. К. Ривьер127атома. При этих условиях электронная корреляция между дырками приводит
к расщеплению CFF-спектра на мультиплеты, которые можно описать в
рамках LS- и промежуточной связи с использованием обозначений термов,
представленных в разд„ 3,2.2.Рис. 3.13. Тонкая структура М4,5^4,5^4.5 0Ж®“Спектра кадмия, связанная с муль-
типлетным расщеплением конечного состояния этого оже-перехода. а —
наблюдаемый спектр вместе с предполагаемым неупругим фоном, обо¬
значенным пунктирной линией; б - расчетные положения и интенсивнос¬
ти различных компонент мультиплета для каждой группы вместе с резуль¬
тирующей теоретической огибающей (сплошная кривая) и с эксперимен¬
тальными точками [15].
1283. Интерпретация спектровМеталлы, для которых "квазиатомный" тип спектра установлен с полной
определенностью, расположены в периодической системе элементов от ни¬
келя до селена, от серебра до теллура и от таллия до висмута; возможно,
имеются и другие металлы с таким же типом спектра. Некоторые металлы,
для которых "квазиатомные" оже-спектры обычно не наблюдаются, могут
давать такие спектры, когда они находятся в сплавах в виде легирующей
примеси малой концентрации [ 141. Так как расщепление некоторых муль-
тйплетов мало, то необходимо регистрировать оже-спектры при более вы¬
соком, чем обычно, энергетическом разрешении; обычно это возможно
только при регистрации недифференцированных спектров в РФЭС-установ-
ках путем счета отдельных импульсов, т. е. используя рентгеновское, а
не электронное возбуждение. Примеры оже-спектров, полученных таким
способом и имеющих хорошо выраженную структуру из-за вышеупомянуто¬
го "квазиатомного" поведения, представлены на рис. 3.13. В обозначени¬
ях на основе jj -связи эти две группы пиков описываются соответственно
как М5N4#5N4t5 и M4N4(5w4^5, при этом не учитывается мультиплетное
расщепление. Под экспериментальным спектром показаны расчетные поло¬
жения и относительные интенсивности мультиплетов, соответствующих де¬
вяти разрешенным конечным состояниям; различить все девять состояний
нелегко, так как энергии состояний 3Р0 и *Рг почти совпадают, однако
можно видеть, что энергетическое положение линий, полученное в резуль¬
тате расчетов с учетом атомной структуры, хорошо согласуется с экспе¬
риментом.З.ЗЛоб. Пики ппазмонных потерьЛюбой электрон с достаточно высокой энергией, проходя через твер¬
дое тело, может возбудить одну из мод коллективных колебаний в море
электронов проводимости. Эти колебания имеют частоты, характерные для
данного вещества в твердотельном состоянии, и поэтому для их возбужде¬
ния необходима вполне определенная энергия. Если электрон потерял ко¬
личество энергии, равное одному из этих характерных значений энергий,
то говорят, что он испытал плазмонные потери. Внутри твердого тела
потери энергии называются "объемными" плазмонами, и если основная
характерная частота объемного плазмона есть со6, то энергия плазмон-
ных потерь есть просто hсо& в Поскольку электроны, испытавшие плазмон¬
ные потери энергии, могут в дальнейшем еще терять энергию аналогич¬
ным способом, то в спектре будет виден ряд пиков потерь, равноудален¬
ных на h соь друг от друга, но с уменьшающейся интенсивностью [ 161,На поверхности упорядоченная атомная решетка твердого тела обры¬
вается и условия возникновения колебаний с частотой ооь больше не вы¬
полняются. Вместо этого может возбуждаться довольно локализованный
тип коллективных колебаний, относящихся к поверхности с основной час-
Д. Бриггс, Дж. К. Ривьер129тотой со, , где со, меньше чем со^. Теоретически можно доказать 116], что для
свободных электронов в металле частота cos равна со& /у/Г9 или более
строго [17] соь /(1 + е)1/2, где е - диэлектрическая проницаемость.
Поэтому при анализе энергии электронов, вырываемых из твердого
тела, можно обнаружить в спектре одновременно с интенсивными пиками
ряд пиков плазмонных потерь. Основной, или * первичный", плазмонный
пик всегда будет наблюдаться, и в зависимости от вещества и эксперимен¬
тальных условий можно также наблюдать несколько уменьшающихся по ве¬
личине пиков кратных плазмонных потерь. В зависимости от условий на
поверхности вещества и энергии "материнского" пика пики поверхностныхРис. 3.14. Тонкая структура KLL оже-спектра алюминия, обусловленная плазмон-ными потерями, при электронном и фотонном возбуждении. Основные оже-
пики и их спектральные термы имеют обозначения kl2 3L2 3(1D),KL2,3L2.3^s )• klil2 з(3^) и Т-Д* Поверхностный плазмон обозначен
буквой s, а последовательные объемные плазмоны — в1 и в2 ; наложе¬
ние объемного и поверхностного плазмонов обозначено (в1 + s) и
(Я2 + S) [18].
1303. Интерпретация спектровплазмонов могут быть заметны, но их интенсивность всегда будет мень¬
ше интенсивности соседнего пика объемного плазмона.Яркий пример оже-спектра, содержащий вышеупомянутые особеннос¬
ти, представлен на рис. 3.14, на котором показаны различные пики потерь,
связанные с KLL-пиками алюминия. На этом рисунке показан KLL-пере¬
ход, возбуждаемый как электронами, так и мягким рентгеновским излу¬
чением. Видно, что при одинаковом энергетическом разрешении форма
оже-пика одна и та же для любого метода возбуждения, одинаковы и плаз-
монные потери.В ЭОС-спектре самый интенсивный пик обычно соответствует упруго
рассеянным электронам и если первичный электронный пучок имеет малый
энергетический разброс, то упругий пик тоже узок, если используется
энергоанализатор, обладающий адекватным энергетическим разрешением.
При этих условиях пики плазмонных потерь, связанные с упругим пиком,Рис. 3.15. Объемные (1) и поверхностные (2) плазмонные потери на чистой (3) ислегка окисленной (4) поверхностях алюминия, связанные с пиком упру¬
гого рассеяния (5) при первичной энергии, равной 100 эВ. Амплитуда
пика поверхностных плазмонных потерь является чувствительным ин¬
дикатором чистоты поверхности [ 19 и 16].
Д. Бриггс, Дж. К. Ривьер131имеют большую интенсивность и хорошо разрешены, а поверхностные плаз-
монные потери hcos , в частности, оказываются полезными для диагностики
чистоты поверхности. Путем уменьшения первичной энергии до типичных
значений 50-300 эВ, при которых проникновение пучка в твердое тело
очень мало и все процессы с потерей энергии происходят на поверхности
или вблизи нее; можно значительно увеличить поверхностные плазмонные
потери. При этом уровень обнаружимости результирующего пика потерь,
связанный с чувствительностью поверхностных плазмонных потерь к заг¬
рязнениям и реакциям на поверхности, таков, что степень чистоты поверх¬
ности можно контролировать, наблюдая за этим пиком по крайней мере с
той же чувствительностью, что и при наблюдении за оже-пиками примесей.Пики поверхностных плазмонных потерь использовались для этих целей,
например, Массиньоном и др, [ 19]. Рис. 3.15, взятый из их статьи, иллюстри¬
рует резкое уменьшение пика поверхностных плазмонных потерь алюминия
при незначительном загрязнении кислородом.3.3.1.7. Ионизационные потериКак было показано на рис. 3.3, первым этапом оже-процесса при элект¬
ронном возбуждении является ионизация остовного уровня первичным элект¬
роном. Как только электрон входит в твердое тело и теряет энергию в од¬
ном или нескольких процессах, любая информация о его происхождении те¬
ряется и он становится неотличимым от других электронов в твердом теле.
Так, при ионизации в твердом теле образуются два электрона, и так как
электрон может получить любую часть общей кинетической энергии, то
любой из этих двух электронов может иметь энергию от (Е -Е{ ) до
нуля. При ионизации уровня Е. кинетическая энергия выше (Ер _ Ei ) не¬
возможна, так как все уровни вплоть до EFf относительно которого изме¬
ряется £f. , заполнены, и, таким образом, возбужденные электроны долж¬
ны переходить только на свободные, разрешенные состояния выше ЕроПоэтому в недифференцированном спектре вторичных электронов иони¬
зационные потери будут проявляться в виде небольшой ступеньки при ки¬
нетической энергии (Ер _ Ei ), за которой следует спадающий хвост,
простирающийся через весь спектр до нулевой энергии. Такую ступеньку
в недифференцированном спектре наблюдать очень трудно, однако ее мож¬
но сделать более четкой, как и в случае оже-пиков, путем обычного диф¬
ференцирования по энергии [20].3.4. Рентгеновские фотоэлектронные спектры3.4.1. Первичная структураНа рис. ЗЛ6 изображен обзорный спектр чистой поверхности серебра,
полученный с использованием излучения MgKa и анализатора, работаю¬
щего в режиме постоянной энергии пропускания (постоянной ДЕ). Наблю-
1323. Интерпретация спектровРис. 3.16. Рентгеновский фотоэлектронный спектр при возбуждении MgKa (в основ¬
ном MgKa<| 2)t записанный при постоянной энергии анализатора, равной
100 эВ ("сателлит *Ag3<2 возбуждается MgKa3 4).дается серия пиков над уровнем фона, который обычно увеличивается с
уменьшением кинетической энергии (увеличение энергии связи) и который
также ступенчато возрастает с низкоэнергетичной стороны каждого ин¬
тенсивного пика. Когда, как в этом случае, рентгеновский источник не-
монохроматичен, его излучение имеет широкое непрерывное распределение
[ тормозное излучение, на которое наложены линии, характерные для ма¬
териала антикатода (анода)]. Это рентгеновское излучение создает фото¬
электронную эмиссию, удовлетворяющую уравнениюЕк = hv -Ев + 9, (3.4)где Ек — измеряемая кинетическая энергия (КЭ) электрона, hv - энергия
возбуждающего излучения, Ев - энергия связи (ЭС) электрона в твердом
теле и — "работа выхода" (обобщенный термин, точное значение которого
зависит как от образца, так и спектрометра, см. приложение 1). Уравне¬
ние (3.4) предполагает, что фотоэлектронная эмиссия представляет собой
упругий процессе Таким образом, любое рентгеновское излучение
(MgKa и т. д., см. ниже) будет давать серию фотоэлектронных пиков,
содержащую информацию о дискретных энергиях связи электронов, присут¬
ствующих в твердом теле.Процесс фотоэмиссии является неупругим, если энергия фотоэлектро¬
на изменяется (обычно уменьшается) в промежутке времени между фото¬
эмиссией из атома в твердом теле и детектированием в спектрометре.
Процессы, связанные с характеристическими потерями энергии, обсужда¬
ются ниже; здесь достаточно отметить, что фоновая "ступенька" к фото-
Д. Бриггс, Дж. К. Ривьер133электронным пикам с низкоэнергетичной стороны обусловлена неупругой
фотоэлектронной эмиссией (потерей энергии внутри твердого тела). Фото¬
эмиссия под действием тормозного излучения дает общий фон, который
преобладает в области спектра, соответствующей низкой энергии связи.
Пики, видимые на рис, 3.16, можно разделить на три основных вида: пи¬
ки фотоэмиссии с остовных и валентных уровней, а также пики оже-
эмиссии под действием рентгеновского излучения (ожв-серии).3.4.101 о Осговные уровниСтруктура пиков остовных уровней на рис. 3.16 является непосред¬
ственным отражением электронной структуры атома серебра. Излуче¬
ние MgKoc (1253,6 эВ) имеет достаточную энергию для зондирования
остовных уровней вплоть до 3s-оболочки. Из рисунка сразу видно, что
пики остовных уровней имеют различные интенсивности и ширины и
что ни один из s-уровней не является дублетом.Дублеты возникают благодаря спин-орбитальной У;-связи, которую
мы рассматривали ранее в разд. 3,2.1, При I > 0 возможны два состоя¬
ния, различающиеся квантовым числом У (У = / ±s) (см. табл. 3.1)о Раз¬
ность энергий этих двух состояний АЕ;- соответствует "параллельному"
и "антипараллельному" расположению векторов спина и орбитального
углового момента электрона. Величина этого энергетического расщепле¬
ния пропорциональна константе спин-орбитальной связи которая за-
висит от среднего значения < 1/г3 > для данной орбитали. Это расщепле¬
ние может составлять несколько электронвольт. Для данной оболочки
АЕ- (п и / фиксированы) увеличивается с ростом Z, а также увеличивает¬
ся с уменьшением I при постоянном п (например, из рис, 3,16 видно, что
для серебра расщепление Ър > 3d).Относительная интенсивность пиков в дублете определяется отношени¬
ем их степени вырождения (2; + 1), В табл. 3.5 представлены отношения
площадей пиков и их (п/,- )-обозначения для спин-орбитальных дублетов.Таблица 3.5. Параметры спин-орбитального
расщепления Относительные интенсивности. Основным параметром, который опре¬
деляет относительные интенсивности пиков остовных уровней, является
сечение фотоионизации атома a • Значения а были рассчитаны теоретичес¬
ки, а также получены из коэффициентов объемного поглощения рентгенов-ПодоболочкаЗначения /.ОтношениеплощадейпиковS1, 2 ,—Р1 3
2, 21 : 2d3 5
1 12 : 3f5 7
2. 23 : 4
1343. Интерпретация спектровских лучей. Чтобы дать общее представление, на рис. 5.14 гл. 5 приведены
расчетные значения Скофилда [21]. Изменения энергии возбуждения так¬
же влияют на значения а, хотя для обычно используемых MgKoc- и А1 Ка «фо¬
тонов это влияние на относительные сечения фотоионизации очень мало.
Пропускание энергоанализатора электронов, зависящее от энергии элект¬
ронов, относится к важным изменяющимся параметрам. Подробно эти эф¬
фекты обсуждаются в гл. 5 в связи с количественными измерениями.Ширина пиков. Ширина пика, которая определяется как полная ширина
на половине высоты (ПШПВ)ДЕ, является сверткой нескольких компонент,
дающих вклад в полную ширину(3.5)где Д£п - естественная или собственная ширина остовного уровня, ДЕ^
ширина источника фотонов (рентгеновской линии) и ДЕд - разрешение
анализатора; все эти ширины представляют собой ПШПВ. При выводе урав¬
нения (3.4) предполагалось, что все составляющие имеют гауссов контур.Вклад аппаратной функции анализатора подробно обсуждается в гл0 2
и 5, он один и тот же для всех пиков спектра при работе анализатора в
режиме постоянной энергии пропускания (ПЭП), однако этот вклад изме¬
няется вдоль спектра при работе анализатора в режиме постоянного отно
сительного разрешения (ПОР) (так как в этом случае постоянной величи¬
ной является ДЕ/Е).Собственная ширина линии остовного уровня, т0 е. разброс КЭ эми¬
тируемых фотоэлектронов непосредственно отражает неопределенность
времени жизни состояния иона, образующегося после фотоэмиссии. Та¬
ким образом, из принципа неопределенности мы получаем для ширины
линии соотношение (в энергетических единицах)(3.6)в котором постоянная Планка h выражена в эВ • с, а время жизни - в се¬
кундах. Ясно, что самые узкие остовные уровни (например, Ag3d) име¬
ют время жизни между 10"14 и 10""15 с, в то время как самые широкие
внутренние уровни (например, Ag3s) - время жизни, близкое к 10~15 с
или даже немного меньшее.Время жизни остовной дырки определяется процессами, следующими
за фотоэмиссией, при которых рассеивается или уменьшается энергия
иона. Вклад может дать любой из следующих трех механизмов: испуска¬
ние рентгеновского фотона (рентгеновская флюоресценция),эмиссия элект¬
рона в оже-процессе или же в процессе Костера - Кронига.Процесс Костера - Кронига является специфическим оже-процессом,
в котором конечный двухзарядный ион имеет одну дырку в оболо*же с тем
же главным квантовым числом (п), что и первоначальный ион. Для рас-
Д. Бриггс, Дж. К. Ривьер135сматриваемого вопроса существенно, что процессы Костера — Кронига
очень вероятны для первоначального распада дырок на остовных уровнях
с малыми квантовыми числами углового момента (например, 2s, 3s и 3р,
4s и 4р и т. д.) и кроме того, они происходят очень быстро. Поэтому эти
остовные уровни шире остовных уровней, которые распадаются за счет
обычного оже-процесса.Можно заметить, что ширины линий остовных уровней легких элемен¬
тов (Is, 2р) увеличиваются с ростом атомного номера. Увеличение плот¬
ности валентных электронов повышает вероятность рассматриваемых оже-
процессов (см. ниже), уменьшая тем самым время жизни остовной дырки.
Этот эффект можно даже наблюдать в спектрах высокого разрешения одно¬
го и того же внутреннего уровня при различном химическом окружении.
Например, атомы N в линейном ионе азида (N-), находящиеся в различных
химических состояниях, дают два N Is-пика (в результате химического
сдвига, см. ниже), из которых более узким является пик от центрального
атома с наименьшей электронной плотностью.3»4.1.2. Вапвнтные уровниВалентными являются те уровни, на которых находятся электроны с
низкой энергией связи (скажем, 0-20 эВ) и которые участвуют в образо¬
вании делокализованных или связывающих орбиталей. Спектр в этой облас¬
ти состоит из многих близко расположенных уровней, образующих зонную
структуру. Можно выделить два случая (рис0 3.17), а именно диэлектрики
и полупроводники (металлы). На рис. 3.17 показана плотность электронных
состояний (на единичный интервал энергии в единице объема) в этих двух
случаях. В случае диэлектриков заполненная валентная зона отделена от
пустой зоны проводимости, .в то время как в случае металлов эти зоныРис. 3.17. Схематическое изображение
плотности состояний для диэлектрика
(а) и металла (б). Штриховкой отмечены
занятые энергетические уровни [ 22].
1363. Интерпретация спектровперекрываются, а самое верхнее занятое состояние называется уровнем
Ферми (EF), Отметим, что Ер не является истинным нулем шкалы
электронной энергии, хотя значения ЭС часто отсчитываются от этой точ¬
ки. Истинным нулевым уровнем является уровень вакуума (Ev), и в пер¬
вом приближении Ef -Ev = 9, где 9 - работа выхода материала.Из рис. 3»17 видно, что при низких энергиях электронная плотность
свободных состояний обладает структурой. В результате, если фотоэлект¬
роны, испускаемые из заполненных уровней, имеют КЭ, которая совпадает
с этой структурной областью, наблюдаемая интенсивность будет опре¬
деляться сверткой плотности заполненных и свободных состояний сов¬
местно с матрицей вероятностей переходоз. Такая ситуация наблюдает¬
ся в ультрафиолетовой фотоэлектронной спектроскопии (УФЭС), и она
приводит к сильной зависимости спектра валентных уровней от энергии
фотонов. В случае РФЭС КЭ валентных фотоэлектронов такова, что ко¬
нечные состояния полностью лишены структуры; таким образом, наб¬
людаемая плотность состояний точно соответствует начальной плотности
заполненных состояний. На рис. 3.16 пик, обозначенный "4<Г, фактичес¬
ки является спектром зоны проводимости, которая образуется в основном
4<2-состояниями. Полный спектр при высоком разрешении, представленный
на рис. ЗЛ8, демонстрирует структуру этой зоны и резкий обрыв элект¬
ронной плотности при Ер0Сечения фотоэлектронной эмиссии валентных уровней значительно
меньше, чем в случае остовных уровней, что, как правило, приводит кРис. 3.18. РФЭС валентный спектр лоликристаллического серебра, возбуждаемый
монохроматическим излучением А1Ка , и его сравнение с теоретической
плотностью состояний: а - грубые РФЭС-измерения; б — эксперимен¬
тальные данные после вычитания гладкого неупругого фона; в, г - сум¬
марная теоретическая плотность состояний после учета двух различных
механизмов уширений линий, которые учитывают конечное время жизни
и эффекты встряски. Область уровня Ферми увеличена, чтобы показать
экспериментальное разрешение [23].
Д. Бриггс, Дж. К. Ривьер137низким интенсивностям. Тем не менее спектры валентных уровней, как это
обсуждается в п. 3.4.2.2, представляют ценность с точки зрения возмож¬
ности проведения анализа.3.4.1.3. Оже-серииКак было показано в п. 3.3.1.1, основной механизм распада состояния
с остовной дыркой состоит в безызлучательных переходах, приводящих к
эмиссии электронов. КЭ электронов, возникающих при переходах Косте¬
ра - Кронига, очень мала и ее обычно не измеряют, однако оже-электроны
могут давать доминирующие пики в "фотоэлектронном" спектре, как это
видно из рис. 3.16. Обозначения оже-пиков детально обсуждались в разд. 3.2.Четыре главные оже-серии можно наблюдать в РФЭС. KLL-cepm впер¬
вые наблюдается для бора и ее можно получить вплоть до натрия (ис¬
пользуя MgKa) или до магния (используя А1К а). При использовании AgLoc
(2984 эВ) можно наблюдать всю серию вплоть до хлора. Как видно из
рис. 3.19, доминирующей линией является KL2lz.Рис. 3.19. KLL оже-спектр паров
натрия [24].LMM-серия впервые наблюдается для серы и она простирается до гер¬
мания (MgKa), селена (А\Ка) или рутения (AgKa). Эта серия очень сложна,
так как линии, обусловленные ионизацией в L2- или £3-оболочках, могут
являться результатом переходов с участием М^ и М^ . Относительные
интенсивности этих групп изменяются с изменением атомного номера (Z).
Пример представлен на рис. 3.20.AfiVAf-серию можно разделить на две части. М45М44М45-серию, которая,
вероятно, впервые наблюдается для молибдена, можно проследить до нео¬
дима, используя MgKa. М45ДГб7№67-серию можно получить, только исполь¬
зуя фотоны с большей энергией (например AgLa), так как теоретически
1383. Интерпретация спектровРис. 3.20. LMM оже-спектр паров цинка [25].наблюдаемые линии редкоземельных элементов слабы и до гафния они
плохо видны. (Для гафния энергия ионизации М4б-оболочки составляет око¬
ло 1660 эВ и поэтому ее нельзя ионизовать с помощью MgKoc или А1£а.)
Типичный M46N45iV46-спектр представлен на рис. 3.21.Расположение главных оже-пиков на шкале ЭС приведено в приложении
7. КЭ оже-электронов определяется уравнением (3.2); таким образом, в
отличие от фотоэлектронов [уравнение (3.4)] КЭ оже-электронов не зависит
от возбуждающего излучения. Если, например, MgKa -рентгеновский ис¬
точник заменяется на А1 Ка -источник, КЭ фотоэлектронных пиков увели¬
чивается на 233 эВ, в то время как оже-пики не смещаются. Для спектров,
построенных непосредственно в шкале ЭС, имеет место обратное. Это поз¬
воляет легко различить два вида пиков в электронных спектрах. Отметим
также, что ширина возбуждаемых рентгеновским излучением оже-пиков,
как это видно из уравнения (3.2), не содержит вклада от ширины линии
рентгеновского источника.Ранее в этом разделе указывалось, какие именно рентгеновские
источники позволяют возбуждать высокоэнергетичные оже-серии (т„ е.
когда энергия начального состояния больше, чем энергия Al&a). Од¬
нако в обычном приборе с одним антикатодом наблюдение главныхРис. 3.21. M4t 5N4< 5N4> 5 оже-спектр цезия
H3Cs2S04 [26].
Д. Бриггс, Дж. К. Ривьер139фотоэлектронных линий и этих оже-линий в одном эксперименте (см.
п. 3.4.2.3) невозможно и в последнее время для этих целей начали исполь¬
зовать тормозное излучение. Как видно из рис. 3.22, используемые в экс¬
периментах величины потоков фотонов, энергия которых изменяется в ши¬
роком диапазоне значений, можно получить с помощью обычного алюмини¬
евого антикатода, особенно если в рентгеновском источнике используется
бериллиевое окно. Вагнер и Тейлор [ 27] использовали этот вариант для
двадцати четырех элементов, оже-линии которых имеют КЭ, лежащие в диа¬
пазоне 1400-2200 эВ.Рис. 3.22. Интенсивность тормозного излучения обычного алюминиевого рентгенов¬
ского источника [ 27]. Отметим, что использование алюминиевого окна
толщиной 2 мкм в грубом приближении эквивалентно использованию берил-
лиевого окна.1 - А1 ка-излучение; 2 - излучение от атомов мишени; 3 - излуче¬
ние, выходящее из анода; 4 - после прохождения окна из Be толщиной
8 мкм, 5 - после прохождения окна из А1 толщиной 6 мкм.3.4.2. Информация, получаемая из первичной
структуры спектров34.2.1. Химические сдвиги остовных уровнейСделанное на заре развития РФЭС открытие, заключавшееся в том,
что неэквивалентные атомы одного и того же элемента в твердом теле дают
пики внутренних уровней с измеряемой разностью ЭС, оказало стимулирую¬
щее влияние на прогресс в этой области. Этой разности ЭС было дано назва¬
ние " химический сдвиг” по аналогии со спектроскопией ядерного магнит-
1403. Интерпретация спектровного резонанса (ЯМР). Неэквивалентность атомов может обусловливаться
несколькими причинами: различием формального состояния окисления,
различием в молекулярном окружении, различием местоположения в решет¬
ке и т. д. Физическая причина химического сдвига иллюстрируется относи¬
тельно простой моделью, которая достаточно успешно используется (в соот¬
ветствующих случаях) для интерпретации данных по химическим сдвигам,
а именно моделью зарядового потенциала [ 28]:(3.7)где Е. - ЭС данного остовного уровня атома i ,Е? - невозмущенная энер¬
гия, qi ~ заряд на атоме i; в последнем члене уравнения (3.7) вычисля¬
ется суммарный потенциал на атоме i, обусловленный "точечными заря¬
дами" на окружающих атомах ;.Если принимать атом за полую сферу, на которой расположены валент¬
ные заряды qi , то классический потенциал внутри этой сферы будет одним
и тем же во всех точках и равен q.frv, где гv - средний радиус валентной
орбитали. Изменение заряда (плотности) валентных электронов на А^. при¬
водит к изменению потенциала внутри сферы на Aq{ /г vo Таким образом,ЭС всех остовных уровней будет изменяться на эту величину. Более того,
с увеличением rv сдвиг ЭС при данном А^ будет уменьшаться. Экспери¬
ментально было действительно показано, что во всех случаях, когда взаи¬
модействие остов-валентная оболочка мало, наблюдаемое положение остов¬
ных уровней данного атома подвержено аналогичному сдвигу ЭС и что
сдвиги ЭС между эквивалентными соединениями уменьшаются, если спус¬
каться по столбцу периодической системы (Aq{ одно и то же, г v увеличива¬
ется). Если сумму в уравнении (3.7) обозначить V{ , то сдвиг ЭС для данно¬
го остовного уровня атома % в двух различных ситуациях есть(3.8)Первый член (~ kLqi ) ясно показывает, что увеличение ЭС сопровождается
уменьшением плотности валентных электронов атома х . Второй член не
следует недооценивать, так как он имеет знак противоположный Ад,- 0 В мо¬
лекулярных кристаллах атомы ; представляют собой атомы, связанные с
атомом i , однако в твердых телах с ионной связью суммирование по этим
процессам распространяется на всю решетку; это тесно связано с энерги¬
ей Маделунга твердого тела и по этой причине V часто ня^ктяют потенциа¬
лом Маделунга.Уравнения (3.7) и (3.8) содержат ряд упрощений Основное упрощение
модели зарядового потенциала состоит в пренебрежении эффектами релак¬
сации, т. е. поляризационное влияние остовной дырки на окружающие элект¬
роны не учитывается как внутри атома (на атоме i), так и вне атома
Д. Бриггс, Дж. К. Ривьер141(на атомах;). Это один из примеров эффектов конечного состояния-терми¬
на, используемого для указания на тот факт, что состояние электрона в
фотоионизованном атоме может отличаться от состояния электрона в на¬
чальном состоянии (обычно основном состоянии атома). Другие эффекты ко¬
нечного состояния обсуждаются далее в этой главе,При сравнении уравнения (3.8) с (3.4) становится ясно, что уравнение
(Зэ8) справедливо только в том случае, если работа выхода веществ, обоз¬
наченных t(1) и е(2) , одинакова. При работе с проводниками величина
"ф" является работой выхода спектрометра и этой проблемы не возникает,
однако в случае диэлектриков величины ф(1) и ф(2) могут различаться.
Более того, диэлектрики приобретают поверхностный заряд при рентгенов¬
ском облучении, и прежде чем проводить точное измерение химических
сдвигов, необходимо сделать поправку на неидентичность зарядки. Этой
проблеме коррекции зарядки или калибровки ЭС посвящено приложение 2.Рис. 3.23 иллюстрирует эффект химического сдвига для трех различ¬
ных типов химического окружения атомов азота в спектре N Is одного
соединения. В этом случае относительные сдвиги легко измерить, однако
для измерения абсолютных энергий связи N Is требуется привлекать
некоторую процедуру калибровки. Четкая тенденция в рассматриваемом
примере заключается в том, что энергия связи N Is увеличивается с уве¬
личением степени окисления атома азота. Из табл. 306 видно, что для азо¬
та это обычно справедливо как в неорганических, так и в органическихРис. 3.23. Is-спектр азота из транс-
[Co(NH2CH2CH2NH2)2(N02)2]N03 [29].
142 3. Интерпретация спектровТаблица 3.6. Is-энергии связи (Вагнер и др. [30])соединениях. В ситуациях, когда состояние окисления одно и то же, общее
правило состоит в том, что ЭС остовного уровня центрального атома уве¬
личивается при увеличении эле - гроотрицательности (способности присое¬
динять электрон) присоединенных атомов или групп,. Много примеров хими¬
ческих сдвигов остовных уровней приводится в последующих главах, а при¬
ложение 4 содержит подробную сводку опубликованных значений сдвигов
для большинства элементов.
Д. Бриггс, Дж. К. Ривьер143Очевидно, возникают многочисленные случаи, в которых различные хи¬
мические состояния приводят к появлению фотоэлектронных пиков, относи¬
тельные химические сдвиги которых достаточно малы, и эти пики значи¬
тельно перекрываются; хотя такие сдвиги можно было бы в принципе изме¬
рить, если они равны - 0,2 эВ. Это становится невозможным, когда перек¬
рывающиеся пики имеют типичную ширину 1 - 2 эВ. Однако если контур
этих пиков известен, то можно пытаться осуществить обратную свертку
огибающей» Эта операция подробно рассмотрена в приложении 3.3.4.2,2. Структура вапвнтной зоныКак упоминалось в п. 3.4.1.2, спектр валентной зоны тесно связан со
структурой плотности заполненных состояний. Это очень полезно при изу¬
чении электронной структуры веществ - фундаментального вопроса для
многих аспектов практического использования приборов и для проверки
точности расчетов зонной структуры. Как показано на рис. 3.18, было дос¬
тигнуто очень хорошее согласие в случае серебра и это является достаточ¬
но типичной ситуацией для всех металлов,, Этим способом интенсивно ис¬
следовали применимость различных моделей электронной структуры спла¬
вов.Рис. 3.24. РФЭС валентные зоны V02 выше и ниже температуры перехода металл-
диэлектрик [31].
1443. Интерпретация спектровРассмотрим еще несколько примеров прямой информации, получаемой
из спектров валентных зон. Двуокись ванадия (V02) является диэлектри¬
ком при комнатной температуре, однако при 65 °С в ней происходит струк¬
турный переход и она превращается в металлический проводник. Рис. 3.24
четко демонстрирует образование запрещенной зоны при более низкой тем¬
пературе, что приводит к исчезновению проводимости. Вольфрамовые брон¬
зы являются семейством материалов с составом NaxWOf, (0,32 < х< 0,93).Металлические свойства указанных материалов полностью противополож¬
ны диэлектрическим характеристикам W08. Рис. 3.25 показывает, что
увеличение содержания натрия приводит к увеличению электронной плотнос¬
ти в "<Г-зоне* Она отсутствует в W03(d°), но ясно видна в ReOs (соответ¬
ствующая величина есть dl), которая обладает металлической проводимостью.В случае полимеров валентные зоны дают структурную информацию,
которую часто нельзя получить при исследовании остовных уровней. В гл. 9
обсуждается эта способность спектров валентных зон полимеров выступать
в качестве "спектров-отпечатков".Рис. 3.25. РФЭС валентные зоны вольфрамовых
бронз NaxW03 и Re03 [32].Зо4л2,Зо Химические сдвиги оже-линийОчевидно, что для оже-процессов, в которых конечные вакансии возни¬
кают на остовных уровнях, изменения химического состояния, вызывающие
химические сдвиги фотоэлектронных линий, будут также приводить к хими¬
ческим сдвигам оже-линий. Однако величина химического сдвига оже-линии
часто значительно больше величины химического сдвига фотоэлектронной
линии. Например, Вагнер и Билон [ 33] в первых работах на эту тему обна¬
ружили, что химические сдвиги оже-линий металлов и их окислов превосхо¬
дят фотоэлектронные химические сдвиги: Mg, 5,0; Zn, 3,7; Ga, 4,2; Ge, 3,5
Д. Бриггс, Дж. К. Ривьер145As, 3,4; Cd, 4,9; In, 1,8 и Sn, 2,5 эВ. Химические сдвиги оже-линий более
подробно обсуждаются в разд. 3.3, однако причины этого явления можно
понять из следующего упрощенного рассмотрения [ 34]. Как обсуждалось в
По 3.4.2.2, разность ЭС двух химических состояний зависит от изменения
энергии остовного электрона и изменения энергий внутриатомной и внеатом-
ной релаксации. Таким образом, для (1$)К-электрона имеем(3.9)где Де (К) - изменение энергии Is-состояния и ДR(K+) - изменение энергий
релаксации (в основном внеатомной) в конечном однократно ионизованном
состоянии. Как объяснялось выше (п. 3.4.2.1),(ЗЛО)и изменение кинетической энергии определяется следующим соотношением;(ЗЛ1)Для KLL оже-процесса изменение кинетической энергии между химически¬
ми состояниями равно(3.12)где (L+ L+) - двукратно ионизованное конечное состояние. Можно доказать,
что R(L+L+) - 4Я(К+), так что из уравнений (3.10) и (3.12) имеем(3.13)и, объединяя уравнения (3.11) и (3.13 ), получим(3.14)Поэтому разность между оже- и фотоэлектронными химическими сдвига¬
ми возникает вследствие различия энергий релаксации в конечном состоя¬
нии различных химических состояний.Можно, например, следующим образом определить параметр а:(3.15)Для электронов а не зависит от статического заряда и является парамет¬
ром, характерным для данного химического состояния, который можно из¬
мерить с большей точностью, чем ЭС остовного уровня или КЭ оже-пика.
Вагнер [ 35] назвал параметр а оже-параметром и определил его как раз¬
ность между КЭ самого интенсивного оже-пика и КЭ самой интенсивной
фотоэлектронной линии0 Неудобство вычисления этой разности состоит в
том, что при этом могут появляться отрицательные значения* Был введен
[36] "модифицированный” оже-параметр, имеющий вид(3.16)
1463. Интерпретация спектровкоторый лишен этого недостатка. Хорошим примером использования этого
параметра является работа Уэста и Кастла [ 37] по материалам, содержа¬
щим кремний, в которой для возбуждения Si Is - и Si KLL-линий использо¬
вался источник ZrLa. Многочисленные оже-параметры приведены в прило¬
жении 4.3.4.2.4. Форма оже-пинийКогда оже-процесс приводит к образованию по крайней мере одной ва¬
кансии на валентных уровнях, интенсивности различных линий оже-серии
могут сильно различаться при переходе от одного соединения к другому.В действительности структура валентной зоны или молекулярной орбитали
влияет на структуру оже-пика. Это прекрасно показано на рис. 3.26, на
котором изображен KLL-спектр F. Во фторе L^sJ-ypoeeHb является по
существу уровнем остовного типа, в то время как £-23(2р)-уровни участву¬
ют в образовании связей. Во фториде натрия окружение фтора аналогично
окружению свободного иона фтора и оказывается, что KLL-структура ана¬
логична структуре Na KLL, в которой все уровни являются остовными.В комплексных ионах и органических полимерах, где ковалентность фтора
возрастает и Ц8-уровни все больше участвуют в образовании связывающих
орбиталей, эти компоненты линий в KLL-серии становятся менее резкими.В случае кислорода KLL-rруппа принимает более разнообразные фор¬
мы, как это показано на рис. 3.27. В частности, отметим, как изменяет¬
ся расщепление (вплоть до 5 эВ) между линиями KLtL2S и KL23L23 и как
изменяется контур KL23L23-линии от практически одиночного пика до
двойного пика. Можно ожидать дополнительных изменений в этом пике,
так как обе вакансии теперь находятся в валентной оболочке, в то время
как Lj-оболочка является опять квазиостовной. Форма KLL-линии кислоро-Рис. 3.26. KLL-спектры фтора в твердых телах
с различной степенью ковалентности
[38].
Д. Бриггс, Дж. К. Ривьер147Рис. 3.27. KLL оже-спектры кислорода для
нескольких твердых веществ [ 39].да вновь имеет характерные изменения при переходе от одного типа соеди¬
нений к другому»3.4.3. Вторичная структура спектров
3.4.3.1, Рентгеновские сателлиты и духиПрежде чем обсуждать особенности истинной вторичной структуры в
спектре, необходимо идентифицировать источники "ложных" пиков низкой
интенсивности. Они возникают по двум причинам.Стандартные рентгеновские источники немонохроматичны. Наряду с
тормозным излучением, упомянутым в разд. 3.4.1, и основной Ка ь2-линией
магниевые и алюминиевые антикатоды дают также ряд линий низкой интен¬
сивности, называемых рентгеновскими сателлитами. Переходами, которые
приводят к появлению Ka t 2 -линии (неразрешенный дублет), являются
2^з/2, i/2 I5* Сателлиты возникают от менее вероятных переходов (на¬
пример, Кр; валентная зона -> 1 s) или переходов в многократно ионизован¬
ном атоме (например, Ка ). Наблюдаемые положения линий и их интенсив¬
ности приведены в табл. 3.7.Рентгеновские духи возникают вследствие возбуждений, причиной ко¬
торых являются примесные элементы в рентгеновском источнике. Самыми
распространенными духами в источнике ЩКа являются \\Ка 1 . Они воз¬
никают вследствие того, что вторичные электроны, образующиеся в источ¬
нике, бомбардируют тонкое алюминиевое окно (которое установлено для
того, чтобы предотвратить бомбардировку образца этими же электронами)*
1483. Интерпретация спектровТаблица 3.7. Высокоэнергетичные сателлитные линии от антикатодовиз Mg и AI [40]РентгеновскаялинияРасстояние от Ка, 2 (эВ) и относительная интенсивность
(Ка, 2 = 100)MgAIКос'4,5 (1,0)5,6 (1,0)/Са38,4 (9,2)9,6 (7,8)Кос410,0(5,1)11.5 (3,3)Кос517,3(0,8)19,8 (0,4)к*ь20,5 (0,5)23,4 (0,3)кр48,0 (2,0)70,0 (2,0)Поэтому это излучение будет создавать слабые пики духов на расстоянии
233,0 эВ в сторону более высоких КЭ от пиков, возбуждаемых основной
MgKa j 2-линией. Старые или поврежденные антикатоды могут давать духи,
возбуждаемые излучением основной CuLa-линией от незащищенного мед¬
ного основания антикатода. Эти духи появляются при более низкой КЭ на
расстоянии 323,9 эВ (556,9 эВ) от пиков, возбуждаемыхMgKalt2(AlKa1#2).B рент¬
геновских источниках с двойным анодом, в которых обычно один из анодов мо¬
жет давать высокоэнергетичное рентгеновское излучение (например, цирко¬
ний, серебро или титан), разъюстировка внутри источника может приводить
к перекрестному взаимодействию между нитями и анодами, что является ещеОДНИМ ИСТОЧНИКОМ Д$<ОВоОкисные пленки на поверхности алюминиевого и магниевого антикато¬
дов являются источником ОКос-духов, которому раньше не придавали осо¬
бого значения. На его важность при регистрации слабых сигналов указа¬
ли Коел и Уайт [ 41], что следовало из их измерений С KVV и О KVV
оже-спектров адсорбированных образцов при рентгеновском возбуждении.
(Жа-пики появляются на расстоянии 728,7 эВ (961,7 эВ) от пиков, возбуж¬
даемых MgKa lf2(AlKa t 2) в сторону более низких КЭ»3.4.3.2. Мультиппетнов расщеплениеМультиплетное расщепление (также называемое обменным или элект¬
ростатическим расщеплением) пиков остовных уровней может возникать,
когда система имеет неспаренные электроны на валентных уровнях. В ка¬
честве примера рассмотрим случай 3s-электрона в ионе Мп2+„ В основ¬
ном состоянии все пять З^-электронов неспарены и имеют параллельные
спины (это обозначается 6S). После испускания 3s-электрона появляется
еще один неспаренный электрон» Если спин этого электрона параллелен
спинам 3d-электронов (конечное состояние 7S), то может иметь место об¬
менное взаимодействие, приводящее к более низкой энергии, чем в случае
антипараллельных спинов (конечное состояние б5 )0 Таким образом, остов-
ный уровень будет представлять собой дублет, и расстояние между этими
Д. Бриггс, Дж. К. Ривьер149пиками есть энергия обменного взаимодействияE = (2S +l)K3s3d, (3.17)где 5 - суммарный спин неспаренных электронов на валентных уровнях( JL в рассматриваемом случае), a K3s3d - обменный интеграл (3s - 3$.
. ^Отношение интенсивностей этих двух пиков равно(3.18)Для 3s -электрона в Мп2+ это отношение равно (+ l)/или 1,4 иV 2 /2£, как было рассчитано, приблизительно равно 13 эВ. В реальном случае
молекулы MnF2 отношение интенсивностей приблизительно равно 1,8, а
расщепление - 6,5 эВ [ 42]. Это расхождение обусловлено эффектами ре¬
лаксации и конфигурационного взаимодействия, которые усложняют описан¬
ную выше простую модель (см. работу [ 43], содержащую детальное обсуж¬
дение этого вопроса). Однако данные по соединениям хрома, представлен¬
ные на рис. 3.28, иллюстрируют тот фак-в, что эта модель правильно пред¬
сказывает тенденцию изменений спинового состояния.Рис. 3.28. Мультиплетное расщепление Зя-пика соединений хрома: Сг(СО)6 является
диамагнетиком (нет неспаренных электронов); Сг(С5Н5)2 имеет два
неспаренных электрона на уровне е2„ ; Cr(hfa)3 имеет три неспарен¬
ных электрона на уровне *2g(hfa = CF3COCHCOCF3). Обратите внима¬
ние на то, как изменяется средняя ЭС при изменении электроотрицатель¬
ности присоединенных лигандов [44].
1503. Интерпретация спектровМультиплетное расщепление уровней, отличных от s -уровня, является
более сложным из-за дополнительного запутывания ситуации связью с ор¬
битальным угловым моментом. Так, в Мп2+ Зр-уровень расщепляется на
четыре уровня. Мультиплетное расщепление проявляется наиболее сильно,
когда оба участвующих уровня образуются в одной и той же оболочке (как
в случае 3s, 3р - 3d), однако эти эффекты в переходных металлах неве¬
лики из-за 2Р - З^-взаимодействия. Хорошим примером служит кобальт, ко¬
торый может находиться в разнообразных спиновых состояниях. Мульти¬
плетное расщепление вызывает уширение (с асимметрией) как 2Р1/2, так
и 2р3/2-пика, что приводит к кажущемуся изменению расстояния между
максимумами пиков. В табл. 3,8 приведено несколько примеров, показы¬
вающих, что дублетное расщепление можно использовать для диагностики
[ 45]. Были опубликованы данные по аналогичным изменениям расстояний
между максимумами пиков в случае соединений хрома [ 46].Таблица 3.8. Изменение 2р-спин-орбитального расщепления для спинового
состояния в комплексных соединениях Со [45]Энергия связи, эВ НеспаренныеСо 2р{12 Со2р3/2 Зр1/2“ЗРз/2 электроныСо(асас)2800,2784,216,03Со(асас)я799,0784,015,00(PEt2Ph)2Co(C6Cl5)2797,4782,115,31(PEt2Ph)2Co (2-метил-1-нафтил)2797,0781,515,51Мультиплетное расщепление велико для As -уровней редкоземельных
металлов (неспаренные электроны на валентных 4/-уровнях) и его можно
также наблюдать в спектрах органических радикалов, у которых свобод¬
ный электрон относительно локализован.3.4.3.3. Сателлиты "встряски” (shake-up)Для валентных электронов в атоме удаление остовного электрона
вследствие фотоэмиссии проявляется в увеличении эффективного заряда
ядра. Этот основной возмущающий фактор приводит к существенной
реорганизации валентных электронов (называемой релаксацией), которая
может заключаться в возбуждении одного из электронов на более высокий
свободный уровень ("встряска"). Энергия, необходимая для этого перехода,
не передается первичному фотоэлектрону, и, таким образом, двухэлектрон¬
ный процесс приводит к дискретной структуре фотоэлектронного пика в
области низкой КЭ (сателлиты "встряски").Детальные исследования, проведенные в случае атомных систем (на¬
пример, газообразного неона), показали, что эти переходы подчиняются моно¬
польным правилам отбора. Было обнаружено, что аналогичные примеры в
Д. Бриггс, Дж. К. Ривьер151твердых телах намного сложнее, за исключением органических систем.
Сопряженные и особенно ароматические системы дают сателлиты "встряс¬
ки" с интенсивностью вплоть до 5 - 10% от интенсивности основного пика.В ароматических системах сателлитная структура, как было показано [ 47],
обусловлена переходами -тт-»п*, включающими две самые высокие запол¬
ненные орбитали и самую низкую незаполненную орбиталь (см. гл. 9).Очень сильные сателлиты наблюдаются для некоторых соединений пере¬
ходных и редкоземельных металлов, которые имеют неспаренные электро¬
ны соответственно на 3d- и 4/-оболочках. Интенсивность сателлита обыч¬
но значительно выше, чем это ожидалось бы для аналогичной атомной
"встряски" (обычно - 10%), и причины этого расхождения дали пищу обшир¬
ной дискуссии [43]. Убедительное объяснение состоит в том, что в конеч¬
ном состоянии происходит значительный перенос заряда лиганд - металл,
так что по сравнению с начальным состоянием присутствует дополнитель¬
ный 3d- или 4/-электрон. Это сразу объясняет, почему системы с заполнен¬
ной оболочкой (например, Си+, 3d10) не дают сателлитов "встряски", в то
время как системы с незаполненной оболочкой (например, Си+, 3d9) дают
эти сателлиты [ 48]. Однако, как указал Федли [ 43], "встряска" является
плохим описанием этого процесса; более точное описание учитывает нали¬
чие сильного взаимодействия в конечном состоянии вследствие релакса-Рис. 3.29. З^-сигнал церия из Се02 : а) - первич¬
ный спектр при возбуждении А1 ка -из¬
лучением; . б - спектр после проведе= ния обратной свертки [49].
1523. Интерпретация спектровции. Сложность такого процесса в случае церия проиллюстрирована на
рис. 3.29.Как и в случае мупьтиплетного расщепления, сателлиты "встряски" име¬
ют диагностическую ценность. Возможность отличить медь (II) от меди (I)
уже упоминалась. Тетраэдрический никель (II) дает сателлиты, а никель (I)
(диамагнитный) не дает [ 50]. Кобальт (И ) дает более интенсивные сател¬
литы в состояниях с большим спином (4F), чем в состояниях с низким спи¬
ном (2D), в то время как кобальт (III) диамагнитен и у него нет никаких
сателлитов [45, 51].3.4.3.4. Асимметричные остовные уровни металловУ металлов в твердом состоянии имеется распределение незаполнен¬
ных одноэлектронных уровней выше энергии Ферми (как показано на
рис. 3.17), которые могут участвовать в явлениях типа "встряски", сопро¬
вождающих эмиссию остовного электрона. В этом случае вместо дискрет¬
ных сателлитов в области низкой КЭ основного пика ожидается, что ос-
товный уровень будет иметь хвост при низкой КЭ. Чем больше плотность
состояний вблизи уровня Ферми, тем более вероятно, что этот эффект бу¬
дет наблюдаться. Это подтверждается рис. 3.30. Такую асимметрию сле¬
дует принимать во внимание при попытке осуществить обратную свертку
сложных огибающих, содержащих пики металлов из подповерхностного
слоя (см. приложение 3).Рис. 3.30. Остовные (4/) и валентные (ВЗ) фотоэлектронные спектры золота и пла¬
тины, записанные с использованием монохроматического AlKcx-излуче-
ния. Обратите внимание на связь между степенью асимметрии остовно¬
го уровня и плотностью состояний вблизи уровня Ферми (ЭС = 0 эВ). [52]
Д. Бриггс, Дж. К. Ривьер1533.4.3.5. Сателлиты "стряхивания" (shake-off)В процессе, аналогичном "встряхиванию”, валентные электроны могут
полностью ионизоваться, т. е. возбуждаться в несвязанное состояние не¬
прерывного спектра. Этот процесс, называемый "стряхиванием”, оставля¬
ет ион с вакансиями как на остовном уровне, так и на валентном уровне»
Дискретные сателлиты "стряхивания” редко различимы в твердых телах,
поскольку а) энергетическое расстояние от основного фотоэлектронного
пика, большее, чем для сателлитов "встряхивания”, означает, что эти сател¬
литы имеют тенденцию располагаться в области широкого неупругого хвос¬
та, и б) переходы с дискретных уровней в континуум дают просто увеличе¬
ние интенсивности (т. е. широкие плечи), а не дискретные пики. Тем не ме¬
нее на хвосте С Is-пиков полимеров была идентифицирована слабая струк¬
тура [53] (после двойного дифференцирования), которую отнесли к "стряхи¬
ванию" и которая отражает структуру валентной зоны полимера.Плазмонные потери уже обсуждались в п. 3.3.1.6. Было обнаружено,
что пики потерь точно такого же типа связаны с остовными уровнями, как
и в случае оже-пиков [ 54].3.4.4. Угловые эффектыВ РФЭС важны два типа угловых эффектов. Первый состоит в увели¬
чении чувствительности при анализе поверхности, достигаемой в случае
электронов, выходящих под малыми углами к поверхности, в то время
как второй проявляет себя в монокристаллических образцах, когда имеет
место дифракция фотоэлектронов.3.4.4.1. Увеличение чувствительности к поверхностному споюПричину этого эффекта легко понять, обратившись к рис. 3.31. Если
А - средняя длина пробега для неупругих потерь (СДПНП) эмитируемых
электронов, то 95% суммарной интенсивности сигнала получается в твердом
теле с расстояния ЗА. Однако глубина по нормали к поверхности, очевидно,
определяется соотношениемd= 3Asinoc, (3.19)Рис. 3.31. Увеличение чувствительности к поверхности
при изменении угла ’’сбора** электронов.
1543. Интерпретация спектровкоторое достигает максимума при а = 90° <> В случае подложки (s) с одно¬
родным тонким поверхностным слоем (о) угловая зависимость интенсивнос¬
ти определяется следующим уравнением:где Л имеет соответствующее значение в случае наблюдаемого фотоэлект¬
рона. В идеальной ситуации эти уравнения приводят к кривым, аналогичным
изображенным на рис. 3.32. Однако реальная ситуация обычно осложняет¬
ся тем фактом, что геометрия системы обусловливает функцию отклика,
которая также зависит от а. Полезное обобщение этих эффектов для одно¬
го конкретного спектрометра было дано Дилксом [ 55]. Это осложнение
преодолевается путем измерений относительных значений 10/IS , так что
аппаратная функция отклика сокращается. Таким образом, как показано
на рис. 3.32, при малых значениях а отношение 1q/Is значительно увели¬
чивается. Пример этого эффекта представлен на рис. 3.33.Главные требования к увеличению чувствительности к поверхности
состоят в том, чтобы поверхность являлась плоской. Шэроховатость поверх¬
ности приводит к усреднению углов выхода электронов, а также к эффек¬
там затенения (как для падающих рентгеновских лучей, так и для испус¬
каемых электронов), так что в большинстве случаев эффект увеличения(3.20)и(3.21)Рис. 3.32. Теоретические кривые угловой зависимости для чистой плоской поверх-
ности и плоской системы подложка/поверхностный слой.
Д. Бриггс, Дж. К. Ривьер155Рис. 3.33. Влияние изменения угла сбора на Зр-спектр
кремния с пассивным окисным слоем. Отме¬
тим относительное увеличение (поверхност¬
ного) сигнала окисла при малом угле (от¬
считываемом от поверхности) [зо].чувствительности к поверхности не наблюдается [ 53].. В большинстве
промышленных спектрометров угол между падающими рентгеновскими лу¬
чами и детектируемыми электронами фиксирован (180° - а —б). Тем не
менее было показано, что при очень малых б эффект увеличения чувстви¬
тельности к поверхности также имеет место, что связано с быстрым обре¬
занием глубины проникновения рентгеновских лучей, когда б приближа¬
ется к углу скользящего падения [ 57].3.4.4.2. Дифракция фотоэлектроновИзмерение зависимости интенсивности остовных электронов, испуска¬
емых поверхностью монокристалла, от угла сбора электронов приводит к
графикам, аналогичным изображенным на рис. 3.34 (и для любого такогоРис. 3.34. Угловая зависимость интенсивности А§3^5/2-линии чистой поверхности
(110): а — с химической полировкой; б - без химической полировки после
ориентации. В обоих случаях окончательная очистка достигалась несколь¬
кими циклами травления ионами аргона и отжига [58].
1563. Интерпретация спектровугла аналогичные графики будут получаться при вращении образца в плос¬
кости поверхности, т. е. путем изменения азимутального угла). Заметная
тонкая структура, наложенная на аппаратную функцию, обусловлена преж¬
де всего дифракцией фотоэлектронов на плоскостях монокристалла. Иссле¬
дования родственных явлений каналирования высокоэнергетичных электро¬
нов в кристаллах и образования зоны Кикучи в ДМЭ указывают [ 36], что
фотоэлектронная эмиссия в направлении, параллельном каждому набору
плоскостей (ihkl), под углом ± Qhkl (угол, соответствующий первому поряд¬
ку брэгговской дифракции) будет приводить к увеличению интенсивности
эмиссии. Здесь hkl являются обычными индексами Миллера, a Qhkl опреде¬
ляется соотношением(3.22)где \е~ дебройлевская длина волны рассматриваемого остовного электро¬
на, a dhki - расстояние между соседними плоскостями.Эффекты фотоэлектронной дифракции, очевидно, усложняют количест¬
венный анализ данных по интенсивностям пиков от поверхностей монокрис¬
таллов, однако они уже с пользой применялись в аналитических приложени¬
ях. На рис. 3.35 показано, каким образом Эванс и Скотт [ 59] использовали
графики дифракции рентгеновских фотоэлектронов (ФЭД) (графики отно-Рис. 3.35. ФЭД-графики: а - пластина (100) GaAs; 6-cnoftZnSe, выращенный на
идентичной Ga As-подложке химическим транспортом ^ водороде. Ось вра¬
щения расположена параллельно < 110>, и угол сбора измеряется относи¬
тельно нормали к поверхности [ 59].
Д. Бриггс, Дж. К. Ривьер157сительной интенсивности остовных уровней в зависимости от угла сбора)
для демонстрации того факта, что рост ZnSe на (100) GaAs происходит
эпитаксиально. Графики аналогичного типа сразу позволяют выявить иден¬
тичность кристаллической симметрии подложки и поверхностного слоя.
Эванс, Адамс и Томас [ 60], используя этот метод, также провели струк¬
турный анализ сложных минералов, представляющих собой сложные силикаты.3.4.5. Спектры, зависящие от времениХотя РФЭС обычно рассматривается как "неразрутающий1’ метод (осо¬
бенно в сравнении с ЭОС и ВИМС), это утверждение нельзя принимать без
доказательств. Довольно очевидной проблемой является контакт материа¬
лов с высоким или сверхвысоким вакуумом спектрометра. Недавние сис¬
тематические исследования [61] нескольких материалов, содержащих крис¬
таллизационную воду, и некоторых гидроокисей показали, что их поведе¬
ние в вакууме можно предсказать, исходя из зависимости между давле¬
нием паров воды и температурой (уравнение Клаузиуса - Клапейрона).
Потеря воды является особенно серьезной проблемой при изучении биоло¬
гических систем.Труднее предсказать повреждения, вызываемые падающим рентгенов¬
ским излучением. В некоторых случаях зависимость от времени РФ-спект-
ров была интерпретирована как следствие структурных и кинетических из¬
менений (например, восстановление платиновых (IV) соединений, разложе¬
ние NaC104) [62]. Некоторые радиационные эффекты происходят практи¬
чески мгновенно при комнатных температурах и поэтому приводят к недо¬
разумениям. По-видимому, первым примером такого явления, которое сле¬
дует рассмотреть, являлась изомеризация на поверхности соединения
(Pb3P)2Pt(C2Cl4) в (Ph3P)2PtCl(C2Cl3) [63]. Охлаждение образца замед¬
ляет изменения и часто позволяет следить за ними i п si tu. Изменения,
индуцируемые рентгеновским излучением, вероятно, происходят более
часто, чем это обычно подразумевается. Обширные литературные данные
относительно появления (окисленного) S 2р-пика при высокой ЭС в спект¬
ре важных с биологической точки зрения Cu -S -протеинов, по-видимому,
недавно были теоретически объяснены тем, что происходят индуцированные
излучением окислительно-восстановительные реакции на участках CuS-по¬
верхности с участием адсорбированной воды, которые приводят к образо¬
ванию сульфогрупп. Такие примеры говорят о том, что в РФЭС на эффек¬
ты, связанные с излучением, следует обращать более серьезное внимание,
чем это делалось до сих пор.Литература1. Eisberg R.MFundamentals of Modem Physics, John Wiley and Sons,New York, 1963, p. 436.2. Sevier K.D», Low Energy Electron Spectrometry, John Wiley and Sons, New
York, 1972, p. 55.
1583. Интерпретация спектров3. Chung M*F*, Jenkins L*H*, Surf, Sci., 21» 353 (1970).4. Coghlan V/Л; Clausing R*E*, Atomic Data, 5, 317 (1973).5. Kim K.S., Gaarenstroom S.JV., Winograd N*, Chem. Phys. Lett., 41, 503
(1976).6. Larkins F.P., Atomic Data and Nuclear Tables, 20, 311 (1977).7. Davies L»E*, MacDonald N*C», et ah, Handbook of Auger Electron Specto-
scopy, Physical Electronics Division, Perkin-Elmer Corporation, USA,
1978.8. McGuire G.E., Auger Electron Spectroscopy Reference Manual, Plenum,New York, 1979.9. Kny J. Vac. Sci. Y. Technol., 17, 658 (1980).10. Kleefeld Levenson L.L., Thin Solid Films, 64, 389 (1979).11. Smith AM., Levenson L.L., Phys. Rev., B16, 1365 (1977).12. Salmeron MBaro AM*, Rojo J.M*, Phys. Rev., В13, 4348 (1976).13. Madden J. Vac Sci. Technol., 18, 677 (1981).14. Weightman P., Rep. Prog. Phys., 45, 753 (1982).15. Aksela H*, Aksela S., J. Phys. B: Atom. Mol. Phys., 7, 1262 (1974).16. Ritchie R.H., Phys. Rev., 106, 874 (1957).17. Stem E*A; Ferrel R.A., Phys. Rev., 120, 130 (1960).18. Dufour G; Mariot J.M., Nils son-] atko Р.Е», Kamatak R*C», Physica
Scripta, 13, 370 (1976).19. Massignon DPellerin F; et al., J, Appl. Phys., 51, 808 (1980).20. Bishop H.E; Riviere /.С. (в печати).21. Scofield J*HJ. Electron Spectrosc., 8, 129 (1976).22. Orchard Л.Р., in: Handbook of X-ray Ultraviolet Riotoelectron Spectroscopy
(ed. D. Briggs.), Heyden and Son, London, 1977, p. 49.23. Barrie AChristensen N*E*, Phys. Rev., В14, 2442 (1976).24. HilligH., Cleff B., Mehlhom W., Z. Phys., 268, 235 (1974).25. Aksela S., Aksela HPhys. Lett., A48, 19 (1974).26. Wagner C.D., in: Handbook of X-ray and Ultraviolet Photoelectron Spectrosco¬
py (ed. D. Briggs), Heyden and Son, London, 1977, p. 256.27. Wagner C.D., Taylor J.A., J. Electron Spectrosc., 20, 83 (1980).28. Siegbahn Кet ah, ESCA Applied to Free Molecules, North-Holland, Am¬
sterdam, 1969.29. Hendrickson D*N; Hollander ]M», Jolly W*L»f Inorg. Chem., 8, 2642 (1969).30. Wagner C*D*, in: Handbook of X-ray Photoelectron Spectroscopy (ed. G.E. Mui-
lenburg), Perkin-Elmer Corporation, USA, 1979.31. Wertheim G.K., J. Franklin Institute, 298, 289 (1974).32. Wertheim G*K*, Campara М., et aU Phys. Rev. Lett., 34, 738 (1975).33. Wagner C.D., Biloen P. Surf. Sci., 35, 82 (1973).34. Wagner C.D., in: Handbook of X-ray and Ultraviolet Photoelectron Spectro¬
scopy (ed. D. Briggs), Heyden and Son, London, 1977 p. 262.35. Wagner С.Z)., Farad Discuss.,Chem. Soc., 60, 291 (1975).
Д. Бриггс, Дж. К. Ривьер15936. Wagner C.D*, Gale L.H», Raymond R*H», Anal. Chem., 51# 466 (1979).37. West R*H., Castle /•£., Surf. Interface Anal., 4, 68 (1982).38. Wagner C.D., in: Handbook of X-ray and Ultraviolet Photoelectron Spectro¬
scopy (ed. D. Briggs), Heyden and Son, London, 1977, p. 266.39. Wagner C»D*, Zatko D»A., Raymond R*H*, Anal. Chem., 52, 1445(1980).40. Carlson T»A., Photoelectron and Auger Spectroscopy, Plenum,New York,
1976. [Имеется перевод: Карлсон Т.А. Фотоэлектронная и оже-спектро¬
скопия. - Л.: Машиностроение, 1981.]41. Koel В*Е%, White JMJ. Electron Spectrosc., 22, 237 (1981).42. Kowalczyk S.P., Ley L., et aU Phys. Rev., В 7, 4009 (1973).43. Fadley C.S., in: Electron Spectroscopy: Theory, Techniques and Appli¬
cations, vol. 2 (eds. C.R. Brundle, A.D. Baker), Academic Press, London,
1978.44. Clark D.T., Adams D.BChem. Phys. Lett., 10, 121 (1971).45. Briggs D; Gibson V*AChem. Phys. Lett., 25, 493 (1974).46. Allen G*C; Tucker P., Inorg. Chem. Acta, 16, 41 (1976).47. Clark D.T; Dilks AJ. Polym. Sci. Polym. Chem. Ed., 15, 15 (1977).48. Frost D.C., McDowell С.Л., Ishitani A,, Mol. Phys., 24, 861 (1972).49. Burroughs P., Hamnett Aet aU, J. Chem. Soc. Dalton Trans., 1686 (1976).50. Matienzo LJ., Swartz W.E., Grim S.O*, Inorg. Nucl. Chem. Lett. 8, 1085
(1972).51. Frost D.Cb, McDowell С.Л., Woolsey LS., Mol. Phys., 27, 1473 (1974).52. Barrie ASwift P., Briggs D*, Preprints, 27th Pittsburgh Conference on
Analytical Chemistry and Applied Spectroscopy, Cleveland, USA, 1967.53. Pireaux ]•]*, Caudano R*, Verbist J. Electron Spectrosc., 5, 267
(1974).54. Siegbahn КJ. Electron Spectrosc., 5, 3 (1974).55. Dilks A*, in: Electron Spectroscopy: Theory, Techniques and Applications,
vol. 4 (eds. C.R. Brundle, A.D. Baker), Academic Press, London, 1981.56. Fadley C.5*, Prog. Solid State Chem., 11, 265 (1976).57. Mehta М., Fadley C.5., Phys. Lett., A55, 59 (1975).58. Briggs D, Marbrow R.A. Lambert R.MSolid State Comm.,26, 1 (1978).59. Evans S., Scott M»D*, Surf. Interface Anal., 3# 269 (1981).60. E-uans S; Adams J.M., Thomas }.M,9 Phil. Trans. Roy. Soc. London,292, 563 (1979).61. Hirokawa КDanzaki YSurf. Interface Anal., 4 (1982).62. Copperthwaite R.G., Surf. Interface Anal., 2, 17 (1980).63. Clark D*T; Briggs £).; Nature Phys. Sci., 237, 15 (1972).64. Thompson M», Lennox R*B*, Zemon D»J., Anal. Chem., 51, 2260 (1979).
Глава 4
ПОСЛОЙНЫЙ АНАЛИЗС. Хофман"4.1. ВведениеАнализ химического соединения, направленный на выявление распреде¬
ления компонент по глубине образца, обычно называется локальным трех¬
мерным анализом. При этом наибольший интерес представляет третье из¬
мерение, перпендикулярное поверхности образца. В принципе для решения
подобной задачи необходимо провести анализ состава тонких сечений (в пре¬
дельном случае имеющих толщину в один моноатомный слой), задаваемых
по шкале измерения глубины. Ее можно решить с помощью как неразрушаю¬
щих , так и разрушающих методов анализа.4.1.1. Неразрушающие методы анализ?Неразрушающие методы основаны на измерении определяющего пара¬
метра (например, интенсивности, энергии), величина которого имеет из¬
вестную зависимость от глубины регистрации. Например, энергетические
потери заряженных высокоэнергетичных частиц пропорциональны расстоя¬
нию, пройденному ими в данном материале. Это свойство эффективно ис¬
пользуется в спектроскопии POP (спектроскопии резерфордовского обрат¬
ного рассеяния первичных ионов, обычно Не4*, 100 кэВ - 5 МэВ, с глубин
0,1 - 30 мкм) [8, 9]. В таких измерениях зависящие от атомного номера
сечения упругого рассеяния несут аналитическую информацию, в то время
как энергетические потери характеризуют глубину, с которой идет регист¬
рируемый сигнал. В случае активационного анализа (АА) для послойного
анализа используются резонансные зависимости сечений ядерных реакций
от энергии падающих частиц: по мере увеличения энергии первичного пучка
возрастает вклад все более глубоких слоев образца. Подобные методы очень
хороши для количественных оценок как концентрации, так и ее распределе¬
ния по глубине с чувствительностями, более или менее сравнимыми с обес¬
печиваемыми электронной спектроскопией. Из-за малых эффективных сече¬
ний неупругого рассеяния легких элементов POP применимо только для
элементов с Z > 10 и ограниченно используется для элементов, распреде-^ S. Hofmann, Max-Planck-Institut fur Metallforschung, Institut fur Werkstoff-
wissenschaften, FRG.
С. Хофман161ленных внутри матрицы тяжелого элемента. В связи с тем что в настоящее
время наиболее широко используются ускорители с энергиями до 5 МэВ,АА применяется для определения легких элементов (Z < 20). Разрешение
по глубине, достигаемое с помощью POP и АА и определяемое в основном
разбросом экспериментальных данных и эффективным энергетическим
разрешением, зависит от материала и толщины слоя и лежит в пре¬
делах 5 - 200 нм [8, 9]. Для неразрушающих методов, использующих рент¬
геновское излучение, таких, как спектроскопия ионно-индуцированного
рентгеновского излучения или электронно-зондовый рентгеновский микро¬
анализ, разрешение по глубине еще хуже (обычно 1 мкм), что препятству¬
ет их использованию в послойном анализе тонких пленок.В электронной спектроскопии интенсивность измеряемого сигнала зави¬
сит от энергии и глубины выхода оже- и фотоэлектронов. Средняя длина сво¬
бодного пробега неупругорассеянных электронов является решающим пара¬
метром при определении глубины выхода. Для данной энергии электрона при
изменении угла эмиссии соответствующая эффективная глубина выхода ме¬
няется от максимального значения при движении перпендикулярно поверх¬
ности почти до нуля [4, 10, 11L Этот метод, применение которого ограничено
глубинами до 50 нм, более подробно рассмотрен в разд. 4.5.1.4.1.2. Разрушающие методыРазрушающие методы послойного анализа схожи с классическими ме¬
тодами механического послойного анализа (косые шлифы с сов¬
ременными усовершенствованиями типа шарового шлифа в сочетании с
методами исследования, позволяющими достичь высокого разрешения) и хи¬
мического послойного отбора проб с последующим химическим анализом
изъятого материала. Кроме проблем, связанных с использованием специфи¬
ческих материалов (например, керамик) и вытекающих из их химических и
механических свойств, к недостатку этих методов относится отсутствие
возможности контроля поверхностных реакций при проведении различ¬
ных технологических операций, что приводит к жестким ограничениям на
достижимое разрешение по глубине. Несмотря на это, прогресс, достигну¬
тый в течение последних лет в электрохимической полировке [ 12], показал,
что можно достигнуть разрешения по глубине порядка 10 нм.Универсальным методом снятия слоев является удаление материала
образца с помощью ионного распыления. Использование этого метода в
сочетании с любым из методов анализа поверхности широко применяется
для послойного анализа. Этой теме посвящен ряд обзоров [ 1 - 7].Распыление является разрушающим методом: образец бомбардируют
ионами, ускоренными в ионной пушке до энергий свыше 100 эВ (обычно
0,5-5 кэВ). Наибольшая часть энергии передается атомам поверхности,
что заставляет их покинуть образец: они "стравливаются”. Таким образом,
образец эффективно делится на две части. Ту часть, которая удалена, мож-
1624. Послойный анализно подвергнуть анализу, например, с помощью ВИМС, а поверхность остав¬
шейся части можно изучать, обратившись к ЭОС и (или) РФЭС.К основным преимуществам послойного анализа, выполненного с ис¬
пользованием ЭОС и РФЭС в сочетании с ионным травлением, относится то,
чтоа) информационная глубина имеет величину порядка 1 нм;б) анализ не зависит от интенсивности распыления;в) влияние матрицы на уровень элементной чувствительности
мало;г) анализируемая область мала по сравнению с областью распы¬
ления, что минимизирует кратерные краевые эффекты.Пункт 11 г” для РФЭС обычно не выполняется, что показано далее в
разд. 4.4.2.4.2. Практическое использование послойного анализа
с ЭОС и РФЭСПоскольку и ЭОС, и РФЭС имеют дело с поверхностью, образующейся
после определенного времени распыления, то и основные особенности ис¬
пользования этих методов в послойном анализе сходны. Однако имеют¬
ся и практические различия, например, такие параметры, как скорость
детектирования, величина фона и пространственное разрешение, обычно
лучше у ЭОС. Поэтому если не оговорено противное, мы будем рассмат¬
ривать в дальнейшем последний метод. Любое промышленное оборудова¬
ние для ЭОС/РФЭС включает электронный энергоанализатор, источник
электронов и (или) источник рентгеновского излучения, ионную пушку и
держатель образца, которые смонтированы в неподвижной камере из не¬
ржавеющей стали, где можно поддерживать сверхвысокий вакуум (< Ю-* Па) „4.2.1. Требования к вакуумуИсходя из средней длины свободного пробега электронов в газах, можно
заключить, что для проведения достоверного анализа распределения элект¬
ронов по энергиям достаточен вакуум лучше, чем Ю~2 Па. Однако необходи¬
мо избегать загрязнений, возникающих в ходе самого процесса анализа
[13-15]. Если считать, что коэффициент прилипания равен единице, то
монослой на поверхности адсорбируется за 1 с при парциальном давлении
Ю-4 Па (« К)-6 мм рт. ст.). Чтобы избежать адсорбции 1% монослоя за ти¬
пичное время измерения (в ЭОС), равное 100 с, парциальное давление хими¬
чески активных газов, таких, как СО, Н20, СхНу, должно быть ниже 10“* Па
(Ю-10 мм рт. ст.), т. е. соответствовать СВВ. Инертные газы (например,
аргон), которые не хемосорбируются, не искажают результатов и при дав¬
лении на несколько порядков больше.
С. Хофман1634.2.2. Ионная пушкаНаиболее часто используемые в оже-спектрометрах ионные пушки
представляют собой простые электростатические устройства, в которых
ионы инертного газа (Аг) возникают вследствие ударной ионизации элект¬
ронами, эмитированными термокатодом и ускоренными обычно до энергии
100 эВ. Положительно заряженные ионы ускоряются до энергии 0,5-5 кэВ
и фокусируются в образце, создавая область распыления диаметром поряд¬
ка 1 -5 мм. Для достижения плотности ионного пучка примерно 100 мкА/см2
давление в ионном источнике пушки должно составлять около 5 • 10-* Па
(5 • 10-® мм рт. ст.). Обычной операцией в системах с ионной откачкой яв¬
ляется заполнение всей камеры аргоном при отключенном насосе. Для обес¬
печения чистоты газа, с помощью которого производится распыление, необ¬
ходимо, чтобы отношение парциального давления примесей к парциальному
давлению газа не превышало Ю~8/5 • 10”3 = 2 • 10"6. Титановый субли¬
мационный насос с криопанелью обычно служит эффективным средством
удаления химически активных газов, что позволяет избежать загрязнения
газа в последующем. По сравнению с такими " статическими11 системами
суммарное загрязнение удается уменьшить, применяя более совершенный
тип ионной пушки, функционирующей в 11 динамическом” режиме с постоян¬
но поступающим потоком распыляющего газа, откачиваемого насосом ос¬
новной камеры. Дальнейшим шагом вперед может служить дифференциаль¬
ная откачка ионной пушки геттероионным насосом или дополнительным
турбомолекулярным или диффузионным насосом. Иэ-за малого (« 1 мм) от¬
верстия на конце ионной пушки в основной (аналитической) камере созда¬
ется давление, почти в 100 раз меньшее, чем давление у выходной диафраг¬
мы ионной пушки. Это является дополнительным преимуществом динами¬
ческого метода, поскольку уменьшается разрушение катода электронной
пушки ускоренными положительно заряженными в процессе распыления ио¬
нами и (или) предотвращается возникновение дугового разряда в источнике
рентгеновского излучения, что часто встречается при полном давлении,
превышающем 10~4 Па.Простые ионные пушки, дающие статическое пятно распыления, могут
быть механически отъюстированы таким образом, что фокус анализатора
(вместе с бомбардирующим образец электронным пучком) будет почти точ¬
но соответствовать центру гауссианы, описывающей распределение интен¬
сивности ионного пучка, что позволяет избежать заметного уменьшения
разрешения по глубине [14]. Неоднородность распределения интенсивности
ионных пучков при размерах порядка 1 мм ограничивает их применение для
проведения послойного анализа с помощью РФЭС.Для точного послойного анализа следует использовать ионную пушку
с возможностью отклонения пучка по осям х/у. Это позволяет точно наме¬
тить анализируемую область и зону распыления (используя для мониторин¬
га вторичную электронную эмиссию, регистрируемую с помощью ЦЗА).
1644. Послойный анализСканирование хорошо сфокусированным ионным пучком по большой площа¬
ди (до 10 х 10 мм) существенно улучшает равномерность распределения ин¬
тенсивности ионного пучка [ 15], позволяя получить плоское дно кратера
распыления, что необходимо для достижения оптимального разрешения по
глубине. Более того, при постоянном токе пучка величина анализируемой
области обратно пропорциональна полной плотности тока пучка, что дает
возможность легко контролировать скорость распыления. Ионное травле¬
ние сканирующим ионным пучком должно предшествовать послойному ана¬
лизу с помощью РФЭС, при котором величина исследуемой области состав¬
ляет несколько квадратных миллиметров.Для предотвращения эффектов затенения пучком при работе с шерохо¬
ватыми поверхностями углы между ионным пучком, электронным возбуж¬
дающим пучком и регистрируемыми электронами должны быть как можно
меньше. В частности, ионный пучок должен проходить вблизи нормали к по¬
верхности образца [16]. Чтобы избежать образования конусов, целесооб¬
разно проводить распыление двумя пушками, расположенными под разными
углами [ 17].4.2.3. Способы проведения анализаОже- или фотоэлектронный анализ может осуществляться либо после
окончания распыления, либо одновременно с распылением. В послойном ана¬
лизе с помощью ЭОС чаще всего используется последний метод вместе с
регистрацией оже-пиков различных элементов с помощью блока мульти¬
плексора и выводом амплитуд (peak-to-peak) соответствующих оже-пиков.В случае применения РФЭС сильный рост вторичной электронной эмиссии
во время распыления затрудняет проведение послойного анализа одновре¬
менно с ионным распылением. Кроме того, для снятия контура пика в РФЭС
требуется большее время измерения, чем в ЭОС, что ограничивает дости¬
жимое разрешение по глубине A za при данной скорости распыления z. Если
Atm - время, необходимое для измерения пика с желаемым отношением сиг¬
нала к шуму, то Aza = z . Д*ш. В отличие от ВИМС, где z и Atm обратно
пропорциональны [18], в послойном анализе с помощью электронной спект¬
роскопии эти характеристики независимы. Поэтому значения Aza могут
быть сделаны пренебрежимо малыми по сравнению с многими другими фак¬
торами, ограничивающими разрешение по глубине, путем уменьшения плотности пер¬
вичного ионного пучка или прерывания распыления. В общем случае из-за большего
значения Д*т это следует делать в случае послойного анализа с помощью РФЭС По¬
слойный анализ с помощью ЭОС с использованием AU3 часто применяют одновре¬
менно с распылением при скорости последнего до 5 нм/мин. Дополнительным пре¬
имуществом большой скорости распыления является одновременное уменьшение за¬
грязнения поверхности (вызванного адсорбцией газа, содержащегося в камере),
что происходит благодаря увеличению десорбции при распылении (см.
п» 4.4.2.1).
С. Хофман1654.3. Количественная интерпретация данныхпослойного анализаНаиболее важной информацией, получаемой во время послойного анали¬
за, являются зависимости интенсивности сигналов, соответствующих де¬
тектируемым элементам, / (т. е. амплитуды оже-пиков peak-to-peak), от
времени распыления, t, т. е. "измеренный за время травления профиль рас¬
пределения элементов" / = f(t). Основной задачей служит получение дейст¬
вительного распределения концентрации элементов по глубине г, с = f(z),
при помощи соответствующей обработки результатов измерений [3-7].Основные этапы решения этой задачи представлены на рис. 4.1. Вна¬
чале необходимо установить взаимосвязи между глубиной кратера травле¬
ния z = f(t) и временем травления, с одной стороны, и интенсивностью
оже-сигнала и локальной концентрацией соответствующего элемента, с дру¬
гой, с = f(z). Таким образом, удается выявить "истинный" профиль концен¬
траций, который совпал бы с первоначально существовавшим, если бы про¬
цесс распыления осуществлялся как гомогенный, при котором распыление
шло^как это должно быть в идеальном случае, "слой за слоем". Однако,
чтобы определить этот первоначальный профиль с = f(z), необходимым вто¬
рым шагом является учет искажений, возникающих в основном из-за обус¬
ловленных распылением мгновенных топографических и композиционных
изменений поверхности образца. Точность, с которой форма полученного
"истинного" профиля концентраций передает форму первоначального профи¬
ля концентраций, может быть определена, если известно разрешение по глу¬
бине [1-7], являющееся ключевым параметром при послойном анализе.
Знание разрешения по глубине или точнее функции разрешения по глубинеРис. 4.1. Методика оценки профилей распыления: преобразование измеренного про¬
филя распыления. /=/(*) в истинный профиль концентраций с = f (z) L 3].1 - вызванные распылением изменения состава и морфологии поверх¬
ности; 2 — количественный анализ поверхности; 3 — скорость распыления.
1664. Послойный анализявляется обязательным для решения задачи обратной свертки профилей
концентраций.Этапы анализа профилей концентраций рассмотрены в последующих раз¬
делах.4.3.1. Калибровка скорости распыления [z = /(*)]Скорость поверхностной эрозии описывается мгновенным значением
скорости распыления z = dz/dt, что позволяет определить среднюю глу¬
бину эродированной части образца как функцию времени распыления:(4.1)Скорость мгновенного распыления z (в м/с) определяется по формуле(4.2)где М - атомная масса, р - плотность (кг/мЗ), na _ 6,02 • 1026 (число
Авогадро), е -1,6 • 10~*19 (заряд электрона), S - выход распыления
(атом/ион), jp - цлотность первичного ионного пучка (А/м2).Для постоянной скорости распыления уравнение (4.1) дает прямо про¬
порциональную зависимость глубины распыления от времени(4.3)В этом случае величина z линейно зависит от времени распыления и только
одна (не считая нулевой) точка необходима для определения постоянной
калибровки z. Эта величина может быть вычислена из уравнения (4.2), ес¬
ли заимствовать выход распыления S из литературных данных [ 19-21] и
измерить jp с помощью цилиндра Фарадея. На рис. 4.2 приведен порядок
величин, который может быть экспериментально реализован. Однако сам
выход распыления S зависит от многочисленных параметров, включая энер¬
гию и массу ионов, угол падения первичного ионного пучка [ 19] и состав
поверхности [13]. Таким образом, такой расчет пригоден лишь для грубой
оценки.Более подходящим методом определения скорости распыления z явля¬
ется измерение (при заданных рабочих параметрах ионной пушки) време¬
ни, необходимого для распыления слоя известной толщины, например мини¬
мального слоя металла или слоя оксида. Для этой цели оказалось очень
полезным использование танталовой пленки, полученной анодированием
[ 22, 23], поскольку ее толщина легко контролируется по величине приложен¬
ного напряжения [24]. Кроме того, благодаря наличию резко выраженной
границы оксид - металл она позволяет проводить быструю проверку раз¬
решения по глубине, обеспечиваемого используемой аппаратурой (см.
разд. 4.4). Таким образом, величина z может быть определена с помощью
С. Хофман167Рис. 4*2» Зависимость мгновенной
скорости распыления ж от
плотности тока первичного
ионного пучка при зна¬
чении М/р * Кг"® м3/моль
и трех различных значени¬
ях выхода распыления S,
соответствующих уравне¬
нию (4« 2) •сопоставления известной скорости распыления Ta2Og и скорости распыле¬
ния исследуемого материала [ 23].Другой возможностью является измерение глубины кратера z0 после
распыления с помощью интерферометра или профилографа [25, 26].В общем случае z изменяется при изменении состава образца, пос¬
кольку, согласно уравнению (4.2), величины М, р и 5 являются функциями
состава и взаимосвязь между временем распыления и глубиной становится
нелинейной [ 27, 28]. Даже при постоянном составе скорость распыления
может изменяться при сравнении нескольких начальных слоев и>за нараста¬
ния структурных и композиционных изменений, пока не будет достигнуто
равновесное состояние поверхности, возникающее, когда глубина распыления
становится сравнимой со средней длиной свободного пробега ионов первич¬
ного пучка [ 29].Поэтому очевидно, что точное определение глубины травления требует
измерения мгновенного значения скорости распыления в процессе травле¬
ния. Этого можно добиться, измеряя in situ толщину остающихся тонких
пленок, например, путем непосредственного измерения массы с помощью
кварцевых микровесов [30], при проведении рентгеновского эмиссионного
анализа [ 29] или путем измерения толщины этих пленок in situ с помощью
лазерного оптического интерферометра [ 31 - 32], например методом, разви¬
тым в последнее время Кемпфом. К сожалению, последние методики непри¬
менимы при работе со стандартным оборудованием.Первое приближение для оценки поправки в соотношении, связывающем
время и глубину распыления в случае наличия зависящей от состава нели¬
нейности, было получено при распылении многослойных структур Ni/Cr,
созданных напылением [ 3, 28], в предположении, что изменение скорости
1684. Послойный анализраспыления пропорционально изменению состава. Для двухкомпонентной
системы А /В полное значение мгновенной скорости распыления г задает¬
ся молярным содержанием ХА, Хв и скоростью распыления zA, zB чис¬
тых компонентДля этого приближения ХА и Хв могут быть получены из нормирован¬
ных оже-сигналов 1А/1\, 1в^в-Ограничения и в некоторых случаях оценки скорости распыления могут
быть получены с помощью рассмотрения результатов количественного ана¬
лиза ЭОС и РФЭС, как это показано в следующем разделе.4.3.2. Калибровка шкалы концентраций [с = /(/ )]Количественные ЭОС и РФЭС рассмотрены в гл. 5. Важной особенностью
снятия профилей концентрации является наличие зависимости между интен¬
сивностью Ii и концентрацией с t- элемента г, которая может быть выра¬
жена в виде явной функции глубины выхода электрона и в случае ЭОС -
коэффициента обратного рассеяния [33 - 38]:где !? - интенсивности чистых элементов, Л£ - "эффективные" глубины
выхода оже-электронов или фотоэлектронов, перпендикулярных поверхнос¬
ти образца, с i (z) - локальная концентрация (в долях моля) на глубине z,
г t > 1 - коэффициент обратного рассеяния (см. гл. 5 этой книги). "Эффек¬
тивная" глубина выхода Xi определяется длиной неупругого рассеяния
[ 35] электронов данной энергии в данном материале X ® и углом q> эмиссии
зарегистрированных электронов относительно нормали к поверхности об¬
разца:Значения Л? обычно лежат между 0,4 и 4 нм [ 35]. Угол <р просто определя¬
ется для полусферических анализаторов. Для анализаторов типа цилиндри¬
ческое' зеркало (АНЗ) может быть определен средний угол эмиссии q> [ 33].
Его зависимость от угла а между нормалью к поверхности образца и осью
ЛИЗ показана на риг. 4.3. ,1ш а * 47,7° часть угла сбора AII3 затеняется
самим образцом, что вызывает отклонение от косинусоидальной зависимос¬
ти (рис. 4.3).(4.4)В соответствии с уравнениями (4.1) и (4.4)(4.5)(4.6)(4.7)
С. Хофман169Рис. 4.3. Зависимость средней величины характеристического параметра глубины вы¬
хода (cos ф) от угла а между осью АЦЗ и нормалью к поверхности образца.
Для а с = 47,7° из-за теневого эффекта относительная интенсивность про¬
пускания анализатора / //0 падает. Пунктирная линия (1) соответствует ко¬
нусу разлета с Дф= 12°.Коэффициент обратного рассеяния трудно уточнять при наличии гради¬
ентов концентрации. Для двухкомпонентных систем приближенное выраже¬
ние было предложено Морабито и др. [6, 37]. В принципе необходимо знать
распределение плотности возбуждающего излучения по глубине, которое
зависит от распределения концентраций. Для двухкомпонентных систем
гм,а = О ~ гв/га)са + гв/гл* Было показано [4, 39], что для профиля
интенсивностей на двухкомпонентной поверхности раздела А/В, зная характе¬
ристическую длину обратного рассеяния (lb)t можно вычислить коэф¬
фициент усиления интенсивности сигнала элемента Л из выражения
[ 1 + гв/га ~ ^ехр -(z0 -z)/lb] (где z0 характеризует положение поверх¬
ности раздела). Эта зависимость подтверждена данными эксперимента [ 4].
Обычно проще провести уточнение коэффициента обратного рассеяния, ис¬
ходя из уточненного значения глубины выхода. За исключением особых
случаев (резкая граница раздела между элементами с большой разницей в
атомном номере), обратное рассеяние электронов в ЭОС является эффектом
второго порядка по сравнению с влиянием глубины выхода, которое обсужда¬
ется ниже.Если пренебречь ri , то уравнение (4.6) справедливо как для ЭОС, так
и для РФЭС. Послойный анализ с математической точки зрения сводится к
смещению нижней границы интегрирования в этом соотношении от нуля к
1704. Послойный анализРис. 4.4. Форма измеренного профиля концентрации тонкого "сандвича” А/В/А,
обусловленная влиянием глубины выхода в соответствии с уравнениями
(4.8а) и (4*86), в предположении наличия идеальной структуры [ 3, 7].мгновенному значению глубины распыления. Таким образом, для заданного
профиля с [г) интегрирование в уравнении (4.6), где правая часть представ¬
ляет собой функцию от z, позволяет получить 1 (z) для случая идеального
непрерывного распыления [3, 7, 39]. Ожидаемый профиль интенсивностей для
типичной ситуации, когда между матрицами элемента А имеется тонкий слой
элемента В ("сандвичевая” структура), показан на рис. 4.4. Интегрирование
уравнения (4.6) в этом случае даетдля z j -z>0nz2-z>0. Отметим, что Ав#л представляет собой глубину
выхода электронов элемента В в Л, а Ав> в - элемента В в В» Для Ав А == Лв, в = * соотношения между интенсивностью и глубиной имеют виддля zt < z < z2.Так как каждый профиль концентраций может быть аппроксимирован
набором тонких слоев толщиной z2 -Zj, уравнение (4.8) дает метод опреде¬
ления интенсивности на глубине z в виде суммы вкладов этих слоев [7,39-42].
На практике достаточно заменить верхний предел интегрирования («>) в урав¬
нении (4.6) величиной zu = z + 5Л. Это обусловлено тем, что разница в резуль¬
тате « 0,7%) будет меньше, чем предел чувствительности ЭОС и РФЭС.(4.8а)(4.86)
С. Хофман171В общем виде решение (4.6) для произвольного профиля можно найти из
(4.8). Оно имеет вид [ 3, 7, 43](4.9)Используя уравнение (4.9), любой измеренный профиль распыления мож¬
но скорректировать с учетом влияния глубины выхода [29, 39, 44, 45]. Как
видно из рис. 4.4, имеет место сдвиг реального профиля 0,7 X в сторону
меньших глубин по сравнению с измеренным профилем, который уширен на
величину AzA « 1,6А [ 5, 7, 39]. Интересно отметить, что максимальный гра¬
диент, который можно зафиксировать, определяется соотношением (4.9)
и поэтому [ 4, 7]:(4.10)Так как dz = zdt, то отсюда определяется нижний предел изменения
мгновенной скорости распыления:(4Л0а)Часто интенсивности при проведении послойного анализа калибруются
с использованием коэффициентов относительной чувствительности [34, 46],
считая, что концентрация пропорциональна мгновенному значению интенсив¬
ности (это означает пренебрежение влиянием X), В этом случае относитель¬
ная погрешность (Ас,- /с, ) л зависит от крутизны измеренного профиля и
совпадает по порядку величины с относительным изменением интенсивности
с глубиной X:(4.11)Используя уравнение (4.9) для описания профиля концентраций, можно
провести количественную оценку измеренных профилей (см. рис. 4.1), ес¬
ли известны эффективная глубина выхода электронов (и коэффициент об¬
ратного рассеяния в ЭОС), нормированные интенсивности и мгновенная
скорость распыления. В особом случае, когда выявляются два пика одно¬
го элемента, лежащие при различных энергиях, и, следовательно, имеют
место различные глубины выхода Х1# Х2 (например, пики ЭОС меди 60 и
920 эВ), если получена зависимость градиента интенсивности от времени
распыления, уравнение (4.9) дает возможность вычислить скорость распы¬
ления с учетом этих глубин выхода [ 47]. Ввиду того, что правая часть
уравнения (4.9) одинакова для /ж(X j) и для /2(Х2), из dz = zdt следует
1724. Послойный анализХотя определенная из (4.12) величина г имеет ограниченную точность,
однако она позволяет быстро оценить порядок величины скорости распыле¬
ния [ 47].На этом этапе мы не рассматривали обусловленные самим процессом
распыления искажения первоначального профиля концентраций, которые
должны быть учтены при его оценке.4.4. Сравнение профиля концентраций, возникающего
при распылении, с первоначальным профилем
концентраций: понятие разрешения по глубине4.4.1. Разрешение по глубине
4.4.1.1 с Определение разрешения по глубинеВлияние средней глубины выхода оже-электронов или фотоэлектронов
на измеряемый профиль приводит к определенному уширению истинного
профиля, как это показано в разд. 4.3.2. До сих пор мы считали, что распы¬
ление протекает как идеальное снятие слоев помонослойно, а углубление
кратера в образец происходит с постоянной скоростью. Экспериментально
это недостижимо, так как ионная бомбардировка неизбежно вызывает из¬
менения в топографии и составе поверхностной области образца. Эти эф¬
фекты, вызванные распылением, при регистрации профиля проявляются в
виде дополнительного уширения, которое можно математически описать с
помощью функции разрешения g(z, z') [ 48, 49], как показано на рис. 4.5.Рис. 4.5. Изображение свертки при послойном анализе с использованием ионногораспыления: каждый тонкий слой истинного профиля с (г) (кривая 1) уши¬
ряется при свертке с g(z -z°) (кривая 2). Измеряемый профиль / (г)
(кривая 3) получается при суммировании вкладов всех подобных слоев
[48].
С. Хофман173Любой бесконечно тонкий сегмент, содержащий истинную концентрацию
исследуемого элемента, при снятии распределения концентрации по глуби¬
не с (г') будет уширен, и сумма всех таких уширений дает нормированную
интенсивность сигнала на глубине распыления z, если нормирована функ¬
ция разрешения(4.13)Нормированная интенсивность I (z) может быть выражена в виде сверт¬
ки [ 3, 48, 49](4.14)Если известна функция разрешения g(z - z'), интегральное уравнение
(4о14) можно подвергнуть обратной свертке и тем самым найти истинный
профиль с,- (z')e Операции, необходимые для этого, рассматриваются в
разд. 4.6.Следует отметить, что уравнение (4.14) также является интегралом
свертки для количественных ЭОС и РФЭС, если g(z -z’) = ехр[ -(z - z')/Af- ]
[уравнения (4.5) и (4.6)]. В этом случае решением уравнения (4.14) являет¬
ся уравнение (4.9) и вид функции разрешения для тонких сечений образца
совпадает с кривой, изображенной на рис. 4.4.Во многих экспериментах по послойному анализу было замечено, что
измеренный профиль может быть аппроксимирован функцией ошибок
[1-7, 26, 39, 48-52]. Этой функцией для g(z -z') является функция оши-Рнс. 4.6. Определение разрешения по глубине Az в случае аппроксимации функции
распределения гауссовой кривой, а - истинный профиль в виде ступенчатой
функции Az = 2ст(0,84 - 0,16), Az* (0,9 - 0,1) = 2,564 ст , Аг0в(обратный
экстремум) = 2,507 ст, Az*#° (ГШ1ПВ функции разрешения) = 2,355а [5, 53];
б- однослойная "сандвичевая" структура л/В/А, где Az определяется
отношением ls/lQ из уравнения (4.15а) [з, 51];- в - многослойная "санд-
вичевая” структура А/в/А/в, где Az определяется отношением / //0
из уравнения (4.156) [з].>
1744. Послойный анализбок Гаусса, которая определяется одним параметром - стандартным откло¬
нением а. Наиболее общим определением разрешения по глубине является
соотношение Az = 2а, что соответствует разности координат по глубине
z при изменении интенсивности сигнала на границе раздела от 16 до 84%.На рис. 4.6, а показаны различные способы, используемые при опреде¬
лении разрешения по глубине. Относительное разрешение по глубине опре¬
деляется выражением A z/z, где z характеризует глубину расположения
анализируемой поверхности. Для постоянной скорости распыления £ относи¬
тельное разрешение по глубине определяется ожидаемым временем распыле¬
ния A z/z = At/f •Разрешение по глубине может быть либо определено экспериментально,
либо оценено в результате вычислений, базирующихся на подходящих моде¬
лях [3].4.4.1.2. Измерение разрешения по глубинеФункция разрешения определяется непосредственно в процессе распыле¬
ния очень тонкого слоя, как похазано на рис. 4.5. Значительно чаще исполь¬
зуется метод, заключающийся в получении прямоугольного профиля с пос¬
ледующим определением производной [ 3, 48, 49, 50]. При аппроксимации
гауссовой кривой количественное значение разрешения по глубине Az можно
получить непосредственно из профиля, полученного при распылении, как
показано на рис. 4.6, а.Для случая слоя "сандвич" толщиной d было показано [51], что уменьше¬
ние максимальной интенсивности /0 (в случае идеального профилирования)
до значения !s (рис. 4.6, б) определяет разрешение по глубинеДля многослойных структур такого рода (например, А/В/А/В...) с пос¬
тоянной для одного слоя толщиной d можно получить подобное выражение,
которое связывает амплитуду интенсивности 1т (как показано на рис. 4.6, в)
с нормированной интенсивностью нераспыленного слоя /0 [ 3, 16, 51 - 53]:Для Az4 d достаточно хорошую аппроксимацию дают первые два чле¬
на. На рис. 4.7 приведен вид /5 /10 и 1т //0 как функций от Az/d [3, 53].Фундаментальным вопросом в практическом определении разрешения по
глубине является его зависимость от глубины распыления. Многослойные
"сандвичевые11 структуры [ 28, 54-58] особенно подходят для решения это-(4.15а)(4.156)
С. Хофман175Рис. 4.7. Диаграмма для определения Az однослойных (7) и многослойных (2) "Санд¬
вичевых" структур известной толщины d% приведенных на рис. 4.6, б, в со»
ответствии с уравнением (4.15 а, б). Аппроксимации для случаев суперпо¬
зиции двух (сплошная линия) и четырех (штриховая линия) смежных слоев
показаны для конфигурации, приведенной на рис. 4.6, в.го вопроса, поскольку зависимость от глубины можно получить из одного
эксперимента, аппроксимируя А г с помощью уравнения (4.15в) [ 3, 4, 16, 28].
Р езультаты подобного анализа для различных систем приведены на рис. 4.8.Рис. 4.6. Зависимость относительного разрешения по глубине от глубины г для раз¬
личных многослойных "сандвичевых" структур, полученная с помощью по¬
слойного анализа с применением распыления образцов ионами Аг+ и(или)N+ и ЭОС.Г-Ni /Сг | Аг+ [54]; 2 - Cu/Ni | Аг+ [ 5в]; 3 - Ni /Cr|N+ [2d; 4-
Cu/Ni I [58]; 5- GaAs -Ga, _XA1X As | Ar+ [55]; 6- Ge/Nb|N* [58].
Литература[1, 7, 14 ,15,
50, 60][18][15][4, 5, 9, 62,63, 108, 109]
[50][3-4, 5, 7,29, 39, 44, 45,
51][16, 17, 54, 56]
[64, 65, 100]
[696 70][77, 107, 111]Воздействие, оказываемое при
проведении послойного анализа,
и вклад в разрешение по глубинеСм. уравнения (4.16 а, б)(С, О)Эффект «запоминания» перво-
начального композиционного
составаАнализируются наравне с друг-
гими компонентами образцаНеоднородное распыление:Az, - гИскажения профиля:i = тСдвиг профиля: 0,7Х;Az х = const * 1,6 XAzr ~ z1/2, ... , zД2С ~ ZНеровности из-за селектив¬
ного распыления
Искажения, вносимые зарядкой
образцаПричины искажения про¬
филя концентрацийАдсорбция остаточных газов
атмосферы камеры
Повторное осаждение ранее
распыленных материаловВлияние стенок кратера
Примеси в ионном пучкеПоспупление нейтралов из
ионной пушкиНеоднородность ионного пучка
по интенсивности
Зависимость интенсивности
ионного пучка от времени
Информационная глубинаИсходная шероховатость об¬
разцаКристаллическая структура и
дефектыСплавы, соединения, двойные
фазыДиэлектрикиАппаратурныефакторыПараметрыобразцаТаблица 4.1. Сводная таблица различных факторов, ухудшающих результаты при проведении послойного анализа, и оценка их
вклада Azf в величину разрешения по глубине
Продолжение табл. 4.1.
[28, 29, 59, 112][71, 78—83][50, 5, 72, 75,110][14, 47, 83—91]
[57, 76, 92—94]
[95, 96, 109][92, 111]Изменение начальной величи¬
ны г; зависимость z отсостава образца
Azk — const(~ Rp)
для z > RpДzs — const, - z1/2 zДzp — const(z > Rp)ДZr = f(Db, Ds, z, t, ...)Дге = f(%, i„, z, t, ...)Искажение результатов ана¬
лиза, электропереносИмплантация ионов первичного
пучкаАтомное перемешиваниеВозникновение шероховатостей,
вызванное распылением
Избирательное распыление и из¬
менение состава соединений
Усиление диффузии и образо¬
вание сегрегаций
Электронно-стимулированная
десорбцияЗарядка диэлектриковЭффекты,
связан¬
ные с
воздейст¬
вием излуче¬
ния
1784. Послойный анализ4,4.2. Факторы, ограничивающие разрешение по глубинеИсточники искажений профилей концентраций при послойном анализе
сведены в табл. 4.1. Их можно разделить на аппаратурные факторы, факто¬
ры, зависящие от характеристик образца, и факторы, обусловленные фунда¬
ментальными процессами взаимодействия ионного пучка (и/или электронно¬
го пучка в ЭОС) с образцом [ 3, 14, 59]. В общем случае у нас нет выбора
в отношении самого образца, однако влияние взаимодействия с пучком и ап¬
паратурных факторов может быть существенно оптимизировано.4.4.2.1. Аппаратурные факторыВажным условием проведения послойного анализа является необходи¬
мость устранения химических реакций образца с окружающей средой в про¬
цессе распыления [ 50]. Это может быть связано с адсорбцией, перенапыле-
нием и примесями в ионном пучке. Чтобы воспрепятствовать адсорбции хи¬
мически активных газов, Образец следует помещать в СВВ (< 10“9 мм рт. ст.
или 10“7 Па). При проведении распыления совместно с непрерывным анали¬
зом на поверхности с помощью ЭОС допустим, что вероятность прилипания
равна единице и вероятность распыления определяется только материалом
самой матрицы. Тогда простые расчеты показывают, что среднюю степень
покрытия поверхности примесями 0 можно оценить (для 0 « 1) как отно¬
шение скоростей адсорбции и десорбции [14, 60](4.16а)где z - скорость распыления в нанометрах в секунду, р - парциальное дав¬
ление в паскалях, 0 - отношение адсорбированных атомов к количеству ато¬
мов на поверхности образца. Уравнение (4.16а) показывает, что 0 будет
значительно ниже чувствительности определения (10~2) для скорости рас¬
пыления 1 А/с, если парциальное давление химически активного газа ниже
р < 10-* мм рт. ст. (10-6 Па). Метыои Ландольт [ 15] показали, что парци¬
альное давление СН4 выше 10“5 Па может серьезно изменить результаты
послойного анализа с применением ЭОС. Следоет отметить, что в атучае
ионной пушки, работающей в замкнутой камере, заполненной аргоном с дав¬
лением 5 • 10-5 мм рт. ст., загрязняющий газ также ионизуется и осажда¬
ется на поверхности образца. Таких примесей в первичном ионном пучке
можно избежать, если применять ионную пушку с дифференциальной откач¬
кой. Другим источником загрязнения поверхности является осаждение ранее
распыленных частиц (вследствие перенапыления на конструкционные части,
близко расположенные к поверхности образца). Поскольку ЭОС или РФЭС
имеют меньшую чувствительность, чем ВИМС, этот эффект для них менее
важен.Следует отметить, что если проводить последовательно распыление и
анализ поверхности, как это обычно бывает при использовании РФЭС для
С. Хофман179послойного анализа, условие устранения влияния адсорбции становится бо¬
лее жестким, так как степень покрытия определяется соотношениемгде f ш- время, в течение которого ионная пушка выключена.Неоднородность по интенсивности ионного пучка в анализируемой облас¬
ти означает, что различные участки образца распылены до разных глубин,
что соответственно влияет на результаты измерений. Это ведет к тому,
что разрешение по глубине Az 2 увеличивается пропорционально глубине
распыления z. Для случая статического ионного пучка с гауссовым распре¬
делением интенсивности и диаметром dlt определяемым ПШПВ, Lzx зависит
от расстояния b между осями ионного и первичного электронного пучка
(при использовании ЭОС) диаметром de. Эта зависимость может быть приб¬
лиженно выражена следующим образом [4, 61, 62]:Более строгое рассмотрение проведено в работе [62]. Уравнение(4.17) показывает, что малый диаметр электронного пучка, который можно
использовать в СОМ при послойном анализе с помощью ЭОС, позволяет по¬
лучить значительные преимущества (de/d1 < 0,01). При использовании
РФЭС для проведения послойного анализа, когда de совпадает с диаметром
анализируемой области (обычно порядка нескольких миллиметров, т. е.
de/dt » 1), результат получается значительно хуже. Таким образом, фоку¬
сировка первичного ионного пучка является обязательной при использова¬
нии РФЭС для послойного анализа [ 63]. Следует отметить, однако, что ней¬
тралы, которые возникают перед электростатическим раст ровым дефлекто¬
ром, могут вызывать слабое возрастание неоднородности ионного пучка по
интенсивности, что может быть исключено только установкой дополнитель¬
ного дефлектора после отклоняющих пластин.Средняя глубина выхода ЭОС электронов или фотоэлектронов является
особенностью, присущей этим методам, и обсуждается в разд. 4.3.2.4.4.2.2. Характеристики образцаСостав и структура образца налагают определенные ограничения на ка¬
чество получаемых при ионном травлении профилей концентраций. Экспери¬
ментально было показано, что первоначальная шероховатость анализируе¬
мой поверхности существенно влияет на разрешение по глубине [ 54, 56].
Математическое рассмотрение этого вопроса недавно было проведено Си-
хом и Ли [ 16], предсказавшими возрастание величины Лzx, пропорциональ¬
ное глубине травления z, и ее рост по мере увеличения отклонения угла
падения первичного ионного пучка от нормали. Для нормального падения бы-(4.166)(4.17)
1804. Послойный анализло предсказано(4.18)где а 0 вычисляется как стандартное отклонение в распределении углов, об¬
разуемых произвольно расположенными элементами неровной поверхности
и плоскостью, соответствующей усредненному положению этой поверхности.
Коэффициент / определяется зависимостью между углом падения ионов и
выходом процесса распыления [20, 21] и изменяется от 0,5 до 1,5 [ 16], Ре¬
зультаты расчетов Сиха и Ли показаны на рис. 4.9 для / = 0,65. Общий вывод
состоит в том, что оптимальные профили при ионном травлении можно полу¬
чить только для плоских полированных образцов и нормальном падении пер¬
вичного пучка.Рис. 4.9. Зависимость относительного разре¬
шения по глубине от угла па¬
дения ионного пучка р для нескольких
значений распределения углов шеро¬
ховатости а задаваемого величиной
а (стандартное отклонение) для
/ = 0,65 (Cu, Pd, Ag, Аи) [16].Наличие зависимости выхода распыления от ориентации поверхности
[19, 21] является фактором, обусловливающим наиболее неравномерное ион¬
ное травление по границам зерен монокристаллических образцов [64]; при
этом ожидается, что разрешение по глубине Az пропорционально глубине
распыления. Обычно при распылении ионами кислорода (или дополнительной
обработке кислородом в процессе распыления ионами аргона) наблюдается
сглаживание поверхности после ионного травления. Это происходит, вероят¬
но, из-за образования квазиаморфного слоя оксида, который распыляется
более однородно [65].Уэлс и др. [ 66, 67] показали, как геометрия поверхности влияет на ее
топографию, возникающую при ионном травлении. С другой стороны, даже
для случая плоских аморфных поверхностей в результате ионной бомбарди¬
ровки из-за наличия зависимости коэффициента распыления от угла падения
С. Хофман181ионного пучка могут возникать конусы травления [ 21, 68]. Эти топографи¬
ческие эффекты могут быть ослаблены при использовании вращения образ¬
ца или распыления двумя ионными пушками с различными углами падения
[17].Ввиду изменения коэффициента распыления, обусловленного измене¬
нием локального состава образца, шероховатость многофазных материа¬
лов в результате ионного травления возрастает. Подобное воздействие
могут оказывать и загрязнения поверхности [ 69, 70].В соответствии с вышеизложенным распыление некристаллических
однофазных материалов, которые аморфизируются в процессе распыления
(например, полупроводников и оксидов), должно было бы приводить к зна¬
чительно лучшим результатам при послойном анализе, чем те, которые
обычно наблюдаются для поликристаллических металлических материалов.
Аналогично распыление последних химически активными ионами часто
приводит к улучшению разрешения по глубине (см. рис. 4.8).4.4.2.3. Эффекты, обусловленные ионной бомбардировкойЯвления, характерные для процесса распыления, как показано в теории
ионного травления Зигмунда [71], по-видимому, обусловливают существование
фундаментальных ограничений достижимой величины разрешения по глубине.
Они вызывают изменение микротопографии поверхности из-за статической
природы процесса распыления поверхностных атомов и изменения состава
вследствие возникновения смеси атомов в пределах области каскада столк¬
новений* Уже первая попытка описания прогрессирующего изменения микро¬
топографии поверхности с помощью модели последовательного послойного
распыления [ 5, 50] смогла объяснить часто наблюдавшуюся зависимость
Az от zV2 [72]. Однако эксперименты с распылением химически активными
ионами [73], и особенно распыление полупроводников и оксидов [74], пока¬
зали, что можно достигнуть постоянного значения разрешения по глубине.
Модельные расчеты последнего времени приводят к выводу, что зависящая
от состава вероятность распыления [75], так же как и определенная степень
подвижности поверхностного слоя [76] в рамках модели последовательного
послойного распыления, приводит к постоянству величины Az на более зна¬
чительных глубинах. Поэтому можно заключить, что уширение профиля кон¬
центраций при распылении, связанное со статистикой распыления, обуслов¬
лено лишь несколькими монослоями образца. Часто наблюдаемая зависимость
разрешения по глубине от z1/2 [ 4, 72] может быть объяснена как следствие
взаимного наложения постоянного разрешения по глубине и линейно зави¬
сящего от глубины травления разрешения по глубине [ 14, 77] (рис. 4.11).Основной вклад в уширение профиля концентраций спектров обусловлен
каскадом столкновений [71]. Частицы, претерпевшие первичные столкно¬
вения, перемещаются в более глубокие слои (эффект "вбивания”), а претер¬
певшие вторичное столкновение усиливают случайный характер распреде-
1824. Послойный анализления смещенных атомов мишени, подвергшихся столкновению ("атомное
перемешивание" или "каскадное перемешивание") [78 - 83]. Расчеты Литмар-
ка и Хофера [ 80] показали, что уширение возникает на переходном слое и
направлено в глубину образца. Это было экспериментально подтверждено
Этцкорном, Литмарком и Киршнером [82]. Уширение возрастает по мере
увеличения дозы ионного облучения вплоть до достижения некоторого пос¬
тоянного значения [ 82]. Для больших глубин вклад атомного перемешива¬
ния достигает постоянной величины и не зависит от z, однако примерно
пропорционален корню квадратному из энергии первичного ионного пучка
[78] и несколько уменьшается с ростом коэффициента распыления [83].
Влияние атомного перемешивания на разрешение по глубине существенно
на глубинах, совпадающих по порядку с областью проникновения ионов
первичного пучка. Из сказанного следует, что возмущения, вносимые эф¬
фектом "вбивания" и атомным перемешиванием, можно минимизировать,
используя тяжелые ионы (например, Хе+) малых энергий (< 1 кэВ).Из-за различий в коэффициентах распыления химических элементов
состав поверхностного слоя многокомпонентного образца обычно меняет¬
ся в процессе распыления [ 83 - 86]. Если коэффициенты распыления ком¬
понент не зависят от их объемной концентрации с ъ , аналогичная характе¬
ристика для поверхности с s обратно пропорциональна соответствующим
коэффициентам распыления S [ 84]:(4.19)где индексы А и В относятся к двум компонентам. Это означает, что по¬
верхность обогащена элементом с меньшим коэффициентом распыления.
Перестройка слоя в процессе распыления до достижения стабильного состоя¬
ния, описываемого уравнением (4.19), приводит к постепенному изменению
состава поверхности, что было аналитически получено Хо и др0 [ 86](4.20)где с sA(t)-мгновенное значение содержания компоненты А, начиная с
начала распыления (t = 0) и до f -> м [csA(t) = с%А в уравнении (4.19).
Параметр т характеризует время, необходимое для стабилизации изменен¬
ного поверхностного слоя, и по порядку совпадает с величиной проникнове¬
ния ионов первичного пучка [ 47, 83, 86, 87]. Изменения в составе поверх¬
ности [ уравнение (4.20)] влияют на профиль концентрации, возникающий
при данной интенсивности ионного пучка и времени распыления, и тем са¬
мым на разрешение по глубине. Более важным вопросом, однако, представ¬
ляется асимметрия профилей концентраций, обусловленная изменением ско¬
рости распыления при изменении состава, обсуждавшаяся в разд. 4.3.1.
Наиболее серьезные искажения,обусловленные селективным распылением,
С. Хофман183Рис. 4.10. Изменение состава анодированного слоя Та2С^ (толщиной 30 нм) при рас¬
пылении ионами Аг+ с энергией 3 кэВ в РФЭС [47]. а - 4/-пики Та в
зависимости от времени распыления; б- профили концентрации, получен¬
ные ионным распылением с помощью РФЭС, соответствующие падению
концентрации Та205 и росту концентрации Та О, Та02 и Та в изме¬
ненном слое вплоть до достижения постоянного соотношения концентра¬
ций.
1844. Послойный анализсвязаны с регистрацией каждого профиля концентрации, когда градиент
концентрации отсутствует (рис. 4.12).В случае химических соединений селективное распыление одной из ком¬
понент приводит к изменению химического состояния образца. В случае
многих оксидов тем самым удается уменьшить степень окисления [ 47, 87-
89]. Этот эффект может быть выявлен при проведении послойного анализа
с использованием РФЭС [ 14, 47]. Например, рис. 4.10 показывает воздей¬
ствие бомбардировки ионами Аг+ на "химический” профиль концентраций
при распылении слоя Та205/Та толщиной 30 нм. На рис. 4.10, а изображе¬
ны 4/ РФЭС -пики Та в зависимости от времени ионного травления. Если об¬
работать пики с помощью операции обратной свертки, принимая во внима¬
ние различную валентность атомов тантала [ 90], а затем проинтегрировать
область пиков, то это позволит получить численную оценку содержания ком¬
понент, появляющихся в образце. Результаты оценки приведены на
рис. 4.10, б, где интенсивности Та 4+, Та2+ и Та0 соответствуют окси¬
дам Ta02f ТаО и чистому танталу, возникающему в модифицируемом слое
окисла [47]. Остающийся Та5+ объясняется наличием неизмененного слоя
Та2Об, соседствующего со слоем, обедненным кислородом. Динамическое
равновесие устанавливается после первоначального переходного процесса,
вызванного селективным распылением кислорода. При разложении соеди¬
нений возникают трудности в интерпретации результатов послойного ана¬
лиза с использованием профилей РФЭС распыления. К счастью, оксиды лег¬
ких элементов (Si02, MgO, А1203) в процессе ионной бомбардировки вос¬
станавливаются крайне незначительно [45, 47, 88, 91].Распыление может также вызвать усиленную диффузию в сочетании с
возбуждением других транспортных процессов, таких, как электроперенос
[ 92] (в случае зарядки диэлектриков под воздействием электронного облу¬
чения) и возникновение сегрегаций [93, 94]0 Индуцированная поверхност¬
ная диффузия может в свою очередь несколько сгладить неровности, воз¬
никшие при ионном травлении, и, следовательно, улучшить разрешение по
глубине [76], однако при этом усиление объемной диффузии приводит к уши-
рению профилей концентраций при послойном анализе [28, 57]. Если распы¬
ление зависит от температуры, то для точного определения степени влияния
на результаты послойного анализа вызванных нагревом процессов перено¬
са, атомного перемешивания или сочетания этих процессов необходимы
дальнейшие систематические исследования*В то время как рентгеновское излучение в РФЭС редко вызывает за¬
метное разложение образца, электронный пучок, используемый в ЭОС,
приводит к вредным последствиям в случае многих химических соединений.
Часто наблюдается вызванная электронной бомбардировкой десорбция хло¬
ра и кислорода [95, 96]. Это может привести к более интенсивному распыле¬
нию в области электронного пучка; следовательно, послойный анализ с по¬
мощью ЭОС приводит к уменьшению разрешения по глубине [96]. Так как
С. Хофман185электронно-стимулированная десорбция (ЭСД) и распыление конкурируют в
поверхностном слое, то воздействие ЭСД можно подавить, уменьшая плот¬
ность тока электронного пучка (например, используя сканирование) и (или)
увеличивая плотность тока ионного пучка.Обычно диссипация энергии в поверхностном слое образца пренебрежи¬
мо мала как при рентгеновском облучении, так и при ионном распылении в
случае послойного анализа (за исключением особых случаев). Однако нагрев
образца под воздействием электронного пучка может привести к увеличению
его температуры до нескольких сотен градусов, особенно в случае тонких
пленок на стеклянных подложках. Расчет роста температуры АТ был про¬
веден Рёллем [97]:(4.21а)где р - мощность входного пучка, г0 - его радиус, h - толщина пленки,
аир- коэффициенты теплопроводности пленки и подложки соответствен¬
но. Для случая массивного образца уравнение (4.21а) может быть заменено
соотношением:(4.216)При уменьшении плотности тока электронного пучка величина АТ падает.
Проверка эффекта разогрева образца электронным пучком при послойном
анализе может быть выполнена путем распыления одного и того же образ¬
ца при различных плотностях тока [ 26, 46].4.4.3. Суперпозиция различных эффектов, влияющих
на разрешение по глубинеЕсли частные эффекты, влияющие на послойный анализ, независимы
друг от друга и могут быть аппроксимированы функциями разрешения,
имеющими форму гауссовой кривой с 2а;- = Az;-, полное разрешение по глу¬
бине Az, измеренное экспериментально, можно описать аналогично оцен¬
ке суммарной ошибки эксперимента [ 72]:(4.22)Уравнение (4.22) является точным только для случая гауссовых функций
разрешения в ряде гауссианов, но можно показать, что оно является доста¬
точно хорошим приближением и для экспоненциальных функций (например,
Д*А=1.6Х)[49, 53].Различные вклады могут быть охарактеризованы по их зависимости от
суммарной дозы ионного облучения или глубины распыления. Из табл. 4Л
видно, каким образом можно поделить наиболее важные Дг . на постоян¬
ные и зависящие от глубины и скорости распыления или от времени. Для
1864. Послойный анализРис. 4.11. Типичная зависимость полного разрешения по глубине от глубины z,предсказанная уравнением (4.23), для суперпозиции вкладов постоянной и
зависящей от глубины составляющих (см. табл. 4.1). в приведенном при¬
мере аппаратурный фактор Az = 0,1 z и характеристика атомного
перемешивания Azfe = 2 нм [14].больших глубин распыления те из Az-, которые увеличиваются с глубиной,
Оудут более существенны, в то время, как для малых глубин распыления
постоянные члены будут ограничивать наблюдаемое уширение профилей
концентраций [ 14, 72]. На рис. 4.11 представлена обычно предсказывае¬
мая зависимость разрешения по глубине от глубины для случая, когда
имеется суперпозиция вкладов аппаратурного уширения (Az ~ z, т. е.Azj/z = 10%) и некоторого постоянного (например, обусловленного атомным
перемешиванием, Azfe = const = 2 нм). В соответствии с аргументами Вер¬
нера [77] в случае суперпозиции вкладов такого типа зависимость вида
Az ~ z1/2 (или A z/z z~1/2), часто наблюдаемая экспериментально
[4, 5, 72], является хорошей аппроксимацией для средних глубин травления*
Знание экспериментального значения разрешения по глубине позволя¬
ет получить качественную картину измеренного профиля. Для получения
формы первоначального профиля концентраций можно использовать проце¬
дуру обратной свертки, как это описано в разд« 4.6.4.5. Специальные методы послойного анализаКроме общих методов послойного анализа существуют некоторые по¬
лезные альтернативные методы, которые требуют специального оборудо-
вания: изменение угла регистрации и энергии эмитированных электронов
в случае использования РФЭС и ЭОС, послойный анализ границы раздела
с помощью СОМ и послойный анализ шаровых шлифов, применяемый сов¬
местно с СОМ .
С. Хофман1874.5.1. Изменение угла регистрации эмитированных
электронов и использование зависимости
глубины выхода электронов от энергииРассматриваемые методы являются неразрушающими и используются
для изучения изменения состава самых внешних атомных слоев твердых
тел. Рассмотрим сначала метод, связанный с изменением угла регистра¬
ции эмитированных электронов. Идея послойного анализа, основанного
на изменении эффективной глубины выхода при изменении угла регистраг
ции эмитируемых электронов, очевидна из общих выражений (4.6) и (4.8),
где <р - угол между нормалью к поверхности образца и направлением
эмиссии оже* или фотоэлектронов. Если Xt- изменяется непрерывным об¬
разом, уравнение (4.6) означает, что интенсивность может быть описана
функцией Лапласа <£ с ci (z) в качестве аргументов [ 4]. Это уравнение яв¬
ляется интегральным преобразованием с нулевыми граничными условиями
при z < 0 и ехр[ — (1/Л,- )z] в качестве граничного условия для z > 0. Та¬
ким образом, уравнение (4.6) может быть записано в видеОтсюда с помощью обратного преобразования Лапласа £-1 можно опреде¬
лить с. (z):В общем виде уравнения (4.23а, б) применимы как для ЭОС (если пренеб-
речь rB)f так и для РФЭС. Проблема состоит в том, что уравнение (4.6)
необходимо разрешить для неизвестного интеграла от с i (z). В некоторых
специальных случаях решение можно найти в таблицах интегралов.Для концентрического полусферического анализатора (ПСА) с со¬
бирающей линзой, который наиболее часто применяется в РФЭС, экспери¬
ментальное определение /(1Д .) можно осуществить, непосредственно из¬
меняя положение образца, как показал Федли [ 10] и позднее Эбел [11, 98].
Однако при неровной поверхности и угле q> > 80° из-за полного отражения
эмитированных электронов могут возникнуть проблемы [10, 11, 98].Из-за ограничений по чувствительности (см. разд. 4.3.2) глубины, дос¬
тупные анализу, ограничены величиной ЗХ0, т. е. 5 нм. Более того, разре¬
шение по глубине ограничено погрешностью при экспериментальном измере¬
нии интенсивности. Наиболее важным приложением метода изменения угла
регистрации эмитированных электронов является определение толщины тон¬
ких слоев, возникших в результате загрязнений [ 4.47], имплантации [ 42]
или сегрегации. В простом случае, когда однородный слой элемента А толщи¬
ной d расположен на подложке из элемента В, нормированные интенсивнос¬
ти [ в соответствии с решением уравнений (4.6) и (4.8)] выражаются в виде(4.23а)(4.236)
1884. Послойный анализ[3,4](4.24а)где ^л а “ сРеДняя длина пробега неупругоотраженных электронов атомов
А в матрице А и в - соответствующая величина, характеризующая про¬
бег электронов атомов В в матрице А„ Для специального случая, когда
AY^°B„4 , отношение (^//д)/^//?) показано на рис. 4.12 в виде функ¬
ции от угла регистрации эмитированных электронов ф при различных зна¬
чениях d/\° [ 47].Рис. 4.12. Отношение уравнений (4.24а, б), UB/i °в)Л1д/1д) = изобРажен*ное в виде функции угла регистрации эмитированных электронов ф при
различных значениях d/\ в предположении, что А = А = \ ).Если для анализа распределения энергий электронов используется АЦЗ,
возрастают сложности, связанные с геометрией анализатора, что ограни¬
чивает полезный диапазон изменения угла регистрации эмитированных
электронов и влияет на интенсивность сигнала вследствие затенения [ 4].
Возможность обойти эти трудности представляется при использовании АЦЗ
с вращающимся барабаном, имеющим щель, которая вырезает сегмент
(например, 4° или 12°) при заданном азимутальном угле V/ из конуса сбо¬
ра [ 4]. Если а - фиксированный угол между нормалью к поверхности об¬
разца и осью АЦЗ, зависимость между азимутальным углом и углом регист-
С. Хофман189рации эмитированных электронов выражается в виде [ 4, 47]
cosq>= sina • sinq)^ • costf+ cos a • совфд,(4.25)где угол анализа АЦЗ фл - 42,3° и а ^ фл. Для a = 30° из уравнения
(4.25) следует, что 0,3 < cos ф< 0,97 для 0< 180°. На рис. 4.13 приве¬
дены результаты эксперимента, проведенного при такой геометрии
(а = 30°, АЦЗ с двойной фокусировкой). В рассматриваемом случае толщина
слоя загрязнений на Nb205, состоящего из гидридов углерода и кислорода,
была определена по результатам измерений пиков С Is и О Is. Здесь пик
О Ь можно было подвергнуть процедуре обратной свертки для разделения
пиков оксида и гидроксида (различающихся по энергии связи на 0,7 эВ).Рис. 4.13. Определение композиционного профиля слоя примеси на Nb2o5 , состоя¬
щей из соединений углерода с кислородом, методом изменения угла реги¬
страции эмитированных электронов (ф) ( АЦЗ с вращающейся щелью) с ис¬
пользованием отношений интенсивностей РФЗОпиков I (0\s оксид//(01* гидро¬
ксид) (1) и I (Сis)// (01 s гидроксид)- (2) в соответствии с рис. 4.12. Посто¬
янное значение последнего отношения указывает на наличие почти одно¬
родного слоя с d/Л (О) = 0,71, откуда получаем толщину d = 1,6 нм при
МО) = 2,3 нм [47].Поделив друг на друга левую и правую части уравнений (4.25а, б) почленно,
можно было найти, как это показано на рис. 4.14, соответствующую вели¬
чину d/\ (О) = 0,71 в предположении, что пик окиси полностью обусловлен
Nb205. Постоянство отношения / (С Is)//(О Ъ гидроксид) указывает на
неизменность состава загрязненного слоя по глубине в пределах погрешнос¬
ти измерений. Отметим, что использование пиков одной и той же подоболоч-
ки позволяет исключать параметр асимметрии при проведении количествен¬
ного послойного анализа с помощью РФЭС. В противном случае необходимо
учитывать дополнительные изменения интенсивности вследствие варьиро¬
вания tfT99].
1904. Послойный анализПодводя итоги, можно сказать, что при проведении послойного анализа
с помощью метода изменения угла регистрации эмитированных электронов
главное преимущество, особенно в случае использования РФЭС, заключает¬
ся в том, что этот метод является неразрушающим и позволяет проводить
абсолютное измерение глубины выхода электронов, выражающейся в едини¬
цах Л°. Однако его применение ограничено очень тонкими слоями < ЗЛ°(4 5 нм) и требует соответствующего экспериментального оборудования
(см. также гл. 3, 9).Второй метод, заключающийся в измерении интенсивности пиков эми¬
тированных электронов при различных энергиях возбуждения, также осно¬
ван на использовании уравнений (4.23) и (4.24), а именно на зависимости
эффективной глубины выхода электронов A°cos <р от энергии. Например,
если мы сравним относительные интенсивности низкоэнергетичного и высо-
коэнергетичного пиков подложки с гомогенным покрытием толщиной d(Ib и
/ в соответственно) с относительными интенсивностями этих же пиков в
случае чистой подложки (/£ и 1°в соответственно), то, как легко показать,4.5.2. Проведение послойного анализа на краю кратера
травления с помощью СОМ с использованием
границ кратеровЗависимость разрешения по глубине от глубины кратера в случае трав¬
ления ионным пучком с гауссовым распределением интенсивности (п. 4.4.2)
может быть использована при проведении послойного анализа на краю кра¬
тера для получения профилей концентрации. Сказанное относится и к обще¬
принятой методике косого шлифа. Как показано на рис. 4.14, а, наклон
кратера может быть использован для того, чтобы преобразовать смещение
хорошо сфокусированного электронного пучка по координате х, параллель¬
ной поверхности образца, в перемещение по глубине (координата z), что
численно выражается соотношением, подобным уравнению (4.17). Пример
определения профиля образца многослойной структуры Ni Cr изображен на
рис. 4.14, б [61]. Аппаратурное разрешение по глубине, определяемое диа¬
метром электронного пучка de [61],. описывается соотношениемгде tga характеризует наклон стенки кратера. Точность определения про¬
филя концентраций зависит от правильности формы кратера, которая в
свою очередь определяется распределением интенсивностей в ионном пучке
(получаемом с помощью цилиндра Фарадея). Однако, принимая во внимание
факторы, зависящие от глубины выхода, состава образца и его топографии,(4.26)(4.27)
С. Хофман191Рис. 4.14. Послойный анализ на краю кратера травления многослойной структуры
Ni Сг [61].а - принципиальная схема проведения послойного анализа с исполь¬
зованием края кратера для случая гауссова распределения интенсивнос¬
ти ионного пучка со стандартным отклонением cxtgam= о,6 zQ/x(v). Схе¬
матически показан профиль многослойной структуры Ni Сг. о - профи¬
ли интенсивностей оже-пика С г (529 эВ) и оже-пика Ni (848 эВ) в об¬
ласти, изображенной на (а),при толщине одного слоя d = 11,5 нм,диамет¬
ре электронного пучка de= 10 мкм, tgam= 1,9*10"4.а также эффекты, обусловленные воздействием ионной бомбардировки, сле¬
дует ожидать, что ограничения, налагаемые на разрешение по глубине,
такие же, как и при обычном послойном анализе [62]. Главным достоинст¬
вом этой методики является простота проведения послойного анализа не¬
посредственно после распыления, что препятствует потере информации и
позволяет улучшить отношение сигнала к шуму.4.5.3. Послойный анализ с использованием
шаровых шлифов и СОМЭта методика [ 101] является особым случаем известного метода косого
шлифа Как и последний, она используется для послойного анализа до глу¬
бин профилей в несколько микронов, когда ионное распыление с использо¬
ванием обычного оборудования требует очень большого времени. Эта мето¬
дика позволяет преодолеть многие трудности, встречающиеся при приготов¬
лении косого шлифа, благодаря использованию вращающегося стального ша¬
ра, покрытого высококачественной алмазной пастой. Геометрия получаемо¬
го кратера схематично показана на рис. 4.15 [ 101]. Поскольку радиус R ша¬
ра известен (обычно 1-3 см), то глубина кратера d вычисляется по форму¬
ле DV8R, где D - диаметр кратера. Горизонтальную координату электрон¬
ного пучка, попавшего на стенку сферического кратера, можно выразить
через глубину в соответствии с геометрией кратера на рис. 4.15. Если счи-
1924. Послойный анализРис. 4.15. Схематическое изображение шарового шлифа (по данным Уэлса
и др.) [101].тать кратер абсолютно гладким, то глубина г связана с расстоянием х, на
которое электронный пучок удален от края кратера, соотношением [ 101]Характерное для этого метода разрешение по глубине описывается выра¬
жениемгде dg - диаметр электронного пучка, т - дополнительный член, учитыва¬
ющий шероховатость поверхности образца. На практике разрешение по глу¬
бине в основном ограничивается именно этой характеристикой образца,
возрастающей в процессе изготовления шлифа. Уравнение (4.29) показыва¬
ет, что относительное разрешение по глубине Лzb/z улучшается с ее рос¬
том, как показали Уэлс, Холл и Сайкс [ 101]. Ли и Сих [ 102] предложили
критерий для использования обычного послойного анализа с помощью обыч¬
ной ЭОС и ионного травления и механического изготовления шлифа для по¬
лучения оптимального значения Az как функции полной глубины распыления,
По отношению к такой характеристике, как разрешение по глубине,
преимущество метода послойного анализа с использованием шаровых шли¬
фов проявляется при построении профилей концентраций на очень больших
глубинах (> 2 мкм). В отличие от обычного послойного анализа с ионным
травлением или послойного анализа с использованием краев кратеров этот
метод обладает тем преимуществом, что имеется абсолютная и линейная
шкала глубин, калиброванная в соответствии с заданной топографией по¬
верхности, хотя для некоторых материалов (очень пластичных или очень
хрупких) могут возникать трудности, связанные с замазыванием или рас¬
трескиванием. Для меньших глубин (< 2 мкм) послойный анализ с исполь¬
зованием ионного травления оказывается более чувствительным методом,
чем в случае механической абразивной обработки, и имеет соответственно
лучшее абсолютное разрешение по глубине.(4.28)(4.29)
С. Хофман1934.6. Построение профилей концентрации с помощью алгоритма обратнойсвертки при послойном анализе с использованием ионного травления.Предположим, что первый шаг в количественной оценке послойного
анализа, т. е. определение истинной (линейной) шкалы глубин (п. 4.3.1) и
шкалы концентраций (п. 4.3.2) уже сделан. Теперь необходимо обсудить все
источники погрешностей при определении истинного профиля концентраций
(разд. 4.4). Задача сводится к решению уравнения (4.14) [ 49, 53](4.30)где c(z’) - истинный профиль концентрации, l(z) - зависимость интен¬
сивности сигнала от глубины в шкале скорректированного профиля концен¬
траций; g(z — z') — функция разрешения. Этот последний член является
результатом воздействия всевозможных искажений в конкретном экспери¬
менте, влияющих на профиль распыления, как это показано в разд. 4.4.Если известен вклад данного искажающего воздействия, то имеется воз¬
можность учесть его отдельно от остальных. Это, в частности, полезно
при учете влияния глубины выхода (вместе с коэффициентом обратного
рассеяния в ЭОС), что определяется формулой(4.31)Если (4.31) подставить в (4.30), то последнее уравнение становится ана¬
логичным (4.6), явное решение которого дается (4.9) для гB(z) = const.
Можно также учесть и коэффициент обратного рассеяния, введя соответ¬
ствующую экспоненциальную функцию [3, 39]. Морабито и др. [6,37] пред¬
ложили первое приближение для количественной оценки, использующее пря¬
моугольную аппроксимацию уравнения (4.6) произведением гв(Хл/Хв ) х
* \ №а/%в) в слУчае двойной системы А /В. В этом случае при измерении
профиля используют калибровочный коэффициент, зависящий от состава.
Следует отметить, что при внесении поправок на обратное рассеяние и глу¬
бину выхода мы оперируем профилем, который уширен в результате воз¬
действия других факторов, рассмотренных в разд. 4.4.Даже если соответствующие различным факторам функции разрешения
являются асимметричными (как, например, для атомного перемешивания
[79 - 83]), их суперпозиция может быть аппроксимирована с достаточной
точностью гауссовой функцией разрешения(4.32)где Az = 2а - разрешение по глубине, определенное в п. 4.4.1.1. В урав¬
нение (4.32) можно подставить соотношение Az = f(z) (см. табл. 4.1).
1944. Послойный анализДля произвольной функции разрешения, как и для гауссовой, решение
уравнения (4.30) не может быть получено в явном виде. Для этого случая
были разработаны два математических подхода [ 103]: метод преобразова¬
ния Фурье и итеративный метод Ван Циттерта [ 104]. Вертгейм [ 105] по¬
казал эквивалентность этих методов. Итеративный метод впервые был
применен для послойного анализа Хо и Льюисом [ 48] • В соответствии с
этим решение после n-й итерации, cn(z) представляется в видегде c°(z) = /(z) (т. е. соответствует измеренному и откалиброванному про¬
филю концентрации). Если итеративный процесс сходится, ожидается, что
limn^oecn(z) = с (z). Алгоритм (4.33) легко реализуется численными мето¬
дами с помощью ЭВМ.Применимость метода Ван Циттерта можно проверить аналогично то¬
му, как это сделали Мадден и Хоустон [106]. Для этого будем считать, что
истинный профиль концентрации с (z) имеет вид функции ошибок. Тогда из¬
меренный профиль концентрации можно получить с помощью свертки с гаус¬
совой функцией g(z -z')[14, 49, 53]:где zQ определяет границу раздела (50% максимальной амплитуды), Az0 -
значение, соответствующее 2а истинного профиля, и Аг - разреше-
шение по глубине, определяющее g(z -z'). Подставляя (4.34) и (4.35) в урав¬
нение (4.30), получаем "измеренный" нормированный профиль/(z):Сходимость можно контролировать, сравнивая ширину Az0 истинного
профиля с шириной профиля после процедуры обратной свертки, проведзнной
п раз, ЛznQu На рис. 4.16 этот фактор сходимости изображен в виде функции
от параметра к = Az0/AZg для 0,05 < к< 1,5 и для 0,2 и 10 итераций. Влия¬
ние к на сходимость очевидно. Из рис. 4.16 видно, что для восстановления
истинного спектра подходят только случаи с к > 1, когда требуется умерен¬
ное количество итераций. Можно показать, что для к < 1 флуктуации возрас¬
тают [ 48, 106] и истинный профиль нельзя восстановить.(4.33)(4.34)(4.35)(4.36)где(4.37)
С. Хофман195Рис. 4.16. Сходимость процедуры обратной свертки Ван Циттерта, характеризуемая
зависимостью отношения Azg (истинного профиля) к Azn (профилю после п итера¬
ций процедуры обратной свертки) от к = Az^/Az^ где определяется функ¬
цией разрешения [49, 53].Применяя это ограничение (к > 1) для измеренного Az в уравнении(4.37), получаем(4.38)Это означает, что получение истинного профиля с (z) возможно лишь
при условии, что уширение Azg составляет менее 70% измеренной величины
Az. Условие, выражающееся уравнением (4.38), можно распространить и на
такую характеристику, как градиент dl /dz нормированного измеренного
профиля /(z), при условии, что мы определяем максимум обратной величи¬
ны наклона для функции ошибок профиля (0 < / < 1), где dz/dl = 1,25 Az.
Тогда соотношение (4.38) принимает вид(4.39^Анализируя отклонение "измеренной1* ширины профиля Az от ее истинно¬
го значения Az0, которое определяется уравнением (4.37) с учетом соотно¬
шений (4.38) и (4.39), мы можем выбрать одну из трех областей (I, II, III),
изображенных на рис. 4.17 [14]. Если при максимальном градиенте измерен¬
ного профиля соответствующая величина составляет менее 10% от Az g для
1964. Послойный анализРис. 4.17. Относительное отклонение (Az -Az0)/Az разрешений по глубине измерен¬
ного (Az) и истинного профиля (Д*0), представленное в виде функции про¬
изведения градиента измеренного профиля d{l /l0)/dz на Az - функции
разрешения, описываемой функцией ошибок [ 14]. Выделенные области со¬
ответствуют случаям: / - когда нет необходимости в процедуре обратной
свертки; и - когда может быть применима процедура Ван Циттерта; III -
когда процедура обратной свертки приводит к неопределенным результа¬
там (необходимо сравнение с измеренным профилем).функции разрешения, то с точки зрения разрешения отклонение измеренного
профиля от истинного составляет менее 1%, а следовательно, вообще не тре¬
буется операция обратной свертки (рис. 4.17, область /). Как описано выше,
такая операция после итеративного метода может быть применена в облас¬
ти И, где на диапазон максимального градиента приходится менее 57% ве¬
личины AПри больших градиентах метод Ван Циттерта не имеет области
сходимости; это означает, что истинный профиль определяется неоднознач¬
нее (область ///). В частности, в этом случае с успехом можно применять
метод, в котором в процессе итераций учитываются предположения об истин¬
ном профиле и используется вычисленный из уравнения (4.30) 11 измеренный”
профиль l{z) с известной функцией разрешения [7, 49, 53]. Прямое сопос¬
тавление с нормированными экспериментальешми данными показывает, на¬
сколько правильны предположения. Если отклонения растут, следует модифи¬
цировать форму предполагаемого истинного профиля, после чего можно по¬
вторять процедуру до тех пор, пока не будет достигнута удовлетворитель¬
ная точность [ 49] (метод "проб и ошибок").В общем случае удобно аппроксимировать истинный профиль после до
вательностью тонких слоев постоянной концентрации c(z;-) и толщины
d = zj + i - Zy и вычислять соответствующую измеряемую нормированную
С. Хофман197интенсивность /;- (z) каждого отдельного слоя по формуле [3,7, 51](4.40)Вычисления с использованием уравнения (4.40) необходимо делать для
каждого слоя c(z -) при значении z;, удовлетворяющем условию - 1,5 Лzg <
< z -Zj; < l,5Azg (т. e. итерации в диапазоне За с точностью 99,7%). Вы¬
бор d зависит от градиента концентрации и желаемой точности с естественным
нижним пределом, равным толщине одного монослоя. После суммирования
по j величин /у (z) для любых z получаем профиль, который можно непо¬
средственно сравнить с нормированным измеренным профилем [3, 49]:(4.41)Чтобы проиллюстрировать две изложенные выше процедуры обратной сверт¬
ки, на рис. 4.18 приведен пример треугольного истинного профиля концент¬
раций [49]. Рис. 4.18, а демонстрирует применение метода Ван Циттерта,
а рис. 4.18, б - метода, когда в итерационном процессе используются раз¬
личные приближения, описывающие истинный профиль концентрации. Из-за
остроты пика и значительного градиента концентрации на профиле получен¬
ный в обоих случаях "истинный" профиль концентрации страдает некото¬
рой неоднозначностью по отношению к погрешности измерений (ср. с об¬
ластью III, рис. 4.17).Если вносить поправки с целью отдельного учета эффекта уширения
вследствие влияния глубины выхода, то такая корректировка также осущест¬
вима в рамках итерационного метода, использующего приближенное описа¬
ние истинного профиля. В этом случае уравнение (4.40) необходимо заме-Рис. 4.16. Сравнение получаемой оценки истинного профиля (1) с функцией разре¬
шения* данной в [ 49]. В качестве первого приближения взят измеренный
профиль /(z)(2). а - метод Ван Циттерта, количество итераций п- 5(3);
б — итеративное приближение к истинному профилю.
1984. Послойный анализнить уравнением, подобным уравнению (4.8) [7, 51](4.42)где А,- зависит от состава (см. п. 4.2.2) для проведения последующей, луч¬
шей аппроксимации [ 7]. Суммирование вкладов отдельных слоев [ уравне¬
ние (4.41)] необходимо так же, как и ранее. Поскольку Af. является эф¬
фективной глубиной выхода, зависящей от геометрии эксперимента, урав¬
нение (4.42) дает возможность использования в случае произвольного про¬
филя неразрушающего послойного анализа, при котором варьируются углы
регистрации эмитированных электронов (см. п. 4.5.1). Полагая А,- = А° Cosq>,
получим I (ф) = 2;* /у (<р) для любого данного угла <р. Это обусловлено тем,
что здесь зависимость от z заменена на зависимость от А . Зависящую от
угла нормированную интенсивность, полученную таким образом, можно не¬
посредственно сравнить с измеренным профилем, также зависящим от угла
(ср. рис. 4.12 и 4.13).Общая схема процедур свертки и обратной свертки показана на рис. 4.19.
Рекомендованные методы (1,11, ///) следует выбирать в соответствии с кри¬
терием, данным на рис. 4.17.Рис. 4.19. Принципиальная схема оценки истинного профиля концентраций соедине¬
ния [49, 53] при помощи процедуры обратной свертки, проведенной с ка¬
либрованными экспериментальными данными (измеренный профиль изо¬
бражен на рисунке сплошной стрелкой), и (или) методом итеративных при¬
ближений к истинному профилю (изображен на рисунке пунктирной стрел¬
кой). 1 - послойный анализ; 2 - обратная свертка; 3- данные послой¬
ного анализа.4.7. ЗаключениеЛюбая количественная оценка данных послойного анализа, полученных
при использовании методов электронной спектроскопии совместно с ионным
распылением, требует количественной оценки интенсивности сигнала и ка¬
либровки шкалы глубины, а также знания функции разрешения по глубине,
что позволяет судить о том, велики ли изменения состава в процессе анали¬
за или пренебрежимо малы. Благодаря высокой скорости анализа, возможности уче-
С. Хофман199та уровня фона и высокой локальности ЭОС высокого пространст¬
венного разрешения имеет существенные преимущества перед РФЭС. В то
время как основные параметры для количественной ЭОС и РФЭС (глубина
выхода электронов и коэффициент обратного рассеяния в ЭОС) можно срав¬
нительно точно определить, прогнозирование большого числа других пара¬
метров (зависящих от аппаратуры и характеризующих взаимодействие ионно¬
го пучка с образцом) остается в значительной степени спекулятивным. Для
оценочного тестирования шкалы глубины и настройки аппаратуры можно ре¬
комендовать использование анодированных слоев Та205/Та с известным
положением границы раздела, которые в ближайшем будущем появятся
как стандартные образцы. Методы измерения глубины травления непосред¬
ственно по месту (например, с помощью оптической интерферометрии, рент¬
геновского анализа), по-видимому, являются единственным способом полу¬
чения точной калибровки шкалы глубины. Тестирование образца с целью выяв¬
ления вызванных ионным пучком изменений можно провести, изменяя усло¬
вия распыления (например, тип ионов, их энергию и угол падения пучка).В отдельных случаях полезным является применение альтернативных обыч¬
ному распылению методов, таких, как послойный анализ на краю кратера,
послойный анализ с использованием шаровых шлифов и с изменением эф¬
фективной глубины выхода.После того как осуществлен первый шаг в количественной оценке (опре¬
делены шкалы концентраций и глубины), остается оценить окончательное раз¬
личие между измеренным профилем и истинным, характеристикой чего может
служить экспериментально определенное разрешение по глубине, или, более точ¬
но, функция разрешения по глубине. Знание этой функции позволяет провести
операцию обратной свертки над нормированным измеренным профилем, чтобы
численным методом получить истинный профиль. С другой стороны, свертка
приближенного истинного профиля и итеративное сравнение его с измерен¬
ным позволяют определить истинный профиль концентраций. В любом случае
ключевым параметром для обеспечения надежности и точности измерения
последнего является экспериментально измеренное значение глубины с из¬
вестным разрешением, которое зависит от тщательной оптимизации условий
проведения послойного анализа.Б лагодарностиАвтор выражает глубокую благодарность за полезные дискуссии д-рам
М.П. Сиху (Теддингтон, НФЛ), Д.М. Санцу и У. Роллу.Литература1* Benninghoven A., Thin Solid Films, 31, 89 (1976).2. Honig R< E., Thin Solid Films, 39, 3 (1976).3. Hofmann S*, Surf. Interface AnaL, 2, 148 (1980).
2004. Послойный анализ4. Hofmann S., Analysis, 9, 181 (1961).5. Hofmann Sin: Wilson and Wilson’s Comprehensive Analytical Chemistry
(ed. A. Svehla), Elsevier, Amsterdam, V.IX, 1979, p.p. 89 — 172.6. Hall PM*, Morabito JM*, Crit. Rev. SoL State Mat. Sci., 8f 53 (1978).7. Hofmann S*, LeVide No. Spec., 3, 259 (1979)*8. Mitchell LV>, Phys. Bull., 30, 23 (1979).9. Holloway Р.Я., McGuire JM., Thin Solid Films, 53, 3 (1978).10. Fadley C*S*, Progr. Sol. State Chem., 11, 265 (1976).11. Ebel M*F*, J. Electron Spectr. Rel. Phen., 14, 287, (1978).12. Maier K*, Kirchheim R*, Tolg G*, Microchim. Acta Supply, 8, 125 (1979).13. Cobum J.W., J. Vac: Sci. TechnoL, 13, 1037 (1976).14. Hofmann S*, in: Proc. Third Int. Conf. on SIMS (Budapest, 1981) (ed.A. Benninghoven et ah), Springer-Verlag, Berlin, 1982, p. 186.15. Mathieu H*J*, Landolt D*, J. Microsc. Spectr. Electron., 3, 113 (1978).16. Seah M.P*, Lea С*, Thin Solid Films, 81, 257 (1981).17. Sykes D.E* et ah, Appl. Surf. Sci., 5, 103 (1980).18. Werner H*W*, Mikcrochim. Acta Suppl., 8, 25 (1979).19. Sputtering by Ion Bombardment, V.I. (ed. R. Behrisch), Topics in Appl.
Phys., Springer-Verlag, Heidelberg, 1981.20. Seah M.P., Thin Solid Films, 81, 279 (1981).21. Oechsner HAppl. Phys., 8, 185 (1975).22. Schoof H*, Oechsner Hin: Proc. Fourth Int. Conf. Sol. Surf, and Third
ECOSS, Cannes (1980) (eds. F. Abeles, M. Croset), Paris, 1980, p. 1291.23. Bevelo A*J*, Surf. Interface Anal., 3, 240 (1981).24. Young LAnodic Oxide Films, Academic Press, London, 1961.25. Morabito JM*, Lewis RtK*, Anal. Chem*, 45, 869 (1973).26. Laty P., Seethanen D*, Degreve F*, Surf. Sci., 85, 533 (1979).27. Cobum J*W*, Kay E., Crit. Rev. Sol. State Sci., 4, 561 (1974).28. Hofmann S*, Zalar A*, Thin Solid Films, 60, 201 (1979).29. Kirschner J*, Etzkom H*W>, Appl. Surf, Sci., 3, 251 (1979).30. Thomas JM., Sharma S*P*, J. Vac. Sci. Technol., 14, 1168 (1977).31. Kempf J*, in: Proc. Second Int. Conf. on SIMS (II) (ed. A. Benninghoven et
al.), Springer-Verlag, Berlin, 1979, p. 97.32. Kempf J*, Surf. Interface Anal., 4, 116 (1982).33. Seah M*P*, Surf. Sci., 32, 703 (1972).34. Palmberg P*W*, Anal. Chem., 45, 549 (1973).35. Seah M*P*, Dench WSurf. Interface AnaL, 1, ? (1979)*36. Jablonski A*, Surf. Interface AnaL, 1, 122 (1979).37. Hall P.M., Morabito JM*, Conley D*K*, Surf. Sci., 62, 1 (1977).38. Seah M.P*, Analusis, 9, 171 (1981).39. Hofmann S*, Mikrochim. Acta Suppl., 8, 71 (1979),40. Pons P., LeHericy J*, Langeron J*P*, Surf. Sci., 69, 565 (1977).41. LeHericy J*, LeVide, -No. Spec., March 3, 307 (1979).
С. Хофман20142. Pijolat М*, Hollinger G*, Analusis, 10, 8 (1982).
43* Iwasaki H*, Nakamura G*, Surf. Sci., 57, 779 (1976).44. Hofmann S*, Talanta, 26, 665 (1979)*45. Thomas JM* III, Hofmann S*, Surf, interface Anal., 156 (1982).46. Wagrier C*D*, Anal. Chem., 44, 1050 (1972).47. HofmannS*, Sanz JMFresenins Z*, Anal. Chem., 314, 215 (1983).48. Ho P*S*, Lewis J*E*, Surf. Sci., 55, 335 (1976).49. Hofmann S*, Sanz JM*, in: Proc. Eighth Int. Vac. Congr., Cannes (1980)
(eds. F. Abeles, M. Croset), Paris, 1980, p. 90.50. Hofmann S*, Appl. Phys., 9, 59 (1976).51. Hofmann S*, in: Proc. Seventh Int. Vac. Congr* and Third Int. Conf. Sol.Surf., (eds. A. Dobrozembsky et al.), Vol. Ill, p. 2613, Berger, Vienna,1977, p. 2613.52. Mathieu H* J*, \Landolt D*, Le Vide, No. Spec., 3, 273 (1970).53. Hofmann S., Sanz JM*, in: Topics of Current Physics (ed. H. Oechsner),
Springer-Verlag, Heidelberg, 1982.54. Hofmann S., Erlewein ]., Zalar Aa, Thin Solid Films, 43, 275 (1977).55. Ludeke R*, Esaki L*f Chang L*L*, Appl. Phys. Lett., 29, 417 (1974).56. Mathieu H*J*, McClure D*E*, Landolt D., Thin Solid Films, 38, 281 (1976).57. Roll K*, Hammer C*, Thin Solid Films, 57, 209 (1979).58. Blattner R*J* et al*, Surf. Interface Anal., 1# 32 (1979).59. Holloway P*H*, Battacharya R*S*, Surf. Interface Anal., 3, 118 (1981).60. Holloway PM*, Stein H*J*, Electrochem. Soc., 123, 723 (1979).61. Zalar A*, Hofmann S., Surf. Interface Anal., 2, 183 (1980).62. Malherbe J*B*, Sanz JMHofmann S*, Surf. Interface Anal., 3, 235 (1981).63. Bradley L* et al*, Appl. Spectr., 32, 175 (1978).64. Hauffe W., in: Proc. Third Int. Conf. on SIMS (III), (Budapest, 1981) (eds.A. Benninghoven et al.), Springer-Verlag, Berlin, 1982, p. 206.65. Tsunoyama К*, et ah, Surf. Interface Anal., 2, 212 (1980).66. Makh S*S*, Smith R*, Walls JM*, Surf. Interface Anal., 2, 115 (1980).67. Smith R*, Valkering T*P*, Walls JM*, PhiL Mag., A44, 879 (1981).68. Chadderton L*D*f Rad. Eff., 33, 129 (1977).69. Wehner G*K*, Haijcek D*J., J. Appl. Phys., 42, 1145,(1971).70. Vossen J*LJ. Appl. Phys., 47, 544 (1976).71. Sigmund P., in: Sputtering by Ion Bombardment (ed. R. Behrisch), Vol. I,
Topics in Appl.Phys., Springer-Verlag, Heidelberg, 1981.72. Hofmann SAppl. Phys., 13, 205 (1977).73. Hofer W*0., Liebl H., Appl. Phys.f 8, 359 (1975474. Helms C*R< et ah, in: The Physics of Si02 and Its Interfaces (ed. S.T. Pan-
telides), Pergamon, New York, 1978, p. 366.75. Seah M*P*, Sanz JM*, Hofmann S*, Thin Solid Films, 81, 239 (1981).76. Erlewein ]*, Hofmann S*, Thin Solid Films, 69, L39 (1980),77. Wemer H*W*, Surf. Interface Anal., 4, 1, (1982).
2024. Послойный анализ78. Andersen Н*НAppl. Phys., 18, 131 (1979).79. Taikang S*, Shimizu R*, Okutani T*, Jap. J. Appl. Phys., 18, 1987 (1979).80. Littmark U*t Hofer W*0*, Nucl. Instr. a Math., 1B8, 329 (1980).81. Sigmund PGras-Marti Л., Nucl. Intsr. a Meth., 182, 25 (1981).82. Etzkom H*W; Littmark UKirschner J*, in: Proc. Symp. on Sputtering
(eds. P. Varga, C. Betz, F.P. Viehbock) Perchtolsdorf, 1980, p. 542,83. Liau Z*L*, Tsaur B*Y», Mayer J*W+, J. Vac. Sci. Technol., 16, 121 (1979).84. Shimizu H*, Ono М., Nakayama К*, Surf. Sci., 36* 817 (1973).85. Mathieu H*J>, Landolt Dt, Surf. Sci., 53, 228 (1975).86. Но P.S. et al, Surf. Sci., 57, 393 (1976).87. Mathieu H*J., Landolt D*, Appl. Surf. Sci., 10, 100 (1982).88. Kelly R; Nucl. Intsr. a Meth., 149, 553 (1978).89. Storp S., Holm RJ. Electron Spectr. Rel. Phen., 16# 183 (1979).90. Holm R*, Storp S., Appl. Phys., 9, 217 (1976).91. Mathieu H*J*f Landolt D*, in: Proc. Seventh Int. Vac. Congr. a Third
Int. Conf. Sol. Surf. (Vienna 1977), Berger, 1977, p. 2023.92. Ohuchi F* et aU, Surf. Interface Anal., 2, 85 (1980).93. Kelly Rin: Proc. Symp. on Sputtering (eds. G. Varga, G. Betz, EP. Vieh¬
bock), Perchtolsdorf, 1980, p. 512.94. Hofmann S-, Mat. Sci, Eng., 42, 55 (1980).95. Thomas SJ. Appl. Phys., 45, 161 (1974),96. Ahn J* et aL, J. Appl. Phys., 46, 4581 (1975).97. Roll К*, Appl. Surf. Sci., 5, 388 (1980).98. Ebel M*F; Wemisch Surf. Interface Anal., 3, 191 (1981).99. Vulli М., Surf. Interface Anal 3, 67 (1981).100. Van Oostrom A*, Surf. Sci., 89, 615 (1979).101. Walls JM4, Hall D. D., Sykes D*ESurf. Interface Anal., 1, 204 (1979).102. Lea СSeah M.P., Thin Solid Films, 81, 67 (1981).103. Carley A,F*, Joyner R*W*, J. Electron Spectr. Rel. Phen., 16, 1 (1979).104. Van Cittert Р.Я., Z. Physik, 69, 298 (1931).105. Wertheim G*KJ, Electron Spetr. Rel. Phen., 9, 239 (1975).106. Madden H*H*, Houston J*Ei, J. Appl. Phys., 27, 3071 (1976).107. Zalar A*, Microchim. Acta, I, 435 (1980).108. Magee Harrington W*L* Honig R*E<, Rev. Sci. Instr., 49, 477 (1978).109. Mathieu H*JLandolt D*, Surf. Interface, Anal., 5, 77 (1983).110. Naundorf V*, Macht P., Nucl. Instr. a Math., 168, 40 (1980).111. Hunt C*P*, Stoddart C*T*H*t Seah M»P*, Surf. Interface Anal., 3, 157
(1981).112. Malherbe J*S., Hofmann SSurf. Interface AnaL, 2, 187 (1980).
Глава 5
КОЛИЧЕСТВЕННАЯ ОЖЕ- ЭЛЕКТРОННАЯ
И РЕНТГЕНОВСКАЯ ФОТОЭЛЕКТРОННАЯ
СПЕКТРОСКОПИЯМ.П. Сих'15.1. ВведениеВ самых ранних приложениях ЭОС предпринимались усилия, направлен¬
ные на повышение точности количественного анализа. Что же касается РФЭС,
то основной целью в прошлом было получение данных по химическому соста¬
ву, так что интерес к применению РФЭС для количественного анализа возник
сравнительно недавно. В настоящей книге количественный анализ состава на
основе названных методик рассматривается в рамках одной главы, поскольку
в обеих методиках используется электронная спектроскопия и многие новые
установки приспособлены для применения обеих методик. Можно надеяться,
что единая методология и система обозначений облегчат работу аналити¬
ков, которым в настоящее время приходится справляться с двумя методика¬
ми одновременно.К решению задачи количественного анализа имеются три различных под¬
хода, применимые, по-видимому, к анализу любого типа: а) вычисление соот¬
ветствующих членов из первых принципов; б) использование опубликованных
справочных данных; в) использование внутренних стандартов или справочных
данных. На практике наиболее эффективным для аналитика может оказаться
сочетание всех трех подходов.В последующих разделах будут описаны основные члены, определяющие
интенсивность сигнала - сначала в ЭОС, а затем - в РФЭС, для простого
случая однородных объемных образцов из сплавов. Это позволит, кроме того,
получить правильную перспективу для применения справочных данных, как
взятых из печати, так и полученных самими исследователями, а также помо¬
жет понять приближения, сделанные при получении этих данных. Однако обыч¬
но анализ поверхности используют в применении к двум группам образцов,
не являющихся однородными. Этому типу анализа посвящены гл. 6-10. Пер¬
вую группу составляют те адсорбаты, в случае которых анализ поверхностиМ.Р* Seah, Division of Materials Applications, National Physical Laboratory,
Teddington, Middlesex, UK.
2045. Количественная ЭОС и РФЭСприменяется как классический метод - для идентификации и измерения суб-
монослойных количеств атомов или мэлекул, расположенных в самом внеш¬
нем слое атомов твердого тела. Во вторую группу входят образцы, в кото¬
рых требуется изучить более толстые слои, так что анализ в них производит¬
ся одновременно с ионной бомбардировкой, позволяющей получить профили
концентрации по глубине. Обсуждение этих двух групп на основе единого фор¬
мализма будет приведено в конце настоящей главы.Интересной отправной точкой служит простое линейное выражение, сог¬
ласно которому интенсивность сигнала 1А от элемента А в твердом теле прос¬
то пропорциональна относительному молярному содержанию ХА этого элемен¬
та на исследуемой глубине. Таким образом,где / J0 - интенсивность сигнала от чистого элемента А. Поскольку, как
правило, величина неизвестна, но может быть известно отношение
то полезнее использовать уравнение (5.1) в преобразованном виде [1]:где суммирование производится по всем компонентам твердого тела. Уравне¬
ние (5.2) дает первый приближенный результат для количественного анализа,
но в следующих двух разделах мы обсудим члены, вносящие вклад в в ЭОС
и РФЭС, что позволит понять вид и порядок величины необходимых поправоч¬
ных членов.5.2. Количественный анализ с помощью ЭОС в случаеоднородных бинарных твердых телЧтобы использовать ЭОС как метод количественного анализа, необходи¬
мо решить две основные задачи: получить правильное уравнение для расчетов,
которым мы будем пользоваться, и установить соответствующую процедуру
измерения.5.2.1. Основные соображенияПадающий пучок электронов, столкнувшись с поверхностью твердого тела,
проникает внутрь, испытывая при этом как упругое, так и неупругое рассея¬
ние, и достигает глубины 1-2 мкм (рис. 5.1). Как упоминалось в гл. 3, эти
электроны, обладающие высокой энергией, способны ионизовать атомы твер-(5.1)(5.2)
М.П. Сих205Рис. 5.1. Схема рассеяния электронов в ЭОС: 1 - оже-электроны, которые могут быть
эмитированы; 2 - электроны высоких энергий; 3 - атомы, ионизованные на
внутреннем уровне X; 4 — обратно рассеянные первичные электроны.дого тела на внутреннем уровне X. Впоследствии возбужденный атом девоз-
буждается за счет заполнения вакансии электроном, переходящим с более вы¬
сокого уровня У, причем избыток энергии уносится за счет эжекции электро¬
на с уровня Z. Таким образом, энергия этого последнего электрона, называе¬
мого XYZ оже-электроном, имеет вполне определенную величину.Самые первые вычисления сечения ионизации о^Е) электронами, обла¬
дающими энергией £, в ЭОС принадлежат Бишопу и Ривьеру [2]. Следуя мето¬
дам, первоначально развитым для рентгеновского микроструктурного анали¬
за, они используют поперечное сечение ионизации Уортингтона и Томлина
[3], которое задается выражением(5.3)где b = 0,35 для К-оболочки и 0,25 для L-оболочки. Функция С зависит от
энергии электронов первичного пучка Ер и возрастает от нуля при Е^ = ЕАХ
2065. Количественная ЭОС и РФЭСдо 0,6 при Ер = (2-3)ЕАХу а затем медленно убывает при дальнейшем уве¬
личении энергии. Приняв в качестве характерного значения С = 0,5, получаем
с помощью приведенного поперечного сечения, что прохождение электронного
пучка через поглощенный монослой дает эффективность ионизации порядка
Ю“4. Таким образом, для покрытия, соответствующего 1% монослоя, если
иметь в виду, что полный коэффициент испускания вторичных электронов для
многих твердых тел есть величина порядка единицы, получаем, что сигнал оже-
электронов должен представлять собой пик, выступающий над фоном, образо¬
ванным в 106 раз превосходящим числом электронов. Проблемы измерений,
возникающие в связи с этим, рассмотрены в гл. 2 и 3. Современные измере-
ния °ЛХ(Е) [4 -7] приводят к лучшему согласию с сечением ионизации, пред¬
ложенным Грижинским [811>, зависимость которого от энергии представлена
на рис. 5.2 (где приведены также некоторые экспериментальные результаты).
Тем не менее абсолютная величина сечения ионизации остается столь же
низкой, как и у сечения ионизации Уортингтона и Томлина.Выше мы рассматривали ионизацию уровня X атомов в поверхностном
слое, вызванную пучком первичных электронов с энергией Е^. Существует
дополнительный источник ионизации этого уровня - за счет электронов вы¬
сокой энергии, претерпевших обратное рассеяние, как показано на рис. 3.2
(гл. 3). Если п(Е) - спектр истинных обратно рассеянных электронов, порож¬
денных одним первичным электроном, то полная ионизация уровня X может
быть представлена выражением(5.4)которое в свою очередь часто записывается в виде(5.5)1) М. Грижинский в рамках задачи двух тел, что при малых значениях энергииналетающего электрона недопустимо, учтя скорость орбитального электрона, полу¬чил формулу, которая в ряде случаев давала неплохое совпадение с экспериментом.Однако это совпадение, как показал тщательный анализ, оказалось практически
таким же, как и в случае использования полуэмпирической формулы Дравина. Необ¬ходимо отметить, что Грижинский при выводе своей формулы использовал ряд уп¬рощений, в результате чего полученное им соотношение не соответствовало изна¬
чально выбранной модели. Впоследствии усилиями В.И. Очкура и р. Стаблера были
получены классические формулы, свободные от недостатков, присущих формуле
Грижинского, и дающие удовлетворительное согласие с экспериментальными дан¬
ными. — Прим. ред.
М.П. Сих207Рис. 5.2. Зависимость сечения ионизации уровня X по Грижинскому от энергии и
результаты измерений для Be [4], С, Si , Ag и Gd [5].где член описывающий обратное рассеяние, зависит от кристаллической
решетки М, в которую входят атомы Л, и от угла а, образованного направле¬
нием падающего пучка электронов с нормалью к поверхности. Оценки члена
обратного рассеяния проводились с помощью вычислений методом Монте-Кар-
ло [2, 6, 9] и эмпирически - Рейтером [10]. Вычисления дают грубое согла¬
сие для глубоких внутренних уровней, а соотношения Рейтера приводят к не¬
верным результатам при низких энергиях (ниже 500 эВ) для внутренних уров*.
ней. Вычисления Симицу и Итимуры [ 6, 7] наиболее полны, поэтому мы их
здесь используем. На рис. 5.3 по материалам их работы показано возраста¬
ние величины гм с увеличением атомного номера Z и ее убывание с ростом
энергии ионизации уровня X для электронного пучка с энергией 5 кэВ, па¬
дающего под углом а = 30° к нормали к поверхности. На рис. 5.4, а показа¬
но изменение гм в зависимости от Z, а на рис. 5.4, б - в зависимости от а.
Наконец, еще один член, дающий вклад в ионизацию внутреннего уровня X,
обусловлен переходами Костера - Кронига. Эти переходы являются очень
быстрыми, но происходят только между оболочками с одним и тем же значе¬
нием главного квантового числа. Так, например, дырка в оболочке L1 запол¬
няется электроном из L2 или L3. Таким образом, слабее связанные уровни
вносят дополнительный вклад в ионизацию и усиливают пики оже-электронов.Теперь мы можем рассмотреть суммарный вклад, вносимый всеми меха¬
низмами в образование сигнала оже-электронов. Ионизованное состояние, воз-
2085. Количественная ЭОС и РФЭСРис. 5.3. Зависимость коэффидента обратного рассеяния гм для некоторых элемен¬
тов, полученная при использовании пучка электронов с энергией 5 кэВ и
углом падения 30° относительно нормали к поверхности [6, 7], от энер.
гии ионизации уровня,
никшее на внутреннем уровне X, распадается с вероятностью, равной вероят¬
ности испускания оже-электрона yXYZ посредством перехода XYZ. Вероят¬
ность того, что возникший оже-электрон пройдет расстояние Л (EAY„7 ), Рав~0 М АлУ2ное средней длине неупругого свободного пробега в данной кристаллической
решетке, равна е~\ Испытав неупругое рассеяние, электрон перестает участ-Рис. 5.4. Зависимость гм от атомного номера z (а) и угла падения электронного
пучка а (б) [б, 7].
М.П. Сих209вовать в образовании пика оже-электронов. Таким образом, поток оже-элект¬
ронов затухает как ехр(-//Л) с увеличением расстояния I от начальной точки.
Характеристическая глубина, с которой могут быть испущены оже-электроны,
равна просто ЛM(EAXYZ) cos0, где 0 - угол эмиссии по отношению к норма¬
ли к поверхности. Этот объединенный член часто называют глубиной выхода.
Значения Л лежат в интервале 2-10 атомных слоев, как видно из кривой,
построенной на основании подборки экспериментальных данных и представ¬
ленной на рис. 5.5 [11]. Именно это важное обстоятельство делает и ЭОС, и РФЭС
поверхностно-чувствительными методами. Испущенный электрон затем де¬
тектируется электронным спектрометром с коэффициентом пропускания
T(Eaxyz) и электронным умножителем с эффективной чувствительностью D(EAXYZ).
Таким образом, ток IAXYZ оже-электронов может быть записан в видеУгде для ясности записи индекс XYZ опущен, /0 - ток в первичном пучке элект¬
ронов, Na(z) - распределение атомов А по глубине z, отсчитываемой от по¬
верхности в глубь образца. Если электронный спектрометр обладает большой
угловой апертурой, то ток следует проинтегрировать по соответствующему
телесному углу.Рис. 5.5. Зависимость Л от энергии электронов [ 11].
2105. Количественная ЭОС и РФЭСВ настоящее время ни один исследователь не пользуется для выполнения
количественного анализа непосредственно уравнением (5.6). Для однородных
бинарных систем можно произвести оценку интеграла, и его значение оказыва¬
ется разным просто Na\m(Ea) cos6. Мы можем еще больше сократить коли¬
чество неизвестных, перейдя к рассмотрению отношения интенсивностей
Iа /1В и сравнив его с отношением тех же величин для стандартных образцов чистых
элементов, / ~// полученных на той же установке. Таким образом,А В(5.7)где R7’R°B~ шероховатоста стандартных образце® чистых элементе®, a и -
их атомные плотности. Отношение R^/R^ можно не принимать в расчет, ес¬
ли отношение установлено по многим образцам или с помощью приготов-А Вленной in situ поверхности излома гомогенного сплава АВ. Уравнение (5.7)
можно еще более упростить, заметив, что(5.8)
откудагде ХА - искомый относительный молярный состав твердого тела иам-
размер атома Л, получаемый из соотношения ЮООрuNa* = Лд , где в своюМММочередь рм - плотность (в кг/м3), N - число Авогадро и Ам - средний атом¬
ный вес атомов кристаллической решетки (разумеется, если кристаллическая
решетка образована атомами Л, то Л просто совпадает с атомным весом Л).
В работе Сиха и Денча [11] средняя длина неупругого свободного пробега
электронов приближенно задается как(5.9)где ам и \м измеряются в нанометрах, а - в электронвольтах. Таким об¬
разом, в результате подстановки (5.8) и (5.9) в уравнение (5.7) получаем(5.10)где фактор, учитывающий влияние матрицы на оже-электроны задает-
М.П. Сих211ся в виде(5.11)(*5.12)Из рис. 5.3 видно, что соотношение tA/гв не очень сильно зависит от энер¬
гии, вследствие этого величина FAAB изменяется незначительно в диапазоне
от Л до В и может с успехом быть принята постоянной для системы АВ*
Описанный подход был впервые применен Холлом и Морабито [12], кото¬
рые вычислили матричные члены двух предельных выражений (5.11) и (5.12)
для 4860 бинарных систем. Вместо использования для вычислений обратного
рассеяния метода Монте-Карло [6, 9], они применили эмпирическое соотноше¬
ние Рейтера [10], однако в качестве дополнительной альтернативы эмпири¬
ческим оценкам Сиха и Денча средней длины свободного пробега
для неупругого рассеяния были приведены теоретические предсказания Пен¬
на [13]. Таким образом, они показали, что в 86% бинарных системс точностью не хуже 5 процентов. Холл и Морабито заметили также, что мат¬
ричные факторы распределены вблизи среднего значения с величиной стан¬
дартного отклонения 1,5. Если пренебречь матричными факторами, то урав¬
нение (5.10) принимает вид (5.2). Величина получаемой при этом погрешности
приведена на рис. 5.6, где представлен график соотношения между истинным
значением ХА , полученным из уравнения (5.10), и приближенным значением
ХА, полученным из уравнения (5.2) для различных значений FAB. Удивитель¬
ным является тот факт, что множество исследователей до сих пор используют
уравнение (5.2), в то время как существует точная оценка матричных факто¬
ров.В качестве примера приведем матричный фактор для углерода в железеи аналогично для алюминия в никеле: Ni = 0,70 для пика алюминия с вы¬
сокой энергией и 0,68 — с низкой энергией. В таблицах Холла и Морабито
эти матричные факторы имеют величины 0,997, 0,999 и 0,589 соответственно.(5.13)
2125. Количественная ЭОС и РФЭСРис. 5.6. Сравнение истинного состава и состава, определенного без учета матрич¬
ного фактора.Разница в этих двух случаях вызвана не членами с обратным рассеянием, а
различием значений Л у Сиха и Денча [11] и у Пенна [13]. Необходимо пом¬
нить, что экспериментальные данные дают фактор разброса 1,3 по Сиху и
Денчу и 1,4 по Пенну, и, таким образом, вычисление матричных факторов с
высокой точностью пока не представляется возможным. В среднем отличие
величины К по Сиху и Денчу от Л по Пенну составляет 0,83, причем ее
среднеквадратичное отклонение составляет 30%»Выше мы предполагали, что стандартный спектр был записан при помощи
того же прибора, который используется для проведения исследований. Если
это не так, необходимо рассмотреть члены Т(ЕА) и D(FA)- Благоприятным
является то обстоятельство, что в настоящее время в большинстве исследо¬
ваний методом ЭОС используются спектрометры, работающие в режиме по¬
стоянного соотношения ДЕ/Е, где ДЕ - энергетическая разрешающая спо¬
собность спектрометра при энергии Е (см. гл. 2). Это означает, что коэффи¬
циент пропускания большинства спектрометров Т(ЕА) просто пропорционален
FА при условии, что отсутствуют рассеянные магнитные поля. В этом слу¬
чае члены Т(Еа) сокращаются и могут не приниматься в расчет. Однако эф¬
фективность детектора различна у разных приборов. Как показали Девис и дрД 1],
коэффициент усиления электронного умножителя увеличивается с ростом Ь'д от
М.П. Сих213нуля до максимума приблизительно при 300 эВ, а затем постепенно снижается-
Этот эффект можно устранить подачей на вход умножителя постоянного на¬
пряжения 150 В таким образом, чтобы на умножитель не попадали электроны
с низкой энергией. При этом ошибки, вносимые колебаниями D(E^), умень¬
шаются до величины менее 5% в энергетическом диапазоне свыше 2000 эВ.До сих пор мы не обсуждали величины, входящие в 1А. Фактически это
рассмотрение может быть проведено для любой величины потока оже-электро-
нов при условии, что он постоянен. В связи с этим возникает проблема, ко¬
торая и рассматривается ниже.5.2.2. Измерение интенсивностиВ прошлом обычным было измерение значения интенсивности peak-to-
peak в дифференциальном энергетическом спектре, как уже упоминалось в
гл. 3. Такой подход вполне оправдан, и с тремя существенными ограничения¬
ми эти значения могут быть использованы для вычисления 1А и /|°. Эти
ограничения состоят в том, что формы пиков в исследуемом и стандартном
спектрах должны быть одинаковы, что энергоанализаторы, использованные
для получения обоих спектров, должны иметь одинаковое разрешение и что в
обоих случаях должна быть использована одинаковая модуляция. В случае
анализа металлических сплавов спектры стандартных образцов легко могут
быть получены на том же приборе. Можно, например, использовать в случае
проведения анализа на спектрометрах с разрешением 0,6% справочник по
ЭОС [1], в котором приведены значения / ~ для энергий электронного луча
3,5 и 10 кэВ. В качестве примера на рис. 5.7 приведен ряд факторов относи¬
тельной чувствительности IКакие же поправки необходимо ввести, если не учитывать ограничений,
связанных с разрешающей способностью прибора и модуляцией? Эти поправ¬
ки связаны с тем, что погрешность, обусловленная наличием рассеянных
магнитных полей [14] и смещением образца С15], в случае использования АЦЗ
очень сильно влияет на разрешающую способность по энергии, и даже при
использовании анализатора той же модели, что и в справочнике, созданном
фирмой Physical Electronics [1], мы можем не достичь той же разрешающей
способности. Кроме того, в этом справочнике точно не описана модуляция,
использовавшаяся при получении стандартных спектров, хотя ее влияние рас¬
смотрено в руководстве по ЭОС Макгайра [16]. Для отдельных синглетных
пиков влияние модулирующего напряжения может быть описано универсаль¬
ным графиком, приведенным на рис. 5.8. Предполагалось, что пик имеет асим¬
метричную форму и что отрицательное крыло может быть описано полушири¬
ной Vi W, как это показано на рисунке. В общем случае положительное кры-
2145. Количественная ЭОС и РФЭСРис. 5.7. Факторы относительной чувствительности для дифференциальных спект¬
ров ЭОС [1]. Цифры и буквы на концах кривых указывают величины энер¬
гии пиков и элементы, которым они соответствуют.ло характеризуется другим значением W, поэтому будет рассмотрено только
отрицательное крыло пика, а интенсивность будет измеряться величиной амп¬
литуды h, отсчитываемой от уровня фона. Таким образом, кривая на рис. 5.8
позволяет осуществить переход от одного типа модуляции к другому для синг-
летных пиков при условии, что ширина W известна. При проведении общих ана¬
лизов с амплитудным (peak-to-peak) значением модулирующего напряжения
5В и разрешением спектрометра 0,5% изменение энергии от 100 до 2000 эВ
перекрывает диапазон от пиков, в случае которых насыщение достигается
при энергиях менее 1 кэВ, до пиков, расположенных на линейной части кри¬
вой, приведенной на рис. 5.8, при энергиях около 2000 эВ.При преобразовании данных, полученных на одном спектрометре, для ис¬
пользования на другом, отличающемся разрешающей способностью, изменя¬
ются и ширина пиков, и их относительные интенсивности. На рис. 5.9 приве¬
дены два спектра одной и той же поверхности, полученные на спектрометрах
с разрешающей способностью 0,3 и 1,2% соответственно. Из графиков видно,
что со снижением разрешающей способности спектрометра острые пики
уменьшаются сильнее, чем более широкие пики, и максимум интенсивности
перемещается с меди на углерод, бее эти изменения учитываются универ¬
сальной кривой,приведенной на рис. 5.8. Пик Меди при энергии 920 эВ сдви-
М.П. Сих215Рис. 5.8. Универсальная кривая зависимости изменения амплитуды одиночных пиков
от амплитуды модулирующего напряжения К [17].гается от V/W = 1,6 до V/W = 0,54 с уменьшением величины hW в 1,65 раза
и соответственно h в 5,5 раза, в то время как для углерода величина V/W
изменяется от 0,71 до 0,65, вызывая потери в интенсивности только в 1,18 ра¬
за. Таким образом, изменение интенсивности пика меди относительно интен¬
сивности пика углерода составило 4,6, что довольно близко к величине 4,3,
полученной из спектра, приведенного на рис. 5.9.Обсудим несколько более подробно рис. 5.8. Если мы рассмотрим какой,
либо отдельный пик в спектре п(Е) или Еп(Е) и удвоим его ширину W при
единичном токе оже-электронов, то ясно, что h уменьшится в 4 раза, так как
величина Ш2 останется постоянной для малого модулирующего напряжения
V, и, таким образом, Ш будет пропорционально V/W, как это и следует для
малых уровней модуляции из рис. 5.8. Рассмотрим теперь изменение отно¬
сительных интенсивностей д вух оже-пиков при 500 эВ и 1000 эВ в соответствии
с кривой на рис. 5.8 в случае измерений, проводимых с помощью АЦЗ с раз¬
решающей способностью 0,5% для случая, когда образец смещен на 0,4 мм.
Ширина пиков будет в общем случае определяться этой разрешающей способ¬
ностью. Указанное смещение образца может легко произойти при исследова¬
нии шероховатых поверхностей или изломов или если исследователь просто этого не
заметил, Сикафус и Холлоуэй [15] показали, что это смещение может ухуд¬
шить разрешающую способность до 0,6%, вызывая при этом изменение значе¬
ний W для двух пиков, например от 2,9 и 5,2 до 3,4 и 6,2 эВ соответственно.
Для модуляции С амплитудой 2 В (peak-to*peak) относительные амплитуды
этих Двух пиков не изменятся; однако при модуляции с амплитудой 5 В пик
2165. Количественная ЭОС и РФЭСРис. 5.9. ЭОС-спектры для загрязненного медного образца, полученные на спектро¬
метре А с разрешением 0,3% и на спектрометре в с разрешением 1,2%
[17].при 500 эВ сдвинется с V/W = 1,7 до 1,5, в то время как пик при 1000 эВ
сместится с V/W = 0,96 до 0,81, что вызовет относительное уменьшение ин¬
тенсивности пика при 1000 эВ на 9% по сравнению с амплитудой пика при
500 эВ.Одним и:з способов решения указанных проблем в будущем могло бы
быть применение модуляции с амплитудой, нарастающей таким образом, что¬
бы величина VЛ\ сохраняла постоянное значение, например 1,5 или 2 во всем
спектре. В этом случае смещение образца будет иметь меньшее значение.
Для энергоанализатора с разрешающей способностью 0,5% модуляция должна
была бы составлять 0,75,или 1% энергии с минимальным значением амплиту¬
ды модуляции 2 В, поскольку ширина большинства пиков колеблется в пре¬
делах от 1 до 1,5 эВ.В заключение необходимо рассмотреть эффект изменения формы пика
в связи с изменениями химического состава среды [ 18 — 201. На рис. 5.10
проиллюстрированы такие изменения формы в случае кремния в дифферен-
М.П. Сих217циальном и интегральном энергетических спектрах. Дополнительные приме¬
ры приведены в гл. 3. Эти изменения формы очень трудно учесть, и в этом
случае для количественного анализа следует использовать пик с высокой энер¬
гией (1606-1619 эВ), при которой пики за счет разрешающей способности
анализатора примут одинаковую форму. Факторы чувствительности, напротив,
должны будут определяться для каждой формы пика.Рис. 5.10 поясняет использование интегрального энергетического спект¬
ра п(Е) или Еп(Е). Ясно, что интенсивность оже-электронного сигнала
должна соотноситься с площадью соответствующего пика в спектре п(Е).Это очевидно, например, из рис. 5.Ю, что позволяет включить в рассмотре¬
ние и пик с более низкой энергией, наблюдаемый для Si02. При этом исчеза¬
ют проблемы, связанные с учетом модулирующего напряжения и разрешаю¬
щей способности энергоанализатора. Исторически используется дифференци¬
альный спектр, как это описано в гл. 3, поскольку для оже-электронов пики
малы и часто их трудно выделить на уровне фона в интегральном энергети¬
ческом спектре. Это означает, что фон будет усиливаться вместе с пиком,
что затрудняет регистрацию пика. Однако при дифференциальной обработкеРис. 5.10. Пики оже-электронов для Si , указывающие на изменение структуры иувеличение сдвига в сторону низких энергий по мере увеличения электро-
отрицательности связи [18].
2185. Количественная ЭОС и РФЭСфоновая составляющая удаляется, что обеспечивает легкость регистрации и
измерения пиков. В настоящее время ввиду все более широкого применения
компьютеров для записи и обработки спектров проблема удаления фоновой
составляющей и выделения пиков оже-электронов в интегральном спектре мо¬
жет быть разрешена. Предпринимаются значительные усилия по разработке
соответствующих программ для вычитания фоновой составляющей. На
рис. 5.11 в качестве примера приведены дифференциальный и интегральный
спектры загрязненного медного образца. Во-первых, необходимо заметить,
что коэффициенты относительной чувствительности, полученные для диффе¬
ренциального спектра, не могут быть использованы при рассмотрении интег¬
рального спектра. Это очевидно, поскольку, например, углерод и серебро да¬
ют пики одинаковых размеров в дифференциальном спектре на рис. 5.11, но
в интегральном спектре соответствующие им пики значительно отличаются
по площади.Первый метод, использованный при создании программы для вычитания
фона, был основан на использовании сплайнов [21]. Однако вскоре стало яс¬
но, что он не позволяет однозначно определить площадь пика, поскольку ре¬
зультат в большой степени зависел от граничных условий. Например, на
рис. 5.11 гладкий фон, на котором наблюдаются пики кислорода, серебра и
углерода, внесет свой вклад в случае кислорода в диапазоне выше 150 эВ и
будет включать приблизительно в два раза больше неупругорассеянных оже-
электронов, чем сам пик. Вследствие этого интенсивность пика кислорода
будет характеризовать многократную длину среднего свободного пробега, в
то время как в случае, когда площадь подсчитывалась только для пика, эта
величина будет, как обычно, характеризовать обычную глубину, такую же,
как и в случае дифференциального спектра. Эти недостатки программ вычи¬
тания фоновой составляющей были подробно рассмотрены [22], тем не менее
важной является возможность использования интегрального спектра, посколь¬
ку для оже-микроскопов с высокой пространственной разрешающей способно¬
стью, работающих с малыми токами электронного пучка и с использованием
техники счета импульсов, может оказаться невозможным проведение изме¬
рений с применением модуляции. Изящный метод анализа источников фона
был применен Сикафусом [23, 24]. Он показал, что фон с низкоэнергетичной
стороны пика состоит из ступеньки, обусловленной неупругим рассеянием
оже-электронов на поверхностных атомах образца, и монотонно растущей
составляющей, связанной с рассеянием оже-электронов, поступающих из тол¬
щи образца. Эта ступенька явно различима на рис. 5.12 в пиках оже-электро-
нов, зарегистрированных в процессе иЬследования медного образца методом
РФЭС. Эта проблема Но существу является точно той же, которая возникает
М.П. Сих219Рис. 5.11. Дифференциальный (а) и интегральный (б) электронные спектры загряз¬
ненного медного образца.в РФЭС и которая будет описана в п. 5.3.2, и кажется весьма вероятным, что
решение, используемое в том случае, будет применимо и Для метода ЭОС-
Это решение заключается в использовании модели фоновой составляющей,
разработанной Ширли [25], согласно которой фон растет при высоких энерги-
2205. Количественная ЭОС и РФЭСРис. 5.12. РФЭС-спектр меди. Заметен ступенчатый фон и пики оже-электронов [22].ях пропорционально площади пика, расположенной над фоном. Более подроб¬
но этот метод описан в п. 5.3.2; в настоящее время он представляется наи¬
более многообещающим для количественного анализа интегрального оже-
спектра.Было также предложено другое решение этой проблемы, основанное не
на факторах относительной чувствительности, а на использовании библиотек
стандартных спектров. В этом методе из рассматриваемого спектра вычита¬
ются соответствующие участки различных стандартных спектров до тех пор,
пока не будут выделены все пики. Это позволяет эффективно получить зна¬
чения 1д/1д, которые могут быть использованы в уравнениях для количест¬
венной оценки, и имеет то преимущество, что при этом выявляются все раз¬
личия в спектре, связанные с химическими изменениями или наличием малых
количеств примесей, в областях перекрытия пиков. К сожалению, до настоя¬
щего времени библиотеки интегральных стандартных спектров не сформиро¬
ваны.Выше мы рассмотрели соотношения и результаты измерений, необходи¬
мые для количественного анализа однородных материалов методом ЭОС.В следующем разделе обсуждаются аналогичные проблемы, связанные с ис¬
пользованием метода РФЭС. Проблемы количественного анализа адсорбатов
и проведения послойного анализа химических соединений рассматриваются
затем совместно для обоих методов РФЭС и ЭОС.
М.П. Сих2215.3. Количественный анализ с помощью РФЭС в случаеоднородных бинарных твердых тепРассмотрение производится аналогично проведенному в разд. 5.2.5.3.1. Основные соображенияНа рисунке, аналогичном рис. 5.1, можно было бы в случае РФЭС показать
характеристическое рентгеновское излучение, проникающее в твердое тело
на много микрон и ионизующее внутренний уровень атома X на всей глубине
проникновения. Эжектированные электроны остова движутся сквозь вещество
образца и в конце концов эмитируются с его поверхности, потеряв некото¬
рую энергию. Интенсивность спектральной линии в РФЭС формируют только
те электроны, которые возникли в зоне, расположенной на глубинеЛм(£ах) cos 0 от поверхности. Как и ранее, ^М(ЕАХ) - средняя длина свобод¬
ного пробега характеристических РФЭС-электронов, обладающих энергией
ЕдХ и эмитированных с уровня X элемента А, находящегося в матрице М,
а 0 - угол эмиссии электронов относительно нормали к поверхности,, Зави¬
симость Еах от Л приведена на рис. 5.5.Аналогично выражению (5.6) можно записатьгде опущен индекс X для уровня X. Здесь <ja (hv) - сечение фотоионизации
внутренней оболочки атома А фотоном с энергией /iv, D(EA) - эффективность
детектирования каждого электрона, прошедшего через спектрометр; смысл
величин у, ф, х, у и г ясен из рис. 5.13, LA(у) - угловая асимметрия интен¬
сивности фотоэмиссии каждого атома, J0(xy) — интенсивность характеристи¬
ческой линии потока рентгеновского излучения в точке (х, у) образца,
Т(хуууЕА) - коэффициент пропускания анализатора, NA{xyz) - атомная плот¬
ность атомов А в точке (xyz). В отличие от ЭОС рентгеновскими лучами
засвечивается значительная площадь образца, отсутствует коэффициент об¬
ратного рассеяния, но существует угловая анизотропия эмиссии фотоэлектро¬
нов. На рис. 5.14 приведены расчетные сечения фотоионизации сJA(hv) [27]
для многих встречающихся спектральных линий с использованием характери¬
стической линии Al&oc, хотя на данном этапе рассмотрения в этой информа¬
ции еще нет необходимости.
2225. Количественная ЭОС и РФЭСРис. 5.13. Геометрическая схема метода РФЭС.Для однородного материала приведенный выше интеграл по z сведется
просто к Na\mEa cos 0. Если в нашем распоряжении имеется стандартный
спектр чистого элемента 1А, полученный на том же приборе, то выражение
(5.14) упрощается следующим образом:(5.15)где - средняя длина свободного пробега электронов в сплаве, и -
коэффициенты, которые для чистых стандартных образцов определяют интен¬
сивность эмиссии с поверхности как функцию чистоты поверхности, NА и
N™ - атомные плотности для чистых стандартных образцов. С использована
ем упрощений при выводе уравнений (5.7) и (5.8) получим
М.П. Сих223Рис. 5.14. Расчетные значения сечения фотоионизации cAhv) для линии А1Ка-С 1s.За единицу принято сечение фотоионизации [ 27].Здесь F* - точный аналог для РФЭС матричных коэффициентов Холла и Мо-
рабито, использовавшихся в случаях ЭОС [12]. Если величина F* равна 1,
то выражение (5.16) сводится просто к (5.2), однако анализ 8000 пар элемен¬
тов показал, что значения F*B группируются около 1 со стандартным откло¬
нением 1,52 [28]. Результат использования полного выражения (5.16) вместо
более простого выражения (5.2) показан на рис. 5.6, приведенном ранее при
обсуждении метода ЭОС.В качестве примера значений F*B мы можем использовать рассмотрен¬
ные ранее системы из углерода в железе и алюминия в никеле. При этому V^CFfe = Ь153 и = 0,812, что отличается от значений, приведенныхдля ЭОС.Выше мы подразумевали, что стандартные справочные данные могут быть
получены на том же приборе. Поскольку это требует приложения определен¬
ных усилий и могут быть допущены различные ошибки, как уже упоминалось
выше, проще использовать опубликованные материалы по /~, которые имеют¬
ся в большом количестве [29 - 42]. Ниже в этой главе мы рассмотрим срав¬
нительные достоинства этих наборов данных, но сначала необходимо рассмот-
2245. Количественная ЭОС и РФЭСреть выражение (5.15) для случая, когда исследования проводились на прибо¬
ре 1, а стандартные справочные данные получились на приборе 2. Если элект¬
ронный спектрометр имеет малую входную апертуру и образец освещен одно¬
родно, из выражения (5.14) следуетПоследний член (проинтегрированное по исследуемой площади пропускание
анализатора) известен как полное пропускание анализатора [43] G^(EA). Та¬
ким образом, выражение (5.16) примет вид(5.19)Как описано в гл. 2, в большинстве работ по РФЭС электронный спектрометр
используется в режиме постоянного значения ДЕ. При наличии соответствую¬
щей аппаратуры электроны, обладающие энергией, пропускаемой анализато¬
ром, затем попадают на умножитель, а эффективность детектирования элект¬
ронов D(Ea) постоянна во всем спектре. Вследствие этого членом D можно
пренебречь.Коэффициент угловой асимметрии L(v), который описывает распределе¬
ние интенсивности фотоэлектронов, эмитированных неполяризованным рент¬
геновским излучением из атомов или молекул, определяется как(5.20)где ^ А ” величина постоянная для данной подоболочки данного атома и рент¬
геновского фотона- Здесь не учтено влияние дифракции, которой подвергают¬
ся электроны при пролете через кристаллическую решетку„ Для крупных крис¬
таллов продифрагировавшие пучки могут иметь ПШПВ максимума дифракции
10° с изменениями интенсивности в первом порядке дифракции [45 - 47]. Од¬
нако для твердых тел с мелкими кристаллами или аморфной структурой эф¬
фектами дифракции можно пренебречь*Значения р, вычисленные для основных переходов всех элементов при исполь¬
зовании широко распространенных источников рентгеновских лучей (А \Ка,
1486,6 эВ; MgKoc, 1253,6 эВ и ZrM£, 151,4 эВ), приведены в таблицах Рейл-
мана, Мсезана и Менсона [44]. Эти таблицы хорошо коррелируют с данными
для газов [48]» Получение таких данных для твердых тел осложнено пробле¬
мами, связанными с методикой измерений [49]„ Ващенко и Нефедов [50] ут-
М.П. Сих225верждают, что анизотропия распределения, описанного выражением (5.20),
для твердых тел мэжет оказаться меньшей при учете влияния упругого рас¬
сеяния, которое разупорядочивает направления движения электронов. Хотя
этот эффект и наблюдается, Бащенко и Нефедов используют в своих вычис¬
лениях сечения, полученные методом Томаса - Ферми, который дает в резуль¬
тате величину упругого рассеяния на два порядка больше, чем наблюдается
экспериментально [11], так что влияние разупорядочения будет скорее всего
незначительным. На рис. 5.15 приведены значения р для линии А\К . В ниж¬
ней части рис. 5.15 показано несколько типов конфигураций наиболее распро¬
страненных коммерческих установок, однако необходимо подчеркнуть, что да¬
же в конкретной модели установки геометрия может отличаться от указан¬
ной, поскольку изготовители меняют положение рентгеновского источника
с целью оптимизации параметров приборов. Величина р обычно имеет значе¬
ние 0,8 или 1,8. На рис. 5.16 приведена угловая зависимость LA (у) для этих
значений р с указанием геометрии для некоторых коммерческих установок.Рис. 5.15. Расчетные значения р [ 44]. Геометрические конфигурации типичных ком¬
мерческих установок приведены в нижней части рисунка.1 “ VG ESCA3; 2 - VG ES CALAB; 3 - AEI - Kratos ES- 200, 300;4 - Varian IEE-15; 5 - PHI 550; б - Leybold.
2265. Количественная ЭОС и РФЭСRic. 5.16. Угловое распределение амплитуд для |3 = 0; 0,8; 1,8 в зависимости от
взаимного расположения источника рентгеновских лучей и спектрометра.Геометрические конфигурации рассмотренных коммерческих установок
приведены на рис. 5.15: 1 - VG ESCALAB AEI ES200 Monochromator, 2 -
HP 5950 А, 3 — Leybold LHS, 4 — Varian IEE 15, 5 — AEI ES200 , 6 —VG ESCA3.Угол входа для указанных направлений обычно не превышает нескольких гра¬
дусов, за исключением устройств, использующих анализатор типа цилиндри¬
ческое зеркало (АЦЗ), где угол изменяется в диапазоне от 45 до 135° в зави¬
симости от конфигурации экспериментальной установки. Очевидно, что спра¬
вочные данные, полученные на установке с одной геометрией, будут отличать¬
ся по амплитуде пиков от данных, полученных на установке с другой геомет¬
рией. Этот член точно описывает результаты, полученные Геттинсом и Коа-
дом [51] для чистого золота, где пик 4р ((3 = 1,63) был на 10% интенсивнее
при измерении на VG ESCA3, чем на АЦЗ фирмы PHI при нормировании по
пику 4f (р = 1,03). Таким образом, роль члена, описывающего угловую асим¬
метрию, достаточно ясна, и остается рассмотреть только пропускание энерго¬
анализатора G(Ea).Расчет пропускания в некоторых случаях достаточно прост, но оценить
влияние неаксиально движущихся электронов, размера и структуры поверхно¬
сти образца, а также неоднородной засветки образца и аберрации электрон¬
ной оптики не всегда представляется возможным. В связи с различием в инди-
М.П. Сих227видуальных требованиях электронная оптика различных приборов может иметь
небольшие отличия. Поэтому необходимо иметь экспериментальные данные
по G(Ea). В 1978 г. комиссия Е-42 при ASTM в этих целях провела опрос
round го bin11 Анализ [ 52], проведенный на основе измерения относительных
интенсивностей, определявшихся по площади под пиком РФЭС-пиков мэди и зо¬
лота, обнаружил большой разброс результатов со стандартным отклонением
25% в энергетическом диапазоне 300 - 1100 эВ для одного прибора, гораздо
худший разброс для второго прибора и несколько лучшие результаты для треть¬
его прибора. Не установлено, является ли причиной разброса сам прибор или
оператор, однако все приборы систематически демонстрируют отклонения от
ожидаемой функции. Недавно проведенный в НФЛ анализ показал, что пред¬
сказанная и измеренная величиныРис. 5.17. Зависимость площади пиков от кинетической энергии электронов для раз¬
личной ширины щелей и энергий анализируемых электронов, обеспечива¬
ющих разрешение 0,5 эВ в случае установки VG ES САЗ [2в].1) Процедура анализа одних и тех же образцов проводится по стандартной мето¬
дике в разных лабораториях с целью выявления систематических ошибок анализа. -
Прим. ред.
2285. Количественная ЭОС и РФЭСРис. 5.1в. То же, что и на рис. 5.17, но для установки VG Scientific ESCALAB. (Вос¬
произведено по данным Хьюза и Филлипса [ 54].)совпадают в пределах 3% в диапазоне энергий от 500 до 1500 эВ для прибора
типаУС Scientific ESCA3 и PHI Model 550 ESCA/SAM. Существует мнение,
что эта простая зависимость сохраняется для приборов типа Varian IEE -
15 [41] и Ley bold LHS 10 [53] при использовании их в режиме постоянного
значения Д£. На рис. 5.17 приведен расчетный график функции пропускания
[28] для прибора типа VG Scientific ESCA3 для больших образцов и разре¬
шающей способности 0,5 эВ. При работе в режиме постоянного отношения
ДЕ/Е для всех перечисленных выше спектрометров С(ЕД) ~ЕД~Исключением из правила (5.21) являются спектрометр VG Scientific
ESCALAB, который имеет менее энергетически-зависимую характеристику
[28, 54], как это показано на рис. 5.18, и AEI ES 200/300. В настоящее вре¬
мя ведутся расчеты и эксперименты для создания базовой методологии опре¬
деления члена G(Ea)D(Ea) для этих и других спектрометров. Как и в случае
ЭОС, измерения / достаточно сложны и в следующем разделе эта пробле¬
ма будет рассмотрена совместно с банками справочных данных для /~.5.3.2. Измерение интенсивности и банки
справочных данныхКак уже обсуждалось в гл. 3, РФЭС-пики появляются на фоне небольшой
постоянной составляющей и потому исиользуются интегральные спектры, при
этом IА определяется просто площадью под соответствующим пиком. Для од-
М.П.Сих229них пиков эта площадь достаточно просто поддается измерению, для других же
встряска, стряхивание и мультиплетное расщепление сигнала могут привести
к появлению особенностей в широком диапазоне энергий, и, таким образом,
измерение площади пика потребует принятия решения о точном определении
вклада фона. В литературе использовались два метода определения фона.
Наиболее популярным и широко применяемым в большинстве коммерческих
систем обработки данных является метод, основанный на проведении прямой
линии между двумя соответствующим образом выбранными точками, например
Р и Q на рис. 5.19. Альтернативой этому методу является метод Ширли [25],
при котором амплитуда фоновой составляющей в точке определяется при по¬
мощи итеративного анализа как величина, пропорциональная амплитуде всей
площади, занимаемой пиком над фоновой составляющей с высокоэнергетич-
ной стороны, ^от метод основан на том предположении, что каждый элект¬
рон в характеристической части пика связан с плоской фоновой составляю¬
щей, отражающей потери. Метод Ширли дает фоновую кривую (рис. 5.19). Вновь
необходимо произвести выбор граничных точек, как на рис. 5.19, однако по¬
ложение точек не так жестко фиксировано. Например, точка Q может быть
перемещена в сторону меньших значений ЭС на 20 эВ, захватив сателлиты
^°34 (см* гл* ПРИ этом ПРИ использовании метода Ширли увеличится пло¬
щадь под пиками меди и СиО. Однако при использовании метода прямых ли¬
ний площадь под пиком уменьшится. Аналогично и в случае точки Р.Рис. 5.19. Пики фотоэлектронов металлической меди (а) и порошка СиО (б), ил¬
люстрирующие методы вычитания фона [28J.
2305. Количественная ЭОС и РФЭССитуация становится даже более неблагоприятной для фона, представ¬
ляющего собой прямую, когда рассматривается смесь двух химических со¬
стояний одного и того же элемента, как это показано на рис. 5.20. На
рис. 5.20,а изображен спектр для различных соотношений железа и Fe203,
а на рис. 5.20, б - спектр с двумя вычтенными фоновыми составляющими
для состава смеси 50/50. На кривой, соответствующей 20% окиси железа в
смеси, приведенной на рис. 5.20, а, положение точки Q установить легко, аРис. 5.20. Смоделированные спектры для смеси Fe и Fe203 при различных соот¬
ношениях компонент (а) и с вычитанием фона при составе смеси 50/50
Fe/Fe2o3 (б). (Воспроизведено по данным Бишопа [ 55].)
М.П. Сих231для Р существуют два варианта. При определении фоновой составляющей по
Ширли величиной Р^ удается получить наилучшее значение площади, тогда
как при использовании величины Р2 площадь несколько меньше. Однако Бишоп
[25], используя метод прямых линий, показал, что при положении Р значение
площади превосходит истинное на 33%, а для Рг площадь меньше истинной на
24%. Как показано на рис. 5.20, 6, Бишоп располагает точку Р вблизи пиков.
Следовало бы доказать, что некоторая часть фона со стороны меньших ки¬
нетических энергий обусловлена электронами, определяющими нарастание
и спад и, таким образом, точка Р должна быть смещена приблизительно на
50 эВ в сторону меньшей кинетической энергии [56]. Бишоп избавляется от
этого фона с помощью фоновой составляющей по Ширли следующим об¬
разом. Если сигнал в i-м канале s. является суммой фона и вкладов пиков
Ь. и р{ соответственно, то по методу Ширли имеем(5.22)где k - постоянная. Бишоп уменьшает величину вклада в связи с тем, что
потери в дальнейшем будут исключены из пика с помощью соотношения(5.23)где подгоночный параметр подбирается так, чтобы обеспечить правильный
наклон графика при малых кинетических энергиях. В настоящее время оста¬
ется нерешенным вопрос: следует ли подбирать значение подгоночного пара¬
метра по методу Бишопа, что исключит из рассмотрения некоторые явления,
связанные с заметными энергетическими потерями, которые хотелось бы
включить в рассмотрение, или, как предлагает Берешейм [56], для получения
оптимальных результатов в рассмотрение следует включить характеристи¬
ческие потери, связанные с рассеянием электронов в кристалле-Дальнейшие уточнения возникают при использовании соотношения Ширли.
В своем основном виде оно подразумевает, что спектр характеристических
потерь для каждого электрона в области малых энергий представляет собой
горизонтальную линию. Это кажется вполне правомерным предположением
для металлов, поскольку в этом случае могут иметь место побочные явле¬
ния, связанные с малыми потерями энергии при возбуждении электронов с
уровня, лежащего чуть ниже уровня Ферми, на уровень, лежащий чуть выше
уровня Ферми. Однако в случае диэлектриков такие переходы невозможны, в
этом случае должен четко наблюдаться электрический интервал после пика
до начала роста фоновой составляющей с низкоэнергетичной стороны пика,
2325 Количественная ЭОС и РФЭСпропорциональный ширине запрещенной зоны. Беглый взгляд на данные, пред¬
ставленные в справочнике по рентгеновской фотоэлектронной спектроскопии
[42], где приведены спектры в широком диапазоне энергий для LiF, BN, по¬
лиэтилена А1203, КВг, CaC03, Sc03, SrF2, Y203, Lil, CsOH, BaO и т. д.,
показывает, что во всех случаях нарастание фоновой составляющей происхо¬
дит примерно на расстоянии 10 эВ с низкоэнергетичной стороны от максималь¬
ного пика в каждой группе, как и следовало ожидать. На рис. 5.21, а изобра¬
жены пики SrF2, для которых наблюдается этот эффект. Для пиков F Is и
Sr 3d ясно различим промежуток, предшествующий нарастанию фоновой со¬
ставляющей, однако пик Sr 3/2 маскирует этот эффект для пика Sr 3Ръ/Г
В этих случаях можно считать фоновую составляющую линейной при отсутст¬
вии дополнительных пиков, заполняющих промежуток, в случае, когда фоно¬
вая составляющая может быть описана какгде п - количество каналов, используемых для описания промежутка.На рис. 5.21, б приведены три различных типа пиков, полученных на од¬
ном и том же спектрометре, соответствующие разным типам фоновой состав¬
ляющей по Ширли. Из сказанного можно заключить, что в настоящее время не
существует единого рекомендованного метода для определения 1Л. Однако(5.24)Рис. 5.21. РФЭС-спектры для S г Ь 2 (а), Си, К«* и У (б). (Воспроизведено по дан¬
ным Вагнера и др. [421.) Видны классическая ступенька Ширли, моди¬
фикация Бишопа и смещенная ступенька соответственно.
М.П. Сих233практически ситуация не так уж плоха, поскольку многие соединения являют¬
ся диэлектриками и модель линейной фоновой составляющей служит достаточ¬
но хорошим приближением. В случае металлических соединений имеют место неболь¬
шие изменения формы пика, так что любое измерение площади пика будет да¬
вать разумные результаты для металлических систем. Результат измерения
должен, конечно, лежать между / и / Однако любой используемый метод
может не всегда давать соответствующие значения 1А, лежащие между зна¬
чениями интенсивности для металлов и диэлектриков.Рассмотрим теперь банки справочных данных, используемые для опреде¬
ления /~. Имеется обширная литература с наборами справочных данных
[29-42], и читателю трудно сделать выбор, какой именно набор данных сле¬
дует применять. При получении этих данных использовались в основном три
различных подхода: измерение стандартных образцов (СЮ) известных соединений в
виде порошков [ 29—32, 36-39, 41 ], измерение СО элементов в виде тонких фольг [34,
36, 40] и расчеты [42]. Расчеты выполнялись с использованием выражения
(5.14), которое для спектрометра с равномерней засветкой гомогенной ми¬
шени и коэффициентом пропускания анализатора, описываемым (5.21), может
быть записано в виде(5.25)где от коэффициента пропорциональности можно избавиться, относя значения
1А к одному конкретному пику, например F Is, и положив величину 1А равной
единице как постоянную. Вагнер и др. [42] в своих расчетах использовали
вычисленные Скофилдом [27] значения cr^(/*v) и результаты Пенна [13] для
значений ЛМ(ЕА). Такой же подход к количественной оценке без использова¬
ния измеренных значений для контрольных образцов был также использо¬
ван Эбелем [57, 58] и Хирокавой и Оку [59, 60], которые сообщают, что полу¬
чена хорошая корреляция между результатами измерений состава сплавов и
известными данными по составу массивных образцов.Описанный подход достаточно просто реализуется, поскольку значения
могут быть легко вычислены для всех уровней всех атомов с использова¬
нием приведенных в литературе значений о, Л и т. д. Однако точность полу¬
чаемых таким образом результатов не превосходит точности использованных
данных, в то время как надежность измерения площади пика в процессе ее оп¬
ределения отражает вклад всех электронов в значение стл(/гу). В общем случае
ири измерениях площади пика в небольшом диапазоне энергии обычно не учи¬
тывают те части спектра, которые должны быть учтены» Они рассматрива¬
ются в гл- 3 как процессы встряски, которые происходят тогда, когда
2345. Количественная ЭОС и РФЭСэлектрон, находящийся в зоне проводимости или в валентной зоне, получает
порцию энергии одновременно с появлением первичного фотоэлектрона и, та¬
ким образом, последний получает пониженную энергию. Интенсивность, поте¬
рянная при таком явлении, зависит от элемента и остовного уровня, вовле¬
ченного в этот процесс, и может, кроме того, зависеть от химического соста¬
ва окружения, особенно для переходных металлов [36]. В переходных З^-эле-
ментах этот эффект сильнее проявляется для линий 2р, чем 3р, и связывает¬
ся с наличием незаполненного d-уровня, поскольку для меди и цинка эффект
наименее выражен. Вычисления, произведенные для разреженных газов [61],
показывают что вероятность встряски падает приблизительно от 18% при
энергии связи 2 кэВ до 16% при 1 кэВ, 14% при 400 эВ, 12% при 200 эВ и рез¬
ко до 5% при 25 эВ. Для металлов вероятность встряски может до¬
стигать 15-30% [62]. Таким образом, значения /~, полученные в результа¬
те вычислений, не могут быть очень точными. Степень их точности можно
оценить путем сравнения расчетных значений с результатами измерений /~.В связи с этим рассмотрим теперь экспериментально полученные справочные
данные.Были проведены подробные исследования этих данных [28], в результате
чего выяснено, что лишь немногие из них сопоставимы. Серии эксперимен¬
тальных данных сравнивались посредством рассмотрения отношения их вели¬
чин к соответствующим теоретическим величинам. Все серии данных, за ис¬
ключением полученных Вагнером [29, 36, 41], Нефедовым [31, 32] и Эвансом
[38, 39], обнаружили при этом значительное расхождение. Однако эти три се¬
рии данных совпадают с разбросом всего лишь 8% для 33 измеренных пиков,
как показано на рис. 5.22. Следует отметить, что экспериментальные данные
получены для порошковых образцов, приготовленных непосредственно перед
измерениями с помощью РФЭС, при этом все образцы имели примесь углеро¬
да. Вследствие этого измеренные значения занижены на величину, учитывае¬
мую коэффициентом exp [- t/hc(EA)cos 0], причем типичная толщина плен¬
ки углерода составляла 0,75 нм. В данные, приведенные на рис. 5.22, были
внесены поправки в соответствии со средней величиной углеродного загряз¬
нения.Систематическое отклонение серий экспериментальных данных от теоре¬
тически предсказанных величин показывает, что обычный метод измерения
площади пиков не даст такую точную количественную оценку при использова¬
нии вычисленных значений какая может быть получена при использовании
одной из трех экспериментальных серий, приведенных на рис. 5.22. Самая
большая серия данных, полученная Вагнером, приведена в приложении 5. Под¬
робный анализ относительных амплитуд пиков для многих распространенных
соединений, исследованных Вагнером [36, 63], показывает, что воспроизводи-
М.П. Сих235Рис. 5.22. Соотношение экспериментально измеренных факторов относительной чув¬
ствительности (ФОЧ) полученных Вагнером (черные значки), Нефедовым
(белые значки) и Эвансом (крестики) расчетной поправкой на наличие слоя
загрязнения.мость количественной оценки имеет величину порядка 10% и что для большин¬
ства пиков наблюдаются значительные отклонения от теоретических значений,
как это показано на рис. 5.23, взятом из работы Вагнера и др„ [41]. Эти от¬
клонения не обязательно связаны с ошибками в сечении Скофилда, просто эти
сечения не отражают результатов измерений. Исследования, проведенные не¬
давно Берешеймом [58], показали, что лучшую корреляцию можно получить при
ограничении пика или пары пиков с низкоэнергетичной стороны точкой, соот¬
ветствующей минимальной энергии фоновой составляющей, вычисляемой по
модели Ширли, даже если эта точка отстоит от пика на 100 эВ. Подход Бере-
шейма может быть не практичен в общем случае, но дает хорошее совпадение
с теоретическими данными.До сих пор мы обсуждали проблемы измерения интенсивностей сигналов
РФЭС и ЭОС и количественного определения на их основе состава однород¬
ных бинарных твердых тел. На практике оба метода применяются э основном
для двух целей: определения содержания поверхностно адсорбированных при-
2365. Количественная ЭОС и РФЭСРис. 5.23. Различия (в процентах) между эмпирическими и теоретическими фактора¬
ми относительной чувствительности, нормированными по F 1s. (Воспро¬
изведено по данным Вагнера и др. [ 41].)месей и проведения послойного анализа in situ при одновременном травле¬
нии ионами аргона. Мы можем теперь рассмотреть эти проблемы в связи с
вышеизложенным в случае однородных материалов.5.4. Количественный анализ состава образцов с тонкослойными
покрытиямиТонкослойные покрытия могут являться адсорбатами, которые
требуется исследовать, или пленкой углеродного загрязнения, влияние кото¬
рой необходимо учитывать при проведении количественного анализа состава
поверхности. Как правило, в исследованиях с применением ЭОС количествен¬
но анализируется состав одного или большего числа адсорбатов на чистой од¬
нокомпонентной поверхности [64, 65]. Аналогичные исследования производят¬
ся и методом РФЭС, однако анализ поверхности, загрязненной слоем углеро¬
да или углеводородов, является при использовании тех же концепций гораздо
более сложной проблемой.В соответствии с идеей, изложенной в п0 5.2.1, сигнал подложки В, не
полностью покрытой монослоем элемента А со степенью покрытия уд, описы¬
вается простой суммой неослабленной эмиссии с (1 — фл) доли поверхности
подложки и ослабленной эмиссии с <рл доли поверхности подложки:
М.П. Сих237а в случав, если слой вещества А имеет толщину d ,(5.26)(5.27)Здесь предполагается, что величина 1А измерена корректно и что /J0 изме¬
рена на том же приборе при той же установке образца. В случае если эти ус¬
ловия не соблюдены, могут быть внесены поправки, описанные выше. Сигнал
от слоя, покрывающего поверхность подложки, в каждом из этих случаев мо¬
жет быть записан соответственно в видегде члены гА и rRt определяющие обратное рассеяние, конечно, равны нулю
для интенсивностей сигналов, измеренных методом РФЭС.Хотя выше мы рассматривали I™ как табулированный фактор относитель¬
ной чувствительности, необходимо иметь в виду, что в связи с шероховато¬
стью поверхности образца и другими переменными, меняющимися при прове¬
дении измерений, на практике можно использовать только отношения ГУ/!? идот. д. В связи с этим, используя выражения (5.26) и (5.28), степень покрытия
можно описать выражением(5.30)Если пики оже-электронов или фотоэлектронов наблюдаются при высоких энер¬
гиях и если величина 9 мала, можно получить приведенное ниже простое со¬
отношение, аналогичное выражению (5.10):(5.28)(5.29)(5.31)где (И - матричные факторы монослоя в случае использования ЭОС и 0х
ли А \в случае использования метода РФЭС задаются в виделв(5.32)
2385. Количественная ЭОС и РФЭС(5.33)и где средняя длина свободного пробега может быть определена при помощивыражения (5.9) [П] или из расчетов Пенна [13], а члены, описывающие обрат¬
ное рассеяние, - с помощью рис. 5.4 или по данным Симицу и Итимуры [6].В качестве примера можно провести оценку фактора QAB для металличе¬
ской системы, представляющей собой слой олова, адсорбированного на желе¬
зе, воспользовавшись данными, приведенными на рис. 5.4, и выражением (5.9),
а также табулированными значениями а:где было принято типичное значение для cos 0 = 0,74 [66]. Полученное значе¬
ние с хорошей точностью совпадает с экспериментально измеренной величи-
ной Q§nFe = 3,82, которая может быть извлечена из данных Сиха [67] с по¬
мощью справочника по оже-электронной спектроскопии [1]0 Автором [65] бы¬
ли составлены таблицы других экспериментальных значений QA для границ зе¬
рен, где все величины увеличены вдвое, поскольку на анализируемой поверх¬
ности излома, проходящего по границам зерен, остается лишь половина веще¬
ства, образующего соответствующие сегрегации.Интенсивность сигнала слоя покрытия d может быть аналогичным обра¬
зом просто вычислена с помощью выражений (5.27) и (5.29), но в более общем
случае она зависит и от сигналов двух типов, обусловленных наличием под¬
ложки1* . Для РФЭС значения /J°, приведенные в приложении 5, соответствуют
образцам со "стандартным" уровнем загрязнения, и в случае исследования
чистых образцов в них должны быть внесены поправки с помощью выражения
(5.27), для чего значение /fi следует заимствовать из приложения 5, чтобы
найти /д - искомый фактор чувствительности. Для метода ЭОС факторы чув¬
ствительности, приведенные в [1], соответствуют чистым образцам.1* Речь идет о том, что в случае тонкослойного покрытия будут регистрироваться
ослабленный (там, где оже- или фотоэлектроны проходят сквозь слой покрытия) и
неослабленный (там, где покрытия нет) сигналы, обусловленные возбуждением оже-
или рентгеновской фотоэлектронной эмиссии в подложке. — Прим. ред.
М.П. Сих2395.5. Количественная оценка результатов послойного анализаПослойный анализ подробно обсуждался в гл. 4, и здесь он будет лишь
кратко рассмотрен. Как показано далее, ионное распыление изменяет состав
образца вследствие двух процессов: атомного перемешивания на глубинах
порядка 1-20 нм, зависящих от рода образца и условий распыления, и селек¬
тивного распыления атомов в одном или двух верхних слоях.Коэффициент распыления для атомов данного рода зависит от энер¬
гии, отдаваемой бомбардирующим ионом, энергии, передаваемой от
атома к атому (каскадные процессы), и энергии связи атомов в поверх¬
ностных слоях [68]. Таким образом, коэффициенты распыления различ¬
ных элементов в чистом виде различаются более чем на порядок,
как это следует из рис. 5.24. Если элементы на модифицированной поверхно¬
сти матрицы А В имеют коэффициенты распыления SA и то в процессе рас¬
пыления гомогенного твердого тела А В состав зоны, модифицируемой приРис. 5.24. Сравнение коэффициентов распыления, вычисленных [бв] для чистыхэлементов (1), и скорректированных экспериментальных данных [69] (2)
[70].
2405. Количественная ЭОС и РФЭСраспылении на глубину zs , изменяется со временем следующим образом [71,
72]:где Хл (0) - это просто ХА, а ХА («>) соответствует конечному составу этой
зоны и *о - время распыления до глубины В Итоге измеренный
состав X™ (оо) для малых ХА связан с коэффициентом распыления соотноше¬
ниемгде <р есть exp (-Zs/\ cos 6) и предполагается, что А и В обладают пиками,
лежащими при близких энергиях, а Л - средняя длина свободного пробега
для неупругого рассеяния для каждого элемента- С использованием
приведенного выше выражения можно определить фактор, характеризующий
изменение матрицы при ионном распылении:и использовать F^B вместо FAB в выражениях (5J0) и (5.16) для проведения
количественного послойного анализа. Однако при этом, к сожалению, невоз¬
можно непосредственно вычислить FA , поскольку SA и SB нельзя отожде¬
ствить с коэффициентами распыления чистых элементов. Это легко понять,
рассматривая простую модель соединения, состоящего из атомов А и В, на
примере структуры галогенидов щелочных металлов, представляющей собой
взаимопроникающие кубические гранецентрированные решетки. В чистом ви¬
де энергии связи еАА и евв атомов Л и В могут изменяться в значитель¬
ных пределах и коэффициенты распыления чистых элементов могут меняться
на порядок. Однако, если А а В имеют одинаковую массу, и поскольку для
обоих атомов в кубической гранецентрированной решетке энергия связи равна
еАВ, они могут иметь одинаковые коэффициенты распыления. Практически эта
проблема гораздо сложнее, поскольку в общем случае суммарный коэффици¬
ент распыления складывается из членов, характеризующих столкновения, ло¬
кальный разогрев и термическое испарение, что ясно показано в эксперимен¬
тальных исследованиях Шимонски, Оверайндера и де Врие [73]. Относитель¬
ное значение этих членов и величины вклада каждого из них зависят от матри¬
цы Проблема изменения состава в процессе распыления до сих пор не решена.
Заинтересовавшиеся этим вопросом читатели могут ознакомиться с обзорами(5.34)(5.35)(5.36)
М.П. Сих241Келли [74] и Кобурна [75], а также другими работами [76-80]. Решением
этой проблемы, наиболее применимым к нуждам практики, в настоящее время
является сравнение профиля концентраций исследуемого образца с профилем
концентраций стандартного образца. Полезно также использование высоко-
энергетичных пиков, поскольку это уменьшает различие между FAR и в
выражении (5.36).Литература1. Davis L.E. et al,, Handbook of Auger Electron Spectroscopy, 2nd ed.,
Physical Electronics Division, Perkin-Elmer Corp., Minnesota, 1976.2. Bishop H.E., Riviere J.C., J. Appl. Phys., 40, 1740 (1969).3. Worthington C.R., Tomlin S.G., Proc. Phys. Soc., A69, 401 (1956).4. Goto К. et al., Surf. Sci., 47, 477 (1975).5. Smith DM, Gallon T.E., J. Phys., D7, -151 (1974).6. Shimizu RIchimura S., Quantitative Analysis by Auger Electron Spectro¬
scopy, Toyata Foundation Research Report 1-006 No. 76-0175, Osaka, 1981.7. Ichimura S., Shimizu R., Surf. Sci., 112, 386 (1981).8. Gryzinski М., Phys. Rev., A138, 305 (1965).9. Jablonski A*t Surf. Interface Anal., 2, 39 (1980).10. Reuter W., in: Proc. Sixth. Int. Conf. on X-ray Optics and Microanalysis (eds.
G. Shinoda K. Kohra, T. Ichinokawa), Univ. of Tokyo Press; 1972, p. 121.11. Seah M.P., Dench W.A., Surf. Interface Anal., 1, 2 (1979).12. Hall P.M., Morabito J.M., Surf. Sci., 83, 391 (1979).13. Penn D.R., J. Electron Spectrosc., 9, 29 (1976).14. Holloway P.H., Holloway DM., Surf. Sci., 66, 635 (1977).15. Sickafus E.N., Holloway DM*, Surf. Sci., 51, 131 (1975).16. McGuire G.E., Auger Electron Spectroscopy Reference Manual, Plenum,New York, 1979.17. Anthony M.T., Seah M.P*, J. Electron Spectrosc. (в печати),18. Madden H.H., J. Vac. Sci. Technol., 18, 677 (1981).19. Netzer F.P., Appl. Surf. Sci., 7, 289 (1981).20. Pantano C.G., Madey T.E., Appl. Surf. Sci., 71, 115 (1981).21. Hesse R., Littmark U., Staib P., Appl. Phys., 11, 233 (1976).22. Seah M.P*, Surf. Interface Anal., 1, 86 (1979).23. Sickafus E.N., Phys. Rev., В16, 1436 (1977).24. Sickafus E.N., Phys. Rev., В16, 1448 (1977).25. Shirley D.A., Phys. Rev., В 5, 4709 (1972).26. Strausser Y.E., Franklin D., Courtney P., Thin Solid Films, 84, 145 (1981).27. Scofield ].Ц., J. Electron Spectrosc., 8, 129 (1976).
2425. Количественная ЭОС и РФЭС28. Seah М.Р., Surf. Interface Anal., 2, 222 (1980).29. Wagner C.D., Anal. Chem., 44, 1050 (1972).30. Jorgensen C.H., Berthou H., Trans. Faraday Disc., 54, 269 (1972).31. Nefedov V.I. et al., J. Electron Spectrosc., 2, 383 (1973).32. Nefedov V.I. at el., J. Electron Spectrosc., 7, 175 (1975).33. Berthou H., Jorgensen C.K., Anal. Chem., 47, 482 (1975).34. Janghorbani M., Vulli М., Starke H., Anal. Chem., 47, 2200 (1975).35. Brillson L.J.S Ceasar G.P., Surf. Sci., 58, 457 (1976).36. Wagner C.D., Anal. Chem., 49, 1282 (1977).37. Adams J.M. et al., Anal. Chem., 49, 2001 (1977).38. Evans S., Pritchard R.G., Thomas J.M., J. Phys., C10, 2483 (1977).39. Evans S., Pritchard R.G., Thomas J.M., J. Electron Spectrosc., 14, 341(1978).40. Vulli М., Starke K., J. de Micros. Spectros. Electron., 2, 57 (1977).41. Wagner C.D. et al, Surf. Interface Anal., 3, 211 (1981).42. Wagner C.D. et al., Handbook of X-ray Photoelectron Spectroscopy, Per¬
kin-Elmer Corp., Minnesota, 1979.43. Heddle D.W.O., J. Phys., E4, 589 (1971).44. Reilman R.F., Msezane A., Manson S.T., J. Electron Spectrosc., 8, 389
(1976).45. Fadley C.S., J. Electron. Spectrosc., 5, 725 (1974).46. Fadley C.S. et al, Surf. Sci., 89, 52 (1979).47. Evans S., Raftery E., Thomas J.M., Surf. Sci., 89, 64 (1979).48. Krause M.O., Phys. Rev., 177, 151 (1969).49. Vulli М., Surf. Interface AnaL, 3, 67 (1981).50. Baschenko O.A., Nefedov V.L, J. Electron Spectrosc., 17, 405 (1979).51. Gettings М., Coad J.P., Surf. Sci., 53, 636 (1975).52. Powell C.J., Erikson N.E., Madey T.E., J. Electron. Spectrosc., 17, 861(1979).53. N oiler H.G., Polaschegg H.D., Schillalies H., J. Electron. Spectrosc., 5,
705 (1974).54. Hughes A.E., Phillips C.C., Surf. Interface Anal., 4, 220 (1982).55. Bishop H.E., Surf. Interface Anal., 3, 272 (1981).56. Berresheim K., Surf. Interface Anal, (в печати).57. Ebel M.F., Surf. Interface Anal., 1, 58 (1979).58. Ebel M.F., Surf. Interface Anal., 2, 173 (1980).59. Hirokawa K., Oku М., Z. Anal. Chem., 285, 192 (1977).60. Hirokawa K., Oku М., Honda F., Z. Anal. Chemie, 28S, 41 (1977).61. Carlson T.A., Photoelectron and Auger Spectroscopy, Plenum, New York,
1978.
М.П. Сих24362. Powell С./., in: Quantitative Surface Analysis of Materials (ed. N.S. McIn¬
tyre), Philadelphia, ASTM STP 643, American Society, for Testing and
Materials, 1978, p. 5.63. Wagner C.D; in: Quantitative Surface Analysis of Materials (ed. N.S. McIn¬
tyre), Philadelphia, ASTM STP 643, American Society for Testing and
Materials, 1978, p. 31.64. Seah M.P., J. Catal., 57, 450 (1979).65. Seah M.P*, J. Vac. Sci. Technol., 17, 16 (1980).66. Seah M.P., Surf. Sci., 32, 703 (1972).67. Seah M.P., Surf. Sci., 40, 595 (1973).68. Sigmund P., Phys. Rev., 184, 383 (1969); Phys. Rev., 187, 768 (1969).69. Laegreid N., Wehner G*K», J. Appl. Phys., 32, 365 (1961).70. Seah M.P., Thin Solid Films, 81, 279 (1981).71. Shimizu H., Ono М., KakayamaK., Surf. Sci., 36, 817 (1973).72. Ho P.S., Lewis J.E., Howard J.K., J. Vac. Sci. Tehnol., 14, 322 (1977).73. Szymonski М., Overeijnder H., De Vries A.E., Rad. Effects, 36, 189 (1978).74. Kelly R., Surf. Sci., 100, 85 (1980).75. Cobum J.W., Thin Solid Films, 64, 371 (1979).76. Kelly R., Lam N*Q., Rad. Effects, 19, 39 (1973).77. Liau Z»L»> Tsaur B.Y., Mayer J.W., J. Vac. Sci. Technol., 16, 121 (1979).78. Mathieu HLandolt D., Appl. Surf. Sci., 3, 348 (1979).79. Chou N.J., Shafer M.W., Surf. Sci., 92, 601 (1980).80. Narusawa Т., Satake Т., Komiya S., J. Vac. Sci. Technol., 13, 514 (1976).
Глава 6
ПРИМЕНЕНИЕ ЭОС В МИКРОЭЛЕКТРОНИКЕP.P. Олсон, П.В. Палмберг,
С. Т. Ховланд, Т.Е. Бреди1>6.1. ВведениеРазвитие полупроводниковой цифровой и аналоговой микроэлектроники
идет по пути увеличения плотности схем. Это определяется требованиями
более высокого уровня интеграции систем, увеличения быстродействия,
снижения рассеиваемой мощности и повышения выхода годных схем. Миниа¬
тюризация достигается изменением архитектуры схем, изменением элемен¬
тов и их структуры, использованием новых материалов и уменьшением про¬
дольных и вертикальных размеров структур» Одновременно с миниатюриза¬
цией в сверхбольших интегральных схемах (СБИС) происходит разработка
новых приборов и структур на основе кремния и полупроводниковых соеди¬
нений для производства датчиков и всех типов электрооптических приборов.По мере того как все аспекты технологии, связанные с ростом плотнос¬
ти схем, приближаются к своим физическим пределам, особенно возраста¬
ет роль аналитического оборудования., Поскольку размеры рассматривае¬
мых структур очень малы, необходим аналитический метод с очень высоким
пространственным разрешением и разрешением по глубине. Таким методом
является сканирующая оже-спектроскопия0 Так как средняя длина свобод¬
ного пробега оже-электронов составляет всего 5-20 А, оже-сигнал форми¬
руется лишь несколькими первыми атомными слоями поверхности образца.
Современный промышленный оже-микроанализатор способен сфокусировать
первичный электронный пучок в пятно диаметром менее 50 нм. Такой прлбор
обладает превосходными возможностями для сканирующей электронной мик¬
роскопии (СЭМ) и обеспечивает элементный анализ состава образцов, поз¬
воляя ясно отобразить и идентифицировать субмикронные особенности
структуры, которые могут быть соотнесены с топографией поверхности, по¬
лученной СЭМ.Электронная оже-спектроскопия (ЭОС) расширяет аналитические воз¬
можности СЭМ совершенно иным способом, чем дисперсионный рентгеновс-^ R.R. Olson, P.W. Palmberg, С>Т. Hovland, Т-Е. Brady, Perkin-Elmer, Physical
Electronics Division, 6509 Flying Cloud Drive, Eden Prarie, Minnesota 55344, USA.
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди245кий анализ (ДРА). ЭОС позволяет зондировать приповерхностный слой тол¬
щиной на несколько порядков меньше 1 мкм - характерной толщины слоя,
анализируемого ДРА (см. гл. 1). Тонкие слои загрязнений, состав припо¬
верхностного слоя, структура тонких пленок и состав тонких переходных
слоев оказывают все более существенное влияние на работу приборов. По
мере уменьшения поперечных и вертикальных размеров элементов схем
отношение поверхности к объему возрастает и роль поверхностей и границ
раздела становится все более важной. Такие параметры, как поверхностные
токи утечки и сопротивления контактов, которые играют второстепенную
роль в структурах большего размера, могут определять рабочие характерис¬
тики современного прибора. Для определения влияния химического состава
поверхностей границ раздела на рабочие характеристики прибора необходим
аналитический метод, чувствительный к характеристикам поверхности, та¬
кой, как ЭОС. Другим отличием ЭОС от ДРА является чувствительность к
элементам с малыми атомными номерами. Все элементы, следующие за ге¬
лием, дают характерные, легко идентифицируемые оже-спектры. Чувстви¬
тельность оже-спектроскопии практически одинакова для всех элементов
периодической системы.Пространственное разрешение ЭОС также намного лучше, чем у ДРА.
Пространственное разрешение ДРА вследствие рассеяния электронов в
твердом теле составляет величину порядка микрона. Пространственное раз¬
решение сканирующей оже-микроскопии в основном определяется диаметром
первичного электронного пучка. При химическим анализе стандартным об¬
разом достигается величина менее 100 нм.Целью данной главы является обсуждение областей технологии произ¬
водства интегральных схем, в которых используется ЭОС, с изложением
нескольких иллюстративных примеров. В конце главы приводится библио¬
графия для читателей, интересующихся другими применениями ЭОС в микро¬
электронике. Среди прекрасных обзоров на эту тему отметим информатив¬
ную статью Томаса [1] и работу Холлоуэя [ 2] с обширной библиографией.6.2. Технология изготовления полупроводниковых ИС6.2.1. Оценка качества исходного материалаВ технологии интегральных микросхем на основе кремния сканирующая
оже-микроскоиия (СОМ) может использоваться как эффективный метод конт¬
роля практически каждой стадии изготовления. Требования к характеристи¬
кам исходного материала - кремниевых пластин — становятся особенно ю
строгими для получения приемлемого выхода годных схем при изготовлении
структур с высокой плотностью элементов на подложках большой площади.
Поверхность пластины не должна содержать загрязнений, таких, как остат¬
ки от полировки, остатки от травления, включения, частицы и пленки,которые
2466. Применение ЭОС в микроэлектроникемогут служить зародышами нарушений при последующей эпитаксии и образо¬
вать дефекты при последующем окислении или на других стадиях, включая
циклы термообработки.Поверхностные примеси могут концентрироваться при окислении в кри¬
тичном переходном слое окисел- полупроводник. Поверхностные загрязне¬
ния могут также неблагоприятно влиять ка адгезию осаждаемых слоев.ЭОС может быть использована для оценки эффективности различных мето¬
дов очистки и идентификации соединений, остающихся на поверхности плас¬
тины после травления, обработки растворителями и деионизованной водой.
Пластины, которые при оптическом контроле обнаруживают помутнения
на поверхности, могут быть проанализированы для выяснения того, что яв¬
ляется причиной: топография поверхности или загрязнения.Метод СОМ очень удобен для идентификации частиц и включений на по¬
верхности пластины. Направляя сфокусированный электронный пучок на
частицу, по ее оже-спектру можно определить химический состав. При сов¬
местном применении СОМ и ионно-лучевого травления можно проводить
анализ тонких пленок (АТП) с очень высоким разрешением по глубине
(порядка нескольких нанометров). Тем самым можно измерять толщину
очень тонких окисных слоев и слоев загрязнений.Для более чувствительного анализа примесей, равномерно распределен¬
ных по поверхности образца, используется дополнительный аналитический
метод - статическая ВИМС (масс-спектрометрия вторичных ионов).
ВИМС-приставки имеются у большинства сканирующих оже-микроскопов.
ВИМС обычно не дает столь детальной количественной информации, как
ЭОС, однако при большой площади анализируемой поверхности обеспечива¬
ет значительно более высокую чувствительность детектирования большинст¬
ва элементов. Используя метод ЭОС в сочетании с более чувствительным
методом ВИМС, можно расширить диапазон регистрируемых концентрацийдо 10-4%.Метод анализа тонких пленок (АТП) можно применять при исследовании
пленочных структур толщиной до нескольких микрон, используя ионный пу¬
чок для травления распылением со скоростью несколько десятков наномет¬
ров в минуту. Если толщина пленки в структуре больше нескольких микрон,
то лучше использовать методы механического секционирования, поскольку
скорость травления распылением в поликристаллических материалах изме¬
няется в зависимости от ориентации кристалла, что вызывает увеличение
шероховатости поверхности. Глубина разрешения, которую можно получить
после травления толстослойной структуры, ограничивается и другими эффектами, та¬
кими, как рост нитевидных кристаллов и образование конических структур
(см. гл. 4).Профили состава по глубине, полученные с помощью метода АТП, очень
полезны. Часто по ним можно определить, что проблемы адгезии, травления
и контакта, связанные, казалось бы, с объемным составом тонких пленок,
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди247в действительности обусловлены составом приповерхностного или промежу¬
точного слоя.С помощью метода ООМ можно исследовать и другие твердые сырьевые
материалы, применяемые в микроэлектронике: мишени, используемые при на¬
несении распылением, и испаряемые материалы. В поликристаллических ме¬
таллах примеси, которые содержатся в материале, обычно находятся на гра¬
ницах зерен. Разрушая образец в сверхвысоковакуумной контрольной каме¬
ре, можно образовать разлом по границам зерен и исследовать свежие по¬
верхности зерен с помощью метода СОМ. Часто на таких поверхностях обна¬
руживаются большие концентрации примесей, которые при объемном анализе
лежат в пределах 10~4 - 10~7%. Примером исследования разлома in situ по-
ликристаллического кремния и корреляции данных СОМ с измерениями то¬
ка, индуцированного электронным пучком, является публикация Казмерского
и др. [3].6.2.2. ОкислениеЭтапы процесса окисления могут также контролироваться с помощью ме¬
тодов ЭОС и АТП. Метод АТП можно использовать для измерения толщины
оксида и таким образом определять различия между скоростями роста ок¬
сида, обусловленные ориентацией или эффектами легирования. Можно также
изучать сегрегацию примесей на поверхности оксида или на границе раздела
между оксидом и полупроводником. Кроме того, многие исследователи ис¬
пользовали метод ЭОС для изучения природы границы раздела Si/Si 02. По
этим вопросам опубликовано множество работ [4-9].Положение и форма оже-линий кремния зависят от того, связан ли дан¬
ный атом с другими атомами кремния, с атомами азота или кислорода
(см. гл. 3 и 5). Этот эффект использовали исследователи, изучающие область
границы раздела. Спектры, однако, не дают прямой информации о состояни¬
ях границы раздела или поверхности. Влияние химического состояния на
о же-спектры использовалось также для исследования силицидов. Многие
другие элементы, такие, как алюминий, магний, фосфор, бор, сера, углерод
и т. д., также проявляют изменения в форме и положении оже-линий.В настоящее время в МОП-структурах делают все более тонкий оксид¬
ный изолирующий слой с повышенными требованиями к напряжениям пробоя,
однородности и целостности оксида. С помощью СОМ можно исследовать ло¬
кализованные дефекты оксида в очень малой области изолирующего слоя
или в пленках нитридов.Метод ЭОС можно также использовать для контроля за окислением
других материалов на поликристаллической кремниевой пластине, нитриде
кремния, сплавных металлических соединениях и силицидах. Этот контроль
повышает выход годных схем в процессе производства и ускоряет анализ при¬
чин отказов.
2486. Применение ЭОС в микроэлектронике6.2.3. Фотолитография, нанесение рисунка схемы и
удаление фоторезистаМетод ЭОС может оказаться очень полезным в процессе фотолитогра¬
фии, даже несмотря на то, что толстые слои органического фоторезиста -
не совсем подходящие объекты для этого метода; эффекты повреждения
электронным пучком и зарядка поверхности создают проблемы при анализе.
Для характеристики таких поверхностей лучше использовать метод РФЭС.
ЭОС, возможно, больше подойдет для анализа неорганических фоторезис¬
тов. С помощью ЭОС можно изучать адгезию фоторезистов до их использо¬
вания и контролировать законченность операции удаления фоторезиста пос¬
ле того, как нанесен рисунок ИС. Остатки на поверхности после операции
удаления, такой, например, как обработка в кислородной плазме, могут
быть легко обнаружены.Томас [ 1] описывает очень интересный опыт, в котором фоторезист
использовался как маска при имплантации большой дозы ионов мышьяка.
После имплантации пластина была обработана кислородной плазмой; одна¬
ко полное устранение резиста оказалось невозможным. Анализ с помощью
СОМ остаточного фоторезиста показал, что в нем содержится большая кон¬
центрация мышьяка. Это говорит о необходимости изменить процесс таким
образом, чтобы добиться полного устранения при травлении.6.2.4. Химическое или плазменное травлениеПри химическом или плазменном травлении можно использовать СОМ
для определения скоростей и однородности травления, а также для контроля за
воздействием дефектов и загрязнений на качество травления. Часто нали¬
чие промежуточного слоя другого состава изменяет характеристики трав¬
ления. Неполное травление может привести к высоким сопротивлениям
контактов или к полному отсутствию контакта с нужной областью. СОМ мож¬
но использовать для идентификации таких стойких к травлению остатков,
которые могут вызвать последующую коррозию и изменение адгезионных
свойств. Возможно появление нежелательных эффектов в результате изме¬
нения химических свойств поверхностей после травления.СОМ высокого разрешения позволяет контролировать перераспределе¬
ние материалов на боковых стенках ступенек и изменения топографии, обус¬
ловленные плазменным или ионно-лучевым травлением. Можно исследовать
однородность легирования (при высокой степени легирования) и окислитель¬
ные свойства поликристаллического кремния при использовании его в зат¬
ворах и межсоединениях.6.2.5. Легирование диффузией или ионной имплантациейМинимальная концентрация примесей, которую можно обнаружить с по¬
мощью ЭОС, составляет около одной тысячной монослоя. Это соответствует
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди249степени легирования 1019 атомов на кубический сантиметр» Предел обна¬
ружения зависит от элемента, его детектируемого оже-перехода, матрицы,
энергии и тока первичного электронного пучка, времени регистрации и про¬
пускания и разрешения спектрометра. Отношение сигнала к шуму в запи¬
санном спектре определяется выражениемгде S/В -отношение величины оже-пика над уровнем фона вторичных элект¬
ронов к уровню фона (зависит от энергии оже-перехода, матрицы, энергии
первичного пучка, разрешения анализатора); Ts - пропускание спектромет¬
ра; I р - ток первичного электронного пучка; т - время накопления сигнала
в одной точке спектра. Достижимая величина отношения сигнала к шуму ог¬
раничивает минимальную регистрируемую концентрацию. Использование
компьютера для обработки спектра и оптимизации тока пучка и времени ре¬
гистрации позволяет увеличить чувствительность и детектировать элемен¬
ты при концентрациях, меньших 1019 см""3. Но этого недостаточно для об¬
наружения легирующих примесей в большинстве структур микросхем. Од¬
нако ЭОС может оказаться полезной при исследовании первой стадии обыч¬
ного двухстадийного диффузионного процесса. Перед высокотемпературной
диффузионной стадией на поверхность наносится легирующая примесь
обычно при более низкой температуре. Для контроля толщины, однороднос¬
ти и концентрации предварительно наносимого слоя можно использовать
метод СОМ. При введении больших доз ионов в приповерхностную область
методом ионной имплантации можно применять метод АТП для определения
однородности и глубинного профиля легирующей примеси.Нежелательные поверхностные примеси могут также вноситься в про¬
цессе ионной имплантации или термической диффузии. При низком вакууме
в установке для ионной имплантации возможно загрязнение остаточными
углеводородами. Причиной загрязнений поверхности пластины может быть
также распыление материала апертур и фиксирующих элементов. В процес¬
се термической диффузии можно использовать СОМ для обнаружения сег¬
регации на поверхности и возможного образования зародышей новых фаз.6.2.6. Системы металпизацииНаиболее шибоким и хорошо зарекомендовавшим себя применением
ЭОС является использование ее для анализа тонких пленок (АТП). При этом
осуществляется травление образца методом ионного распыления, а состав
поверхности, образующейся в центре области травления, определяется с
помошью ЭОС. Измеряя величину оже-сигналов как функцию времени ион¬
ного травления, можно построить график зависимости состава образца от
глубины. Эта информация бесценна при анализе состава, толщины и од-
2506. Применение ЭОС в микроэлектроникенородности тонких пленок. Изучая распределение состава по толщине плен¬
ки при металлизации сплавами, можно наблюдать эффекты сегрегации и от¬
жига в многокомпонентных пленках, примеси в тонких пленках, а также вы¬
явить состав, структуру и химические характеристики границы раздела пе¬
реходов металл - полупроводник как в омических контактах, так и в диодах
с барьером Шоттки. Электрические свойства этих переходов, такие, как вы¬
сота барьера, могут быть соотнесены с результатами анализа зависимости
состава от глубины [ 10 - 12].Метод АТП также используется для:а) исследования контактов или металлизации силицидов, чтобы опреде¬
лить, полностью ли прореагировал тугоплавкий металл с кремнием;б) оценки стехиометрии пленки, образующейся при одновременном осаж¬
дении нескольких соединений;в) контроля возможного сквозного проникновения алюминия через мел-
козалегающий эмиттерный переход;г) проверки совместимости слоев в сложных многослойных материалах,
в структурах с многослойной металлизацией и контроля межслойной диффу¬
зии и эффективности диффузионных барьеров. На рис. 6.1 изображен полу¬
ченный методом АТП профиль по глубине тонкопленочной структуры, осаж¬
денной распылением и состоящей из золота по хромовой подложке на крем¬
нии [13]. Слой хрома должен был служить диффузионным барьером дляРис. 6.1. Профиль состава по глубине тонкопленочной структуры золото - хром на
кремнии сразу после осаждения, полученный с помощью метода АТП.
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди251предотвращения взаимной диффузии золота и кремния. На рис. 6.2 показан
профиль по глубине после прогрева структуры в течение деух часов при тем¬
пературе 300 °С. АТП показывает, что барьер оказался неэффективным.Чтобы создать новую структуру, в установке для осаждения методом распы¬
ления было намеренно увеличено парциальное давление азота в течение все¬
го времени осаждения хрома. Это привело к образованию структуры, изобра¬
женной на рис. 6.3. Слой хрома с добавлением азота является более эффект¬
ным диффузионным барьером. Профиль по глубине этой структуры после теп¬
ловой обработки, полученный методом АТП, показан на рис. 6.4. Целый ряд
материалов, таких, как титан, вольфрам, молибден, хром, платина, никель
и т. д., используется сейчас в системах металлизации, и для их успешного
исполнения будут необходимы аналитические методы. ЭОС используется
также для исследования контактов между слоями в многослойных системах,
таких, как омический контакт металла с поликристаллическим кремнием,
и для оценки роли химических характеристик границы раздела в проблемах
адгезии между слоями.Как упоминалось выше, площадь, исследуемая ЭОС, определяется глав¬
ным образом диаметром первичного электронного пучка. Чтобы определить
свойства материала, необходимо знать его микроструктуру. Это привело
к развитию сканирующих оже-электронных анализаторов с малым диамет-Рис. 6.2. Профиль по глубине той же тонкопленочной структуры (рис. 6.1) после про¬
грева в течение 2 ч при 300°С. Диффузионный барьер оказался неэффектив¬
ным.
Рис. 6.3. Профиль по глубине тонкопленочной структуры золото - хром со специаль¬
но введенным азотом на кремнии сразу после осаждения.Рис. 6.4. Профиль по глубине той же структуры (рис. 6.3) после прогрева в течение
2 ч при температуое 300°С.
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди253ром пучка. Изображение в оже-электронах показывает двумерное распре¬
деление элементов по поверхности.Для создания оже-изображения распределения элементов сфокусирован¬
ный электронный пучок перемещается по поверхности образца, и в каждой
точке прямоугольной матрицы точек измеряется превышение интенсивности
оже-линии над фоном неупругорассеянных электронов. Вычитание уровня
фона является существенным моментом, так как отношение оже-линии к
фону в энергетическом спектре всех вторичных электронов, испущенных об¬
разцом, принципиально мало, причем уровень фона сильно зависит от топо¬
графии образца. Оже-сигнал, измеренный таким образом, используется за¬
тем для модуляции изображения на экране электронно-лучевой трубки от
черного до белого, так что более светлые области соответствуют более
высоким концентрациям элемента.Метод СОМ использовался для исследования сегрегации в системе
металлизации алюминий - медь (4 вес. % меди). В работах [ 14 - 16] пленки,
полученные методом осаждения распылением, исследовались с помощью
оже-анализа большой площади. Зависимости состава от глубины для пленок,
не прошедших термообработку, указали на значительную сегрегацию меди
на границе раздела A^,ee) Cu(4) /Si02 и наличие некоторого взаимодействия
Al -Si02. Было обнаружено, что нагревание этих пленок до температуры
500 °С или несколько выше уменьшает наблюдаемую сегрегацию меди и де¬
лает ее профиль более однородным. В последующих работах аналогичные
пленки исследовались с помощью сканирующего оже-микроанализатора вы¬
сокого разрешения (СОМ) для обнаружения возможных неравномерностей
материала, которые были пропущены при анализе большой площади. Пленки
А1(96) Си(4)на S Ю2 были обследованы до и после нагревания до 500 °С.На изображении СЭМ (рис. 6.5) и изображениях СОМ (не показаны) поверх¬
ности непрогретого образца видно отсутствие особенностей и однородность
распределения пленки алюминий - медь. Профиль по глубине (рис. 6.6) пока¬
зывает однородность распределения меди по толщине пленки и наличие неко¬
торой сегрегации меди на границе раздела приблизительно на 8500 А ниже
поверхности. Изображения СЭМ и СОМ на границе раздела (рис. 6.7 и 6.8 соот¬
ветственно) в основном не содержат особенностей и однородны вдоль поверх¬
ности.Профиль по глубине после нагревания до 500 °С (усредненный по пло¬
щади сканирования электронного пучка 400 мкм2) изображен на рис. 6.9. Он
показывает наличие сегрегации меди на поверхности, однородное распреде¬
ление меди по толщине пленки и отсутствие сегрегации меди на границе раз¬
дела. Используя только эти данные, можно заключить, что прогрев улуч¬
шает качество пленки, так как наблюдавшаяся межслойная сегрегация ме¬
ди исчезла. Однако на изображении СЭМ поверхности пленки после прогре¬
ва (рис. 6.10) видна значительная неоднородность, включая некоторые осо¬
бенности диаметром около 1 мкм. Соответствующие изображения СОМ для
2546. Применение ЭОС в микроэлектроникеРис. 6.5. Вторично-электронное изображение поверхности А1-Си-депозита до тер¬
мической обработки.Рис. 6.6. Профиль по глубине А1 - Си-депозита, полученный при большой площади
анализируемой поверхности, до термической обработки. Видно повышен¬
ное содержание меди на границе раздела Al -Cu/S i02.
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди255Рис. 6.7. Вторично-электронное изображение пленки А1 -Си до термической обра¬
ботки, после распыления образца ионами аргона на глубину примерно
11 ООО А. Обратите внимание на то, что небольшие конические структуры
указывают на направление падающего ионного пучка.меди и алюминия (рис. 6.11) показывают, что на самом деле в алюминиевой
пленке произошла сегрегация меди. Отношение атомных концентраций меди
и алюминия в областях с большим содержанием меди, полученное с помощью
точечного оже-анализа, равно примерно 1/2, т. е. значительно больше, чем
соответствующее объемное отношение.Для определения пространственной природы сегрегации проводилось ион¬
ное травление пленки с одновременным анализом различных областей вдоль
кратера распыления. Изображение СЭМ и соответствующие изображения СОМ
для мер, алюминия и кремния, полученные в пленке на глубине примерно
7500 А, показаны на рис. 6.12 и 6.13. Изображение в оже-электронах крем¬
ния завершает картину, показывая, что черные области на карте алюминия,
не содержащие большого количества меди, заполнены Si 02. Анализ в отдель¬
ных точках опять показал, что отношение атомных концентраций меди и алю¬
миния в областях с большим содержанием меди равно примерно 1/2. Фазовая
диаграмма алюминий - медь указывает на образование тета-фазы данного
сплава, сегрегирующейся в более чистом алюминии. Аналогичные серии
данных (не показаны) были получены для пленок, нагретых до температуры
150, 200, 300 и 400 °С. Даже после отжига при 150 °С в течение всего
30 мин были замечены некоторые признаки сегрегации меди.
2566. Применение ЭОС в микроэлектроникеРис. 6.8. Сканирующие изображения в оже-электронах алюминия (а) и меди (б) облас¬
ти, показанной на рис. 6.7. Элементы распределены однородно.
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди257Рис. 6.9. Профиль по глубине пленки А1 -Си, полученный при большой площади ана¬
лизируемой поверхности, после термической обработки (прогрев в течение
30 мин при 500 °С). Обратите внимание на кажущееся однородное распреде¬
ление меди по глубине.Рис. 6.10. Вторично-электронное изображение поверхности пленки А1 -Си после тер¬
мической обработки. Обратите внимание на наличие пустот и сегрегацию
новой ^азо1.
2586. Применение ЭОС в микроэлектроникеРис. 6.11. Сканирующие изображения в оже-электронах алюминия (а) и меди (б) об¬
ласти, показанной на рис. 6.10. Заметьте, что новая фаза богата медью.
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди 259Рис. 6.12. Вторично-электронное изображение пленки А1 -Си после термической об¬
работки на дне кратера распыления. Обратите внимание на различную
скорость распыления на зернах фазных фаз.Итак, видно, что зависимость состава от глубины, полученная при ана¬
лизе большой площади поверхности образца, прошедшего тепловую обработ¬
ку, указывает лишь на незначительное изменение распределения меди после
прогрева. Однако результаты сканирующей оже-микроскопии высокого прос¬
транственного разрешения демонстрируют гораздо более сложную картину
сегрегации и доказывают необходимость получения изображения в оже-элект¬
ронах. Действительно, нагревание вызывает образование тета-фазы А12Си;
которая распространяется от границы раздела Si02 до поверхности. Это го¬
ворит о том, что более полное, детальное исследование, в котором система¬
тически изменялись и температура, и время отжига, привело бы к лучше¬
му пониманию кинетики системы. Более того, с помощью такого исследо¬
вания по степени сегрегации меди в алюминии можно определить, какой
термической обработке подвергался образец. Аналогичный анализ можно
провести для систем алюминий - кремний и алюминий - медь - кремний,
а также для других систем металлизации.
260 6. Применение ЭОС в микроэлектронике
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди 261Рис. 6.13. Сканирующие изображения в оже-электронах алюминия (а), меди (б) и
кремния (в) области, показанной на рис. 6.12.6.2.7. Химическое осаждение из газовой фазы и
плазменное осаждениеВ методах химического осаждения из газовой фазы и плазменного
осаждения АТП также широко используется для исследования осажденных
пленок. Информация о зависимости состава от глубины очень полезна
при анализе тонких пленок сложного состава, таких, как SiOxNy, в которых
соотношение кислорода и азота изменяется по толщине пленки. Эта ин¬
формация дополняет такие методы, как оптическая эллипсометрия, в ко¬
торой результат измерения является интегралом по гораздо большей пло¬
щади и по глубине пленки, и необходимо вводить средний оптический коэф¬
фициент преломления. Ниже приведен пример типичного использования ЭОС
в этой области технологии интегральных схем (ИС).Фосфосиликатное стекло (ФСС) часто используется в производстве
полупроводниковых приборов для создания изолирующего или герметизи¬
рующего слоя, так как его свойства могут улучшать надежность [ 18, 19].В то же время было показано, что слои ФСС с высоким содержанием фос¬
фора могут ускорить коррозию и вызвать другие проблемы понижения на¬
дежности [20 - 23]. Поэтому необходимо тщательно контролировать кон¬
центрацию Рз05 в слое ФСС-
2626. Применение ЗОС в микроэлектроникеМетоды, обычно используемые для измерения концентрации Р205 , такие,
как химический анализ, электронное зондирование, рентгеновская флуорес¬
ценция и нейтронный активационный анализ, дают информацию об объем¬
ной концентрации и часто применяются для контроля за процессом [ 24]. Эти
методы, однако, не могут дать детальной информации о распределении фос¬
фора в пассивирующем слое. Чтобы установить, какое количество Р205 со¬
держится в определенном месте или на определенной границе раздела, необ¬
ходимо использовать сканирующую оже-электронную спектроскопию. В этом
методе размер зондируемой области сравним с геометрическими размера¬
ми элементов микроэлектронного прибора и анализируемый объем достаточ¬
но мал, чтобы можно было изучать характеристики поверхностей и границ
раздела.Ниже описаны аппаратура для применения ЭОС при анализе фосфосили-
катного стекла, методы получения количественных данных, потенциальные
проблемы и некоторые приложения. В данных исследованиях для представ¬
ления и интерпретации оже-спектров использовалась производная энергети¬
ческого распределения электронов. Образцы представляли собой слои фоо
фосиликатного стекла, содержащие от 2 до 20 вес. % Р205. Объемный сос¬
тав определялся с использованием жидкостных химических методов или
рентгеновской флуоресценции.При проведении оже-анализа диэлектрических образцов, таких, как фос-
фосиликатное стекло, возможна зарядка образца. Влияние эффекта заряд¬
ки на результаты анализа зависит от того, до какой величины доходит по¬
верхностный заряд. Небольшой поверхностный заряд приводит к сдвигу
спектральных линий по шкале энергии. При неоднородной величине сдвига
происходит уширение оже-линии, осложняющее надежные измерения ампли¬
туды пиков. При большом поверхностном заряде наблюдение спектра мо¬
жет оказаться вообще невозможным. Для данных исследований важно, что
оже-линия фосфора находится в области малых энергий (120 эВ) и, следо¬
вательно, легко искажается небольшими поверхностными зарядами. Кроме
того, фосфор обладает высокой подвижностью в стекле, и большие заряды
на поверхности могут вызвать его миграцию. Было обнаружено, что для
предотвращения зарядки образца важно устанавливать его под углом сколь¬
жения по отношению к электронному пучку. Это приводит к увеличению выхо¬
да вторичной электронной эмиссии из образца. В данных исследованиях об¬
разцы укреплялись под углом 30°. При укреплении образцов под углом 60
или 90° (перпендикулярно электронному пучку) происходила значительная
зарядка, затрудняющая получение информативных спектров оже-электронов.При анализе микроэлектронных приборов и других толстых (> 1 мкм)
диэлектрических структур укрепление образца под углом скольжения (нап¬
ример, 30°) может оказаться не всегда достаточным для полного устране¬
ния зарядки образца. В этих случаях может потребоваться некоторая допол¬
нительная его подготовка. Несколько методов, успешно использовавшихся для
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди263уменьшения зарядки образца, основаны на улучшении электрического контак¬
та крепежных приспособлений с образцом. Плоские образцы могут укреплять¬
ся под тонкой металлической пластиной (маской), имеющей маленькое окно
(^3 мм). В таком случае образец удерживается маской, которая обеспечива¬
ет более надежный электрический контакт около анализируемой области и
минимизирует облучаемую площадь диэлектрического материала. Образцы,
имеющие неровную поверхность, могут быть обернуты в тонкую металличес¬
кую фольгу, подогнанную по форме к образцу. Маски из индиевой и алюминие¬
вой фольги успешно использовались для уменьшения зарядки образцов при¬
боров пакетированной конструкции и других объектов нерегулярной формы.В данных исследованиях установлено, что при использовании этих методов
большинство образцов ФСС можно анализировать с помощью ЭОС.Другой круг вопросов в данном исследовании связан с воздействием
электронного пучка на образец. Было показано, что высокие плотности то¬
ка электронного пучка могут вызвать разрушение образца. Так, в Si02 мо¬
жет происходить разрыв связей Si -О, приводящий к появлению на поверх¬
ности свободного кремния. Однако эту проблему можно преодолеть, исполь¬
зуя меньшую плотность тока [25, 26]. Было установлено также, что при
анализе фосфосиликатных стекол при использовании электронного пучка
с высокой плотностью тока могут изменяться состав и распределение
Р202. При достаточно высоких плотностях тока наблюдалось движение фос¬
фора к области, облучаемой электронным пучком. Происходило накаплива¬
ние фосфора у поверхности и его десорбция. Этот процесс продолжался до
полного удаления фосфора из объема в пределах диффузионной длины Р205
от облучаемой области [27, 28]. Эти эффекты ионизации, десорбции и дви¬
жения Р205 к поверхности могут внести значительную путаницу в данные,
полученные при высоких уровнях тока. Для их уменьшения следует исполь¬
зовать малые плотности тока электронного пучка. В данных исследовани¬
ях, а также в других работах [27, 28] было показано, что плотность тока
должна составлять менее КМ А/см2.В данной работе ионное распыление образца производилось ионами Аг +
с энергией 5 кэВ. Во всех известных случаях ионный пучок приводил к
уменьшению количества Р205, вызывая заметный химический сдвиг в оже-
спектре [ 28]. Этот эффект уменьшения наблюдался даже при использовании
ионных пучков меньшей энергии. Однако он, по-видимому, имеет постоян¬
ную величину и не препятствует количественному определению содержания
Р206 в стекле.Были получены эталонные спектры образцов Si02 и пленок фосфосиликатного
стекла с известной концентрацией и произведено их сравнение со спект¬
рами серебра, который имеет фактор относительной чувствительности, рав¬
ный 1. Этот метод количественного анализа спектров оже-электронов опи¬
сан в монографии [ 17]. Эталонные спектры были получены при несколь-
2646. Применение ЭОС в микроэлектроникеРис. 6.14. Факторы относительной чувствительности фосфора, кремния и кислорода
в ФСС.ких значениях энергии электронного пучка при плотности тока примерно
Ю”4А/см2 на распыляемых образцах. Полученные факторы чувствитель¬
ности представлены на рис. 6.14. Затем несколько образцов фосфосиликат*
ного стекла с известным по данным объемного анализа (с помощью рент¬
геновской флуоресценции и жидкостных химических методов) содержанием
Рг05 были проанализированы с помощью ЭОС. Для количественной интер¬
претации спектров использовались найденные факторы чувствительности.
Обнаружено, что количественные данные, полученные с помощью ЭОС, в
пределах 10% согласуются с данными объемного анализа (рис. 6.15). Важ¬
ным результатом количественного анализа пленок ФСС является наблюде¬
ние нестабильности пленок, содержащих высокую концентрацию Р205, во
влажной среде. При нормальных условиях окружающей среды происходит
уменьшение концентрации фосфора в пленке с течением времени. В данной
работе была обнаружена долговременная нестабильность пленок с содержа-Объемный составЭОСР2О5 вес.%Р, вес.%Р, ат.%Р, ат.%9,24,02,42,610,24,42,62,814,36,23,83,617,57,64,64,821,59,45,85,8Рис. 6.15. Сравнение объемного состава образцов ФСС с составом, определенным с
помощью ЭОС.
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди265нием Р2Об свыше 15 вес.%. На рис. 6.16 представлен профиль фосфора в
образце, первоначально содержащем примерно 6,0 ат. % (22 вес. %) фосфо¬
ра,, После нескольких месяцев хранения концентрация фосфора у поверх¬
ности пленки уменьшилась.Прибор был вскрыт и проанализирован на содержание фосфора в зави¬
симости от глубины. Полученный профиль состава (рис. 6.17) показывает
наличие трех различных слоев. Первый слой является пассивирующим и со¬
держит примерно 1,2 ат. % фосфора. Под ним находятся еще два слоя: снаг
чала слой, содержащий примерно 3,0 ат. % фосфора и ниже слой двуокиси
кремния без фосфора. Та же самая процедура может быть использована для
изучения распределения фосфора в зависимости от глубины в приборе.Из этих исследований можно сделать следующие выводы:а) ЭОС можно использовать для определения содержания фосфора в
пленках фосфосиликатного стекла и получения информации о распределе¬
нии фосфора в зависимости от глубины;б) контроль плотности тока электронного пучка позволяет довести до
минимума разрушение образца пучком;Рис. 6.16. Профиль по глубине фосфора в пленке ФСС, полученный с помощью мето¬
да АТП. Обратите внимание на пониженное содержание фосфора у поверх¬
ности.
2666. Применение ЭОС в микроэлектроникеРис. 6.17. Профиль по глубине пленки ФСС (в), увеличенное изображение области ма¬
лых концентраций, показывающее распределение фосфора по глубине (б).в) при тонком распылении будет происходить разрушение ионным пуч¬
ком, однако это не представляет серьезной проблемы для проведения коли¬
чественных измерений;г) при надлежащей подготовке образца можно избежать в большинстве
случаев его зарядки;
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди267д) вышедшие из строя приборы можно анализировать для получения
информации, полезной с точки зрения улучшения надежности.Контроль целостности покрытий на выводных рамках и коррозии вслед¬
ствие наличия поверхностных загрязнений на выводах и корпусах - это еще
одна область применения ЭОС для анализа.6.2.8. Герметизация и прикрепление кристаллаОкончательным этапом изготовления интегральной схемы является
упаковка готового кристалла. Как при проволочном, так и при ленточном
монтаже проблемы образования контакта, его прочности и электрической
целостности определяются химией поверхности контактной площадки и сое¬
динительных проводников. Тонкие слои окисей или загрязнений щелочными
металлами могут препятствовать образованию контакта. Зарегистрировать
их можно только при использовании метода, чувствительного к составу
поверхности.Состав тонких пленок, диффузия и химия поверхности также играют
важную роль и на другой стадии - при прикреплении кристалла к телу кор¬
пуса. В работе Хоге и Томаса [ 29] приведены примеры прикрепления крис¬
талла с использованием преформы из AuSi эвтектоида. Показано, что успех
данной методики в достижении высокой механической прочности и высокой
теплопроводности соединения зависит от принятых правил обращения с
преформой и химией ее поверхности. Очень тонкие слои двуокиси кремния
препятствуют образованию надежного соединения. Бреди и Ховланд [ 30]
привели пример использования СОМ для изучения диффузии никеля через
пленки золота в тонкопленочной системе медь - никель - золото, которая
применялась для прикрепления кристаллов к керамической подложке в гиб¬
ридных интегральных схемах. Было показано, что никель из промежуточно¬
го слоя, предназначенного для образования диффузионного барьера между
золотом и медью, диффундирует вдоль границ зерен золота к поверхности,
где в результате реакции с атмосферой образует очень тонкий слой окиси.
Этот слой толщиной 5 нм препятствует присоединению покрытых золотом
кристаллов к подложке гибридной интегральной схемы.6.3. Другие области микроэлектронной технологииСовременные методы анализа поверхности используются также в мик¬
роэлектронной технологии на некремниевой основе, где проблемы материа¬
лов столь же или еще более сложны. Применение оже-спектроскопии высо¬
кого разрешения будет играть определяющую роль в развитии производст¬
ва интегральных схем на основе GaAs, AlGa As и других полупроводнико¬
вых соединений, а также на основе перехода Джозефсона, где используются
очень тонкие пленки.
2686. Применение ЭОС в микроэлектроникеВ методах выращивания кристаллов, таких, как молекулярная эпитак¬
сия, обычно используется электронная оже-спектроскопия для непосредст¬
венного контроля чистоты подложки, термической десорбции углерода и
кислорода с подложек Ga As, для контроля состава пленки и изучения ки¬
нетики внедрения легирующей примеси. Диффузию между тонкими слоями
и вдоль поверхности можно исследовать с помощью методов АТП и СОМ
соответственно.6.4. ЗаключениеПостоянное улучшение рабочих характеристик приборов и повышение
уровней интеграции систем в микроэлектронике будут продолжаться. В прошлом
увеличение плотности схем осуществлялось в основном благодаря из¬
менениям в архитектуре схем и приборов, а не за счет какого-либо су¬
щественного изменения абсолютной ширины линий на интегральных схемах.
Сейчас ситуация изменяется. Характерные масштабы структур уменьшают¬
ся, а для этого требуются более тонкие пленки окислов и меньшие ширины
линий. Это создает дополнительные трудности для технолога, который дол¬
жен использовать новые методы и материалы. Когда ему необходимо уви¬
деть и понять, что происходит, требуется современная аналитическая ап¬
паратура, пространственное разрешение которой соответствует масштабу
структур.Методы анализа поверхности могут использоваться не только для раз¬
работки процесса травления. Инженер-технолог, обеспечивающий работу
производственной линии, нуждается в помощи при устранении неполадок
на линии* Если выход годных схем падает до нуля, возникает кризисная
ситуация, и инженеру необходимо аналитическое оборудование, с помощью
которого можно быстро обнаружить причину неисправности.В последние несколько лет основными требованиями в микроэлектрони¬
ке стали качество и надежность. Сейчас становится ясно, что нельзя дос¬
тигнуть высокого качества, контролируя только готовое изделие. Качество
достигается благодаря постоянным усилиям по многим направлениям и
соответствует степени оптимизации процесса изготовления интегральных
схем. Качество требует наличия обратной связи между всеми этапами про¬
изводства и конструкцией микросхемы, организацией и ходом процесса произ¬
водства. Современные методы анализа поверхности, такие, как СОМ, бу¬
дут использоваться все шире при обеспечении этой обратной связи для оп¬
тимизации надежности готовых изделий и выхода годных схем.ЭОС, за исключением нескольких частных случаев, еще не стала общепри¬
нятым методом технологического контроля. По мере совершенствования это¬
го метода и появления более дешевого и высокоавтоматизированного обору¬
дования ситуация будет медленно меняться.
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди269БлагодарностиЭта глава посвящается памяти Томаса Бреди. Авторы выражают своюпризнательность Дж. Ф. Моулдеру, Д. М. Беркстренду, Д.Ф. Паулу, Г. Ле¬
монсу из фирмы Signetics, Р. Макдональду из фирмы Intel и Р.С. Новицкииз Exxon Enterprises.Литература1. Thomas Simon, Proceedings of Test and Measurement Expo (1982).2. Holloway P*H; Characterization of electron devices and materials by sur¬
face-sensitive analytical techniques, Appl. Surf. Sci., 4, 410 — 444 (1980).3. Kazmerski L.L., Jamjoum O., Ireland P./., Whitney R*L*, A study of the
initial oxidation of poly crystalline Si using surface analysis techniques,J. Vac. Sci. Technol., 18, 960 - 964 (1981).4. Schwarz S-Л*, Helms C*R*, Spicer W*E*, Taylor N*J* High resolution
Auger sputter profiling study of the effect of phosphorus pileup on theSi — Si02 interface morphology, J.Vac. Sci. Technol., 15, 227 — 230 (1978).5. Adams Smith T*E*, Chang C.C*, The growth and characterization of
very thin silicon dioxide films. J. Electro chem. Soc., 127, 1787 — 1794
(1980).6. Wildman H*SBartholomew R*F; Pliskin W*A*, Revitz M*, Variations in
the stoichiometry of thin oxides on silicon as seen in the Si LVV Auger
spectrum, J. Vac. Sci. Technol., 18, 955 — 959 (1981).7. Helms C*R*, Johnson N*M*t Schwarz S*A*, Spicer 1У.Е., Studies of the ef¬
fect of oxidation time and temperature on the Si — Si02 interface using
Auger sputter profiling. J. Appl. Phys., 50, 7007 — 7014 (1979).8. Helms C*R•, Strausser Y<E*, Spicer W*E*, Observation of an intermediate
chemical state of silicon in the Si/Si02 interface by Auger sputter profi¬
ling, Appl. Phys. Lett., 33, 767 - 769 (1978).9. Olevskil S»S», Repko V»P•, Use of Auger Spectroscopy to Investigate Si02
Films, pp. 1192 — 1194, Scientific Research Institute of the Horological
Industry, September 1979.10. Grinolds H*R*, Robinson G*K%, Pd/Ge contacts to я-type GaAs, Solid Sta¬
te Electronics, 23, 973 — 985 (1980)*11. Grinolds H*R*, Robinson G*Y*, Study of Al/Pd2 Si contacts on Si, J* Vac*
Sci. Technol., 14, 75 -78 (1977).12. Erickson L*P; Waseem A*, Robinson G.Y*, Characterization of ohmic con¬
tacts to InP, Thin Solid Films, 64, 421 — 426 (1979).13. Nowicki R*S*, Moulder J*F*, Studies of CrN — Au as an improved die at¬
tach metallization system, Thin Solid Films, 83, 209 — 216 (1981).14. Mader S,, Herd S*, TTiin Solid Films, 10, 377 (1972).15. Learn A.]., Thin Solid Films, 20, 261 (1974).
2706. Применение ЭОС в микроэлектронике16. Denison D.RHartsough L*D., J. Vac. Sci. Technol., 17, 1326 (1980).17. Davis L.E; MacDonald iV.C*, Falmberg P»W. et ah, in: Handbook of Au¬
ger Electron Spectroscopy, Physical Electronics Division, Perkin — Elmer
Соф., Eden Prairie, Minnesota (1976).18. Kerr D*R*, Logan J*SBurkhardt P.]*, Pliskin W»A», IBM J.; 18, 376
(1964).19. Schlacter M.MSchlegel E*S., Keen #.S. et ah, IEEE Trans, on Electron
Devices, ED 17 - 12, 1077 (1970).20. Paulson WtM», Kirk R*W,, Proc. Twelfth Rel. Phys. Symp., 172 (1974).21. Comizzoli R.B., RCA Rev., 37, 483 (1976).22. Maeda К*, Sato Ban YJpn. J. Appl. Phys., 16, 729 (1977).23. Nagasima N*, Suzuki H; Tanaka К*, Nishida S.,J. Electrochem. Soc., 121,
434 (1974).24. Adams AX*, Murarka S.P., J. Electrochem. Soc., 126, 334 (1979).25. Thomas S., J. Appl. Phys., 45, 161 (1974).26. Johannessen J*SSpicer W*E., Strausser Y*EJ. Appl. Phys., 47, 3028
(1976).27. Inoue TJpn. J. Appl. Phys., 16, 851 (1977).28. Queirolo G.r Pignatel G.l/., J. Electrochem. Soc., 127, 2438 (1980).29. Hoge С%Еь Thomas S,, Some considerations of the gold — silicon die bond
based on surface chemical analysis, Eighteenth Annual Proc. Rel. Phys.
(1980).30. Brady T*E; Hovland С.Г., Scanning Auger microprobe study of gold — ni¬
ckel — copper diffusion in thin films, J.Vac. Sci. Technol., 18, 339 (1981).БИБЛИОГРАФИЯОбзорыHolloway P*H», Application of surface analysis for electronic devices, Appl.
Surface Analysis ASTM STP, 1980, 5 - 23 (1980).Lowry R*K; Hogrefe A*W*, Applications of Auger and photo-electron spectro¬
scopy in characterizing IC materials, Solid State Techn., January 1980,71 - 75 (1980).McGuire G.E; Choosing between ESCA and Auger for surface analysis, NBS
Special Publication 400 — 23, pp. 175 — 182 (March 1976).McGuire G*E*, Holloway P»H», Use of X-ray photoelectron and Auger electronspectroscopies to evaluate microelectronic processing, Scanning Electron
Microscopy I, 173 — 202 (1979) «Wagner N»K%, Scanning Auger analysis of III — V semiconductor heteroepitaxial
interfaces, pp. 165 — 169.
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди271РаШеBrinker £)./., Wadlin W*H*, Wang E.YLegge R.N., Auger study of the inter¬
face in evaporated tin oxide — GaAs MIS solar cells, J. Electrochem. Soc»,
128 (9), 1968 - 1971 (1981).Christou A., Anderson W.7V, Sieger K.J., Environmental effects on GaAsMESFETs, International Electron Devices Meeting, Washington, D.C.,
ррь 389 - 393 (1978).Davis G.D*, Sun T.S., Buchner S*P*, ByerN*Et, Anodic oxide composition and
Hg depletion at the oxide — semiconductor interface of Hgj _xCdxTe,J. Vac. Sci.Technol., 19 (3), 473 - 476 (1981).Duchemin J*P•, HirtzJ,P•, Razeg/ii M* et aU, GalnAs and GalnAsP materials
grown by low pressure MOCVD for microwave and optoelectronic applica¬
tions, J. Crystal Growth, 55, 64 — 73 (1981).Elabd H; Steckl A*J*, Structural and compositional properties of the PbS—Si
heterojunction, J. Appl. Phys., 51 (1), 726 — 737 (1980).Elabd H*, Steckl Л.Д, Auger analysis of the PbS— Si heterojunction. J. Elect¬
ronic Materials, 9, 525 — 549 (1980).Ingrey S. /•, Shepherd F*R., Westwood Thermal diffusion of chromium in
poly crystalline CdSe films, J. Vac. Sci. Technol., 17 (1), 481 — 484
(1980).Kuwano H; Miyake S•, Kasai TDry cleaning of Si surface contamination by
reactive sputter etching. Jpn. Appl Phys., 21, 529 — 533 (1982).Lieske N., Hezel R*, Chemical composition and electronic states of MNOS
structures studies by Auger electron spectroscopy and electron energy-loss
spectroscopy, Inst Phys., Conf. Series No. 50, Chapter 3, pp. 206—215 (1980).Matsuura М., Ishida. М», Suzuki А*, Нага К», Laser oxidation of GaAs, Jpn.J. Appl Phys., 20,L726 - L728 (1981).Pemas JErman M», Theetan J.B. et ah, Characterization of thin films of
amorphous GaP using optical and electron spectroscopy, Thin Solid
Films, 82, 377 - 391 (1981).Shin-ichi Takahashi, Influence of supersaturated melt for InP growth on InP —
InGaAsP interface of double-heterostructure laser wafers, J. Appl Phys.,
52 (10), 6104 - 6108 (1981).Профили no глубине легирующих примесей, введенных ионной имплантациейNagahama КNishitani К», Ishii М., Thermally stable metallization to InSb,
Japn. J. Appl Phys., 20, 1171 — 1172 (1981).Park У.5. Grant J*THaas T.W., The determination of sulfur-ion-implantation
2726. Применение ЭОС в микроэлектроникеprofiles in GaAs using Auger electron spectroscopy, J. Appl^ Phys., 50,809 - 812 (1979).Park Y.S., Theis W*M*, Grant J*T*, Characterization of ion implants in GaAs
by AES and GDOS, Appl Surf. Sci., 4, 445 - 455 (1980).Pignatel G*U*f Queirolo G., Further insight on boron diffusion in silicon ob¬
tained with Auger electron spectroscopy, Thin Solid Films, 67, 233 — 237
(1980).Schwarz S*A*, Helms C*R; Spicer W*E», Taylor N*J*, Abstract: Auger sputter
profiling study of phosphorus pileup at the Si — Si02 interface. J. Vac.Sci. Technol., 15, 1519 (1979).Омические контактыAnderson Jr*, Christou A*, Davey J»E., Smooth and continuous ohmiccontacts to GaAs using epitaxial Ge films. J. AppL Phys*, 49, 2998 —3000 (1976).Aydinli A,, Mattauch R*J*, Au/Ni/Sn,Ni/n-GaAs interface: ohmic contact
formation, J. Electrochem. Soc.,128, 2635 — 2638 (1981).Barnes Р.Л., Williams R,S*y Alloyed tin-gold ohmic contacts to n-type indium
phosphiode, Solid State Electronics, 24, 907 — 913 (1981).Watanabe М., Kishimoto AL, Hiratsuka Y*, Mitani S*, Mori T*, Study of contact
failures caused by organic contamination on Ag — Si contacts, IEEE
Trans, on Components, Hybrids, and Manufacturing Technology, CHMT-5,
No. 1, 90 - 94 (1982).Системы металлизацииBaker J*E; Blattner R.JNadel S. et ah, Thermal annealing study of Au/Ti — W
metallization silicon, Thin Solid Films, 69, 53 — 62 (1980).Chang С.Л., Chou /V-/#, Ambient effects on the out-diffusion of GaAs through
thin fold films, J.Vac. Sci. Technol., 17, 1358 - 1359 (1980).Chang C»C; Quintana G., Diffusion kinetics of Au through Pt films about 2000
and 6000 A thick studied with Auger spectroscopy, Thin Solid Films,31, 265 - 273 (1976).Christou A., Jarvis L., Weisenberger W*H^ Hirvonen J*KSEM Auger spectro¬
scopy and ion backscattering techniques applied to analyses of Au/ref-
ractory metallizations, J. Electronic Materials, 4, 329 — 345 (1975).Dini J*W4, Johnson H*ROptimization of Gold Plating for Hybrid Microcircuits,
pp. 53 — 57, Sandja National Laboratories (January 1980).Ghate P.B., Blair J»C*, Fuller C*RMcGuire G*E», Applications of Ti: W bar¬
rier metallization for integrated circuits, Thin Solid Films, 53, 117 - 128
(1978).Grunthaner P.Jt, Grunthaner F*J*, Scott D»M* et* ah, Oxygen impurity effects
at metal/silicide interfaces: formation of silicon oxide and suboxides in
the Ni/Si system, J.Vac. Sci. Technol., 19, 641 - 648 (1981).
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди273Guo K*J*, Wiley /.D*, Perepezko /.#. eU aL, Amorphous metal diffusion bar¬
riers, Proc* Second Conf. on High Temperature Electronics and Instrumenta¬
tion, Houston, 7 — 8 Dec. (1981).Hall PM; Panousis N*T*, Menzel P*R., Strength of gold-plated copper leads
on thin film circuits under accelerated aging, IEEE Trans, on Parts, Hyb¬
rids and Packaging, PHP-11, No. 3, 202 - 205 (1975),Lee W-S-, Skinner D»K*, Swanson J*G*, Structural evidence for a low temperatu¬
re interaction of aluminium and InP, Thin Solid films, 70, L17 — L19
(1980),Lee C*P*, Tandon J*L*, Stocker P-Alloying behaviour of Au — Ge/Pt ohmic
contacts to GaAs by pulsed electron beam and furnace heating, Electro¬
nics Letters, 16, 849 — 850 (1980).Luzzi G-, Papagno L-, Vacuum-deposited thin films studied by Auger spectrosco¬
py, J. Vac. Sci. Technol., 16, 1004 — 1006 (1979).McGuire G-E-, Jones /-F-, Dowell H*J*, The Auger analysis of contaminants
that influence the thermocompression bonding of gold, Thin Solid Films,45, 59 - 68 (1977),Magee T-/-, Peng J*, Si epitaxial regrowth and grain structure of Al metalliza¬
tion on (100) Si, Jo Appl Phys., 49, 4284 - 4286 (1978)eMorkoc H; Cho Л-У-, Stanchak CM*, Drummond T«J<,, Properties of Au/W/GaAs
Schottky barriers, Thin Solid Films, 69, 295 — 299 (1980).Nowicki Harris JMNicole М-Л-, Mitchell LVStudies of the Ti—W/\u
metallization on aluminium, Thin Solid Films, 53, 195 — 205 (1978).Ogawa M*, Alloying behaviour of Ni/Au — Ge films on GaAs, J* Appl. Phys.,51, (1), 406 - 412 (1980).Oura K*, Okada Kishikawa Y-, Hanawa Т., Surface structure of epitaxial,Pd2Si thin films, Appl, Phys, Lett., 40, 15 138 - 140 (1982).Shankoff Х-Л., Chang C-C-, Haszko S*E<, Controlling the interfacial oxide layer
of Ti-Al contacts with the СЮ3_ H3P04, J* Electrochem. Soc., 125,467 - 471 (1978).Thompson Cropper D*R*, Whitaker В-1У-, Bondability problems associated
with the Ti — Pt — Au metallization of hybrid microwave thin film circuits,
IEEE Trans, on Components, Hybrids, and Manufacturing Techn. CHMT-4,
No. 4, 439 - 445 (1981).Wildman Я-S-, Schwartz G»C*, Auger profiling and electrical resistivity studies
of the interface between evaporated Al — Cu layers, J* Vac. Sci. Technol.
20, 396 - 399 (1982).Wiley 7-D-, Perepezko Nordman High Temperature Metallization
System for Solar Cells and Geothermal Probes, Sandia National Labora¬
tories, December 1980.Wiley J.D; Perepezko /.//-, Nordman Kang-Jin Guo, Amorphous metal¬
lization for high-temperature semiconductor device applications, Proc.First High Temperature Electronics Conf. Tucson, March 1981, pp. 35 — 38
(1981)„
2746. Применение ЭОС в микроэлектроникеБарьеры ШотткиAmith Л-, Mark P., Schottky barriers on ordered and disordered GaAs (110),J. Vac. Sci* Technoh, 15, 1344 - 1352 (1978).Aydinli AMattauch R*JGaAs Schottky barrier interface by AES, Proc. of
the Southeastern Conf., pp. 14 — 17 (1980)*Chang C-C«, Murarka S*P*, Kumar V-, Quintana СInterdiffusions in thin-film
Au on Pt on GaAs (100) studied with Auger spectroscopy, J. Appl. Phys.,
46, 4237 - 4243 (1975).Crider C*A-, Poate J»M*, Growth rates for Pt2Si and PtSi formation under UHV
and controlled impurity atmospheres, Appl. Phys. Lett., 36* 417 — 419
(1980).Gamer C»M*, Su С.У., Saperstein W-Л* et- al, Effect of GaAs or GaxAlj _ x
as oxide composition on Schottky-barrier behaviour, J.AppL Phys., 50,3376 - 3382 (1979).Grinolds H*R*, Robinson G-У-, Low-temperature sintering of Pd/Ge films on
GaAs, AppL Phys. Lett,, 34, 1, 575 - 577 (1979),Hokelek Robinson G-У-, Aluminum/nickel silicide contacts on silicon,Thin Solid Films, 53, 135 - 140 (1978),Lee BW*, Jou LMark P-, Yeh J»L<, So E-, Surface composition and charac¬
teristics of oxide-free Gaj _xAlxAs (110) Schottky barriers, J. Vac. Sci.
TechnoL, 16, 514 - 516 (1979),McGuire G*E*, Wisseman W»R*, Diffusion studies of Au through electroplated
Pt films by Auger electron spectroscopy. J. Vac. Sci. Technol. 15,1701 - 1705 (1978),McKinley A-, Parke AJN*, IWilliams RM*, Silver overlays on (110) indium pho¬
sphide: film growth and Schottky barrier formation, J. Phys. C* Solid
St, Phys., 13, 6723 - 6736 (1980).Ogawa MAlloying reaction in thin nickel films deposited on GaAs, Thin
Solid Films, 70, 181 - 189 (1980),Oura КOkada S., Hanawa TThermally induced accumulation of silicon on
palladium silicide surfaces as studied by Auger electron spectroscopy.
Appl, Phys, Lett,, 35, 705 - 706 (1979),Robinson G-У-, A study of metal-semiconductor contacts on indium phosphide,
Rome Air Development Centre, RADC-TR-81-169, July (1981),Ruths P<F; Ashok S*, Fonash 5-/-, Ruths ]MA study of Pd/Si MIS Schottky
barrier diode hydiogen detector, IEEE Transactions on Electron Devices,
Ed-28, 1003 - 1009 (1981),Schrmd P-E,, Ho P*S*, Foil H-, Rubloff G-W-, Electronic states and atomic
structure at the Pd2Si — Si interface, J Vac Sci. TechnoL, 18, 937 —943 (1981).Skinner D*K*, New method of preparing (100) InP surfaces for Schottky barrier
and ohmic contact formation. J. Electronic Materials, 9, 67 — 78 (1980).
P.P. Олсон, П.В. Палмберг, С.Т. Ховланд, Т.Е. Бреди275Szydio N•, Oliver J•, Behaviour of Au/InP Schottky diodes under heat treat¬
ment, J. Appl. Phys., 50, 1445 - 1449 (1979).Tongson L*L, B%E Knox B. £., Sullivan T.E., Fonash S., Comparative study
of chemical and polarization characteristics of Pd/Si and Pd/Siox/Si
Schottky-barrier-type devices, J. Appl. Phys., 50, 1535 — 1537 (1979).Пассивация и герметизацияInada Т., Mixva HKato S., Kobayashi £., Annealing of Se-implanted GaAs with
an oxygen-free CVD Si3N4encapsulant. J. Appl, Phys., 49, 4571 — 4573
(1978).Inada Т., Ohkubo Т., Sawada S., Chemical vapor deposition of silicon nitride,J. Electrochem. Soc., 125, 1525 - 1529 (1978).Ito Т., Nazaki Т., Ishikawa H., Direct thermal nitridation of silicon dioxidefilms in anhydrous ammonia gas, J. Electrochem. Soc., 127, 2053 — 2057
(1980).Leig)iP*A*, Investigation of CVD Silicon Nitride Encapsulation for Gallium
Arsenide, British Telecom Research Laboratories, 29 July 1981.Vaidyanathan K»V*, Helix M»J*, Wolford D.J* etaL, Study of encapsulants for
annealing GaAs, J. Electrochem. Soc., 124, 1781 — 1784 (1977).WangXReyes A*-Mena ALichtman DInterface composition studies ofthermally oxidized GaAs using Auger depth profiling. J. Electrochem. Soc.,
129 (1982).Yoshimi T*, Sakai H%, Tanaka К♦, Analysis of hydrogen content in plasma sili¬
con nitride film, J. Electrochem. Soc., 127, 1853 — 1854 (1980).Молекулярно-пучковая эпитаксияAlexandre F•, Raisin СAbdalla MJ• et ah, Influence of growth conditions ontin incorporation in GaAs grown by molecular beam epitaxy, J. Appl. Phys.,
51, 4296 - 4304 (1980).Arthur ]*R; LePore Quantitative analysis of AlxGaj _ xAs by Auger elec¬
tron spectroscopy, J. Vac. Sci. Technol., 14, 979 — 984 (1977).Dupuis R*Dt, Moudy L»A*, Dapkus P*D*, Preparation and properties ofGaj _ xAlxAs—GaAs heterojunctions grown by metallorganic chemical va¬
pour deposition, Inst. Phys. Conf. Ser. No. 45, Ch. 1, pp. 1—9 (1979).Gamer CMSu C.Y., Shen Y*D* et ah, Interface studies of AlxGaj _xAs — GaAs
heterojunctions, J. Appl. Phys., 59, 3383 — 3389 (1979).Ichimura S., Aratama M.y Shimizu RAn interpretation of quantitative analysisof Arthur and LePore for AlxGaj _ xAs by using AES, J. Vac.Sci. Technol.,
18, 34 (1981).
2766. Применение ЭОС в микроэлектроникеJohannessen J*S., Clegg Foxon C.T., Joyce B*A*, Interface composition
profiles of MBE grown GaP films on GaAs substrates, Physica Scripta 3,
1980, 440 - 443 (1980).Ploog K*, Surface studies during molecular beam epitaxy of gallium arsenide,J. Vac. Sci. Technol., 16, 838 - 846 (1979).Ploog К*, Fischer A, Surface segregation of Sn during MBE of n-type GaAs
established by SIMS and AES, J. Vac. Sci. Techol. 15, 255 — 259 (1978).Wood C*EtC*. Joyce В.Л., Tin-doping effects in GaAs films grown by molecular
beam epitaxy, J. Appl. Phys., 49, 4854 — 4861 (1978).Si 02 и граница раздела Si02-S iChuang T*J*, Electron spectroscopy study of silicon surfaces exposed to XeF2
and the chemisorption of SiF4 on silicon, J. Appl. Phys.. 51, 2614 — 2619
(1980).Strausser Y.E., Helms Schwarz S., Spicer W»E>, Factors affecting depth
resolution and chemistry in Auger electron spectroscopy depth profiles of
MOS and MNOS structures, Thin Solid Films, 53, 37 (1978).
Г язва 7
ОЖЕ-СПЕКТРОСКОПИЯ В МЕТАЛЛУРГИИ
М.П. Сих"7.1. ВведениеМеталлургия - не новая отрасль знания, она развивается умеренными
темпами в течение последних нескольких тысяч лет путем проб и ошибок,
отвечая требованиям соответствующих цивилизаций. В настоящее время
сложные продукты металлургии встречаются во всех областях современной
жизни: и в домашнем хозяйстве, и в промышленности. Из года в год
улучшаются свойства металлоизделий, отвечающие требованиям конкретной
области применения - коррозионная стойкость, сохранение свойств при вы¬
соких и низких температурах, износостойкость и удельная прочность - в
соответствии с растущей конкуренцией на рынках сбыта. В связи с усложне¬
нием материалов и ужесточением требований прогресс достигается только
за счет более глубокого понимания поведения материала в многопараметри¬
ческой среде» В частности, за последние годы в связи с повышением стои¬
мости энергии разработка материала с более высокой жаропрочностью для
лопаток турбин может привести к созданию более эффективного авиацион¬
ного двигателя, что в свою очередь дает значительные преимущества дви-
гателестроителям, авиастроителям, использующим эти двигатели, и авиа¬
линиям, на которых эти самолеты летают. Вторая область - материальные
ресурсыо Здесь проблемы поиска возможных альтернатив для элементов,
имеющих ограниченные или неопределенные источники, и степень повторно¬
го использования материала или его регенерации становятся все более
настоятельными.Значительный вклад в развитие всех областей металлургии был внесен
применением методов анализа поверхности, в частности оже-спектроскопии.
ЭОС успешно использовалась для изучения коррозии [1 — 3] , окисления
[4, 5], цементации [6] и азотирования [7], пайки [8], спекания [9], проблем
порошковой металлургии [ 10], механической обработки [ 11], износа [ 12] и
смазки [ 13], при разработке твердых [ 14] и тяжелых сплавов, а также для
решения широкого круга общих проблем межфазного разрушения. НекоторыеM.P Seah, Division of Materials Applications, National Physical, Laboratory,
Teddington, Middlesex, UK.
2787. Оже-спектроскопия в металлургиииз этих проблем обсуждены в гл. 10; при решении многих из них использо¬
ван метод построения профилей концентрации, как показано в гл. 4. Интер¬
претация проблем, связанных с послойным анализом, может быть до неко¬
торой степени рассмотрена как дальнейшее развитие метода микрорентге-
носпектрального анализа, но при более высоком разрешении по глубине,
тогда как изучение и интерпретация проблем межфазного разрушения явля¬
ется отдельной областью, которая стала возможной только в связи с раз¬
витием оже-спектроскопии.В этой главе мы сосредоточим внимание на проблемах, связанных с
поверхностями раздела, в частности на разрушении, рассматриваемом в
качестве модельного примера того, как элементы с концентрациями мень¬
ше монослоя, согласно классическим теориям адсорбции, поглощаются
на внутренних поверхностях раздела, таких, как границы зерен и межфаэ-
ные границы, и вызывают катастрофическое разрушение конструкционных
материалов. Для доказательства перспективности использования метода
рассмотрим известный случай катастрофического межзеренного разруше¬
ния ротора турбогенератора электростанции в Хинкли Пойнте. При испыта¬
нии турбины в режиме перегрузки в 1969 г. ротор разрушился сам и раз¬
рушил большую часть турбины [ 15]. Ротор, послуживший причиной аварии,
был повторно собран, как показано на рис. 7.1. Первоначально турбинные
лопатки были расположены по периферии диска, а ротор насажен на вал,
проходящий через его ось. Фрактограмма поверхности излома, полученная
с помощью растрового электронного микроскопа, представлена на рис» 7.2
[16]. Видно, что разрушение произошло по границам исходных зерен аусте-
нита с малой деформацией и низкой энергоемкостью. Такой тип разрушения
называется хрупким, и, с точки зрения инженеров, ротор, склонный к хруп¬
кому разрушению, эквивалентен ротору из стекла! Описанное поведение не
характерно для данной легированной стали и ее термической обработки,
так как некоторые другие роторы из этой же стали были надежны в эксплу¬
атации. Причиной такого рода хрупкого разрушения является сегрегация
имеющихся в стали примесей по границам зерен. Концентрация этих приме¬
сей колеблется от отливки к отливке, и, если не принять специальных мер
предосторожности, может превысить значение, при котором становится
неизбежным катастрофическое разрушение.Для более детального понимания этой проблемы необходимо рассмот¬
реть границу зерна на атомном уровне. Модельный двумерный кристалл из
пузырьков Брэгга и Ная, показанный на рис. 7.3, хорошо иллюстрирует
степень несоответствия у границ двух упорядоченных двумерных кристал¬
лов. Ясно видно, что если атом несколько большего диаметра замещает
атом матрицы (т. е. находится в растворе), то это связано с затратой
соответствующей энергии деформации, тогда как атом может заместить
определенные атомы матрицы у границы зерна и в то же время снизить
М.П. Сих279Рис. 7.1. Вновь собранный ротор из стали 3 Сг 0,5 Мо, послуживший причиной разру¬
шения турбины в Хинкли Пойнте.энергию деформации этой границы. Такое смещение растворенных атомов
из тела зерна и скопление их по границам зерен, на свободной поверхнос¬
ти или других поверхностях раздела называют равновесной сегрегацией.
Степень развития равновесной сегрегации контролируется термодинами¬
кой процесса - соответствующим изменением свободной энергии сегрегации.
Более полная схема процесса показана на рис. 7.4. Здесь твердое тело со
свободной поверхностью и внутренними дефектами выдерживается в замк¬
нутой камере в изотермических условиях при температуре, обеспечиваю¬
щей достижение равновесного состояния за счет протекания диффузионных
процессов. В этом случае химический потенциал растворенных атомов пос¬
тоянен в пределах всей камеры, эти атомы находятся в твердом растворе,
в паровой фазе, а также распределены по внутренним дефектам, как пока¬
зано на рис. 7.4.
2807. Оже-спектроскопия в металлургииРис. 7.2. Микрофрактограмма поверхности разрушения ротора (растровый электрон¬
ный микроскоп, ширина снимка 200 мкм) [1б].Перераспределение растворенных атомов между границами зерен и
кристаллической решеткой было впервые рассчитано Маклином в 1957 г.[18], хотя блестящие межкристаллитные изломы сталей, разрушившихся
благодаря присутствию мышьяка, фосфора и серы, наблюдали еще в 1984 г.[19]. Маклин разработал программу работ для Национальной физической
лаборатории (НФЛ) по измерениям сегрегаций растворенных элементов в
меди и железе с использованием расчетных зависимостей изменения по¬
верхностной [20] и зернограничной [21] энергий вследствие сегрегации.Эта работа дала очень хорошие результаты, хотя велась очень медленнои кропотливо и была ограничена двойными системами. Положение сущест¬
венно изменилось при использовании ЭОС для непосредственной идентифи¬
кации сегрегирующих элементов; ранее существовавшие ограничения от¬
пали и появилась возможность проведения измерений при пониженных тем¬
пературах, в большей степени соответствующих проблемам, связанным
с поведением материалов. Очень важна также возможность использования
ЭОС для выявления присутствия сегрегирующих элементов при разрушении
М.П. Сих 281Рис. 7.3. Пузырьковый двумерный кристалл Брэгга - Ная, показывающий границу
между двумя плотноупакованными упорядоченными кристаллами.промышленных материалов. Единственным недостатком использования ана¬
лиза поверхности при изучении зернограничных сегрегаций является необ¬
ходимость разрушения образца по границам зерен в приборе. Это не
представляет трудности для материалов, подверженных отпускной хруп¬
кости, однако в некоторых случаях межкристаллитное разрушение проис¬
ходит только при соответствующих условиях под действием циклического
нагружения [22] или температуры [23, 24], что не легко моделировать в
оже-спектрометре. Если межкристаллитное разрушение в приборе получить
не удается, то измерение зернограничных сегрегаций невозможно.На рис. 7.5 приведены элементы, для которых были измерены зерно¬
граничные сегрегации в железе. Очень возможно, что при соответствующих
условиях и многие другие элементы будут сегрегировать из твердого раст¬
вора в железе в его двойных и тройных сплавах. При экспериментальном
изучении сегрегаций на поверхности в стали наблюдались сегрегации бора, переходных
и щелочных металлов. Аналогичные результаты получены для меди и никеля,
и весьма вероятно, что сегрегации в технически чистых материалах явля¬
ются правилом, а не исключением.В следующем разделе будут рассмотрены характерные признаки зерно¬
граничных, межфазных и поверхностных сегрегаций, определяемые с по-
2827. Оже-спектроскопия в металлургииРис. 7.4. Места адсорбции в кристаллических твердых телах [17].Рис. 7.5. Элементы, сегрегирующие к границам зерен в железе, концентрации кото¬
рых были измерены с помощью ЭОС. Элементы, вызывающие охрупчивание,
указаны в заштрихованных квадратах.мощью ЭОС. Затем описывается теория сегрегаций: движущие силы процес¬
са сегрегации и кинетика процесса, выраженная членами, определяющими ско¬
рость достижения равновесия. В последнем разделе рассмотрено влияние
сегрегаций на основные параметры материала и, следовательно, их общее
влияние на металлургические свойства, а также предложения по устране¬
нию вредного влияния сегрегаций.
М.П. Сих2837к2. Характеристика сегрегацийМеталлурги проявляют большой интерес как к зернограничным, так и к
поверхностным сегрегациям. Была предпринята попытка [ 25] использовать
более простые методы измерения поверхностных сегрегаций для получения
данных о зернограничных сегрегациях. Хотя в принципе это и возможно,
пока еще не получены точные численные значения соответствующих членов,
которые позволили бы достичь значимой корреляции между этими двумя
типами сегрегаций, за исключением простейших двойных систем [ 26]. Дру¬
гим подходом, используемым некоторыми исследователями, является упро¬
щение задачи зернограничного разрушения и сведение ее к разрушению об¬
разцов вне СВВ-системы [ 27]. Это приводит к очень сомнительным и не
позволяющим получить численные значения результатам и может быть ис¬
пользовано только для выявления возможных сегрегирующих элементов
[28].Методика количественного анализа в оже-спектрометре для измерения
концентрации сегрегирующих элементов описана в гл. 5 и здесь упоминает¬
ся лишь кратко. Из уравнения (5.31) следует, что(7.1)это выражение может быть еще более упрощено приближенно до(7.2)где <рл - концентрация элемента, сегрегирующего на поверхности или у
границ зерен, IА и 1В - интенсивности, приведенные в справочнике [29],
т = 0,5 для поверхностей и 1 для границ зерен, иобозначения всех членов в уравнении (7.3) приведены в гл. 5. Для удобства
значения КА в для элементов, сегрегирующих в железе, отнесенные к пику
железа при 703 эВ, представлены в табл. 7.1. Эти значения рассчитаны из
уравнения (7.3) с использованием данных по неупругому среднему свобод¬
ному пробегу Л (£) Сиха и Денча [ 30], а также членов, описывающих обрат¬
ное рассеяние/ (Е) Итимуры и Симицу [31], и коэффициентов относитель¬
ной чувствительности IА > приведенных в [29]. Напомним, что при количест¬
венном анализе принята такая плотность укладки сегрегирующих атомов,
как если бы они составляли чистый элемент. Точность данных по неупруго-
2847. Оже-спектроскопия в металлургииТаблица 7.1. Значения КАВ для элементов, образующих зернограничные
сегрегации в железе, полученные при использовании электронного луча с
энергией 5 кэВ под углом 30° к нормали к поверхностиЭлементэВKABЭлементэВ*ABЭлементэВKabLi433,2Co7757,2Sn4301,8Be1043,8Ni8486,9Sb4542,6В1794,4Cu9207,9Те4833,7С2724,6Zn99410,3I5115,6N3794,4Ga107013,9Cs56314,0О5032,8Ge114716,9Ba734,1F6473,0As122821,2La781,9Na9907,6Se131528,6Ce822,8Mg453,2Br139639,2Hf1857,1Al682,4Rb136569,1Та1791,2Si921,9Sr164976,9W1697,3P1201,5Y174688,8Re1768,7S1521,0Zr1475,8Ir17112,7Cl1810,9Nb1674,3Pt641,9к2521,3Mo1863,5Au691,9Ca2913,0Ru2732,3Hg761,9Sc3404,9Rh3021,8Tl842,1Ti4183,7Pd3301,1Pb942,3V4733,4Ag3511,4Bi1012,5Cr5294,6Cd3761,4Mn5427,5In4041,6му среднему свободному пробегу (стандартное отклонение) составляет 30%,
так что точность рассчитанных значений К не может превышать этой ве¬
личины.7.2.1. Измерения на границах зерен
7.2.1.1. Разрушение образцаИзмерения зернограничных сегрегаций проводятся либо с целью выяс¬
нения причин разрушения в процессе эксплуатации, либо в рамках програм¬
мы исследований разрабатываемых сплавов. В первом случае используют
детали в состоянии поставки, а во втором случае требуется термическая
обработка,, В последнем случае все образцы подвергают термической обра¬
ботке вне СВВ-системы при соответствующей температуре под нагрузкой
или без нее в течение необходимого времени. В зависимости от требований
материал может находиться либо в виде заготовки и нагреваться на возду¬
хе, либо подвергаться механической обработке до конечных размеров и по¬
следующему нагреву в аргоне или в вакууме. Затем образцы быстро охлаж¬
дают до комнатной или более низких температур и в случае необходимости
механически обрабатывают до требуемых размеров. Механическая обработ-
М.П. Сих285ка после термической обработки на воздухе удаляет окалину, а также слой
материала, насыщенный кислородом, продиффундировавшим по границам
зерен. Если необходимо измерить сегрегации, существовавшие при темпе¬
ратуре нагрева, то охлаждение должно быть достаточно быстрым, чтобы
ограничить перемещения сегрегирующих атомов. Разумеется, большинство
образцов можно хранить при комнатной температуре бесконечно долго.Для множества систем коэффициент объемной диффузии увеличивается в
10 раз при повышении температуры на каждые 50 °С. Таким образом, даже
в малых образцах, подвергнутых предварительной механической обработке
и закаленных в замороженном рассоле (обеспечивающем наиболее высокую
скорость охлаждения в металлургии), измерения сегрегаций в железе могут
быть проведены при температурах до 900 °С. Это ограничивает исследова¬
ния железа только а-фазой.Подготовленные образцы помещают затем в систему оже-спектромет-
ра, в которой создается сверхвысокий вакуум путем обезгаживания до
достижения герметичности (см. гл. 2)» Обезгаживание при 200 °С не вызы¬
вает перераспределения диффундирующих элементов замещения, но препят¬
ствует возникновению водородной хрупкости и обеспечивает полное пере¬
распределение элементов внедрения. После этого большинство образцов
охлаждают в жидком азоте и разрушают при ударном нагружении при этой
температурво Все материалы с о» Ц. к^-решеткой и некоторые с г. ц« к.-ре-
шеткой становятся хрупкими под действием высокой скорости ударной де¬
формации и низких температур. Затем с помощью сфокусированного элект¬
ронного луча, сканирующего по поверхности, получают карты распределе¬
ния концентрации соответствующего участка поверхности излома аналогич¬
но получению изображения в растровом электронном микроскопе» Карты
оже-электронов с пространственным разрешением около 1 мкм (исключение
составляют более дорогие приборы, в которых может быть достигнуто раз¬
решение 0,1 мкм) позволяют выбрать участки, представляющие интерес, с
тем чтобы произвести на них точный количественный анализ степени сег¬
регации элемента» При разрушении образцов требуется соблюдать осторож¬
ность, так как в некоторых сталях механизм разрушения изменяется от
вязкого (при температурах выше комнатной) до межкристаллитного в ин¬
тервале от 0 до - 40 °С и затем до разрушения сколом» Такие образцы
должны быть разрушены в соответствующем температурном интервале, а
не просто при предельно возможном охлаждении. При исследовании сварных
соединений в случае, когда образец разрушается вне области, представля¬
ющей интерес, можно использовать столик с приспособлением для разруше¬
ния при растяжении [ 32]» Для высокотемпературных исследований были
сконструированы столики с приспособлениями для высокотемпературного
растяжения [ 33]» Хрупкое разрушение стали можно вызвать, помещая обра¬
зец в водородные среды [34] или используя катодную зарядку образца [35].
2867. Оже-спектроскопия в металлургииВ последнем случае точные режимы зарядки зависят от исследуемого об¬
разца. В общем случае зарядка осуществляется при пропускании тока плот¬
ностью 20 мА/см2 между платиновыми электродами при комнатной темпе¬
ратуре в течение двух часов через 0,5 М-раствор H2S04 с добавкой 0,3 г/л
отравляющей примеси, например As203. Во многих случаях допускается
длительное хранение образцов с повышенным количеством водорода при
температуре жидкого азота, очень кратковременное хранение при ком¬
натной температуре и не допускается дегазация.Необходимо добавить несколько слов по поводу предосторожностей при
выполнении рассмотренных выше процедур. После разрушения поверхность
металла, например железа и вольфрама, обладает высокой реакционнойРис. 7.6. Внутри кристаллитный и межкристаллитный сколы в а-железе (растровый
электронный микроскоп, ширина снимка 0,75 мкм).
М.П. Сих287способностью и в случае недостаточно высокого вакуума углеродные и кис¬
лородные пики будут линейно увеличиваться со временем. Это может су¬
щественно затруднить прецизионные измерения содержания этих важных
элементов. Хлор может быть внесен в результате загрязнений пальцами,
азот - при попадании воздуха в систему. При наличии устройств для вос¬
произведения изображения с достаточной разрешающей способностью мож¬
но четко различить области внутрикристаллитного скола и межкристал-
литного разрушения, как показано на рис. 7.6« На тех и других об¬
ластях возможно появление различных загрязнений, тогда как сег¬
регирующие элементы могут располагаться только по границам зерен. Заг¬
рязнения попадают на поверхность излома не только из прибора, но и из
самого материала за счет диффузии по поверхности. Так, совершенно не¬
растворимые во многих металлах щелочные металлы и элементы, такие,
как свинец и висмут, образуют очень мелкие выделения практически чис¬
того металла. После разрушения металл из этих выделений, оказавшихся
на поверхности, может диффундировать по всей поверхности, в одинаковой
мере покрывая зоны скола и межзеренного излома и затрудняя точное
измерение сегрегаций [ 36]. Для устранения этого эффекта имеется только
одно средство: выдержка образцов во время анализа при температуре жид¬
кого азота<> Поверхностная диффузия играет очень важную роль при про¬
ведении экспериментов на столиках с устройством для высокотемператур¬
ного растяжения. В этом случае разрушение происходит медленно и обраг
зец может находиться при высоких температурах до начала охлаждения в
течение часа, почему обработка результатов всех измерений должна произ¬
водиться с большими предосторожностями.7.2J.2. Характеристика областей сегрегацииНа рис. 7.7 показан спектр оже-электронов от границ зерен ротора, выз¬
вавшего описанную выше аварию в Хинкли Пойнте. Ротор был в состоянии
отпускной хрупкости и в соответствии с количественным анализом, приве¬
денным в табл. 7.1, содержал по границам зерен 45% моноатомного слоя
фосфора. При рассмотрении такого спектра сразу же возникает ряд вопро¬
сов: а) как изменяется концентрация фосфора по глубине от границ зерен;
б) насколько представителен один спектр от границ зерен в стали; в) распре¬
деляется ли сегрегирующий элемент при разрушении поровну на каждую из
двух половин излома; г) сегрегируют ли еще какие-либо элементы из имею¬
щихся выделений, например карбидов, и если сегрегируют, активны ли они в
данном изучаемом процессе разрушения?Ответ на первый вопрос, касающийся локализации сегрегирующих эле¬
ментов относительно плоскости излома, может быть получен при травлении
ионным пучком поверхности излома в приборе. На рис. 7.8 показаны резуль¬
таты, полученные для многих металлических систем и соединений. Экспо-
2887. Оже-спектроскопия в металлургииРис. 7.7. Оже-электронный спектр межкристаллитной плоскости, показанной на
рис. 7.2. Представлена зернограничная сегрегация 0,45 моноатомного
слоя фосфора.ненциальный спад в характерном интервале 0,3 - 0,6 нм согласуется с рас¬
четными данными для распыления атомов, расположенных в самом наружном
атомном слое поверхности [37]. Таким образом, из рис. 7.8 ясно, что атомы
сегрегирующих элементов занимают у границы зерна зону шириной только
в один атом и связи именно этих атомов разрываются в процессе разрушения.
Анализ обеих половин излома показал, что в общем случае при разрушении
сегрегирующий элемент распределяется поровну по двум поверхностям из¬
лома [ 38], однако нельзя ожидать, что общее утверждение будет справедли¬
во во всех случаях. Действительно, как показали Джонсон и Смарт для шаро¬
видного графита в чугуне [ 39], при разрушении по межфазным поверхностям
раздела сегрегирующий элемент стремится остаться на поверхности с более
высокой реакционной способностью-Осторожность нужна при интерпретации профилей концентрации распы¬
ляемых элементов, которые могут находиться в виде выделений у границ
зерен, например пиков от хрома и молибдена в спектре на рис. 7.7. Часть
атомов хрома и молибдена может находиться в виде сегрегаций, а часть -
в составе дисперсных карбидов Сг23С6 и Мо 2С„ Важно знать концентрацию
сегрегирующих элементов. Эту информацию можно получить из профиля кон¬
центрации, показанного на рис. 7.9. Источниками остаточных сигналов пос-
М.П. Сих289Рис. 7.8*. Профиль концентрации элементов, сегрегирующих по границам зерен, по¬
лученный с использованием ЭОС и травления ионами аргона. Сплошная
кривая показывает ожидаемый результат для атомов, находящихся точно
в плоскости границы зерна.1 - S В Fe — 10_2%S; 2 - р в стали AISI 5140; 3- Sb в Fe-22%Sb;
4- S в стали AISI 5140; 5 - Sn в Fe -4%Sn; 6 - Bi В Cu -0,02%Bi ; 7 -
Y2°3 В A1203 - 1% Y203; 8 - Ca b MgO; 9 — К в W; 10- P в W; 11 -
Те В Cu.ле продолжительного распыления считаются крупные карбиды, а начальный
участок спада на кривых для хрома и молибдена, сопровождающий участок
спада на кривой для фосфора, указывает на концентрацию элемента в сегре¬
гации. Количественный анализ спектров [ 40] показал, что 7 и 5% площади
границ зерен покрыты дисперсными карбидами Сг23С6 и Мо2С соответст¬
венно, 17 и 12% площади соответственно заняты сегрегациями в виде моно-
атомных слоев хрома и молибдена» Однако при анализе не учитывалось
дифференциальное распыление, рассмотренное в гл. 4. Было показано
[ 41, 42], что состав поверхности материала стехиометрического состава в
результате селективного распыления меняется по экспоненте от исходной
2907. Оже-спектроскопия в металлургииРис. 7.9. Усредненный профиль концентрации для десяти границ зерен в роторной
стали 3 СгО,5Мо [40].до новой, измененной, концентрации* Изменение состава происходит при
распылении нескольких верхних атомных слоев и может иметь место как в
карбидах, так и во всех остальных материалах [ 43]. Все модели распыления
указывают на то, что металл обедняется карбидами, так что некоторые или
все показанные на рис* 7.9 кривые спада для хрома и молибдена могут быть
следствием селективного распыления. Четкое разделение селективного рас¬
пыления и сегрегаций в настоящее время невозможно; для этого необходи¬
мы эталонные кривые, снятые с разрушенных в высоком вакууме карбидов
стехиометрического состава.Анализ многих межзеренных поверхностей показывает, что сегрегирую¬
щие элементы располагаются в общем случае достаточно равномерно, и гра¬
ницы зерен в этом отношении ведут себя идентично [ 32, 44], как видно из
гистограмм на рис. 7.10, относящихся к сегрегациям в железе. Сюда же
включена и гистограмма распределения для стали, из которой был изготовлен
упомянутый выше ротор, разрушившийся в Хинкли Пойнте, Очень большое
количество границ зерен позволяет ожидать равномерного распределения
сегрегирующих элементов по границам зерен, при этом разрушение должно
М.П. Сих291Рис. 7.10. Гистограммы распределения зернограничных сегрегаций серы и олова вжелезе при 600 ЬС [ 32], висмута в меди при 700 °С [ 45] и фосфора в ста¬
ли ЗСг 0,5Мо при 450 °С [17].происходить по произвольным высокоэнергетическим границам у плоскостей
с высокими индексами. Однако в некоторых случаях наблюдаются большие
колебания уровня сегрегаций, и поскольку начало разрушения определенно
связывают с границами, имеющими наиболее высокую концентрацию сегреги¬
рующего элемента, эту концентрацию можно оценить путем подгонки дан¬
ных к распределению Вейбулла [46]. Такому распределению удовлетворяют
сегрегации висмута, как показано на рис. 7.10.Одной из причин возможного различия уровня сегрегаций на различ¬
ных границах является различная степень разупорядочения на этих грани¬
цах. Ватанабе, Китамура и Карасима [ 47] при очень тщательном изучении
системы железо - олово обнаружили, что при увеличении угла наклона гра¬
ницы от 0 до 60° уровень сегрегации увеличивается в 20 раз, однако мини¬
мума концентраций сегрегаций на границах совпадения не наблюдали. С дру¬
гой стороны, в системе железо - фосфор Сузуки, Абико и Кимура [ 48] вы¬
явили корреляцию между интенсивностью сегрегации и индексами поверх¬
ности разрушения, которая выражена более четко, чем корреляция между
интенсивностью сегрегации и полной разориентировкой границы. Таким об¬
разом, на большинстве неупорядоченных границ наблюдается наиболее высо-
2927. Оже-спектроскопия в металлургииРис. 7.11. Сегрегация фосфора в железе с 1% фосфора, а — расположение сегрегиру¬
ющих атомов (темные квадраты) в процессе разрушения; б- уровень сег¬
регации относительно ориентировки каждого зерна по отношению к его гра¬
нице [48]. Высота оже-пика для Р (% Fe)1 - менее 40; 2- 40- 50; 3- 50-60; 4 - более 60.кий уровень сегрегаций, и, более того, после разрушения сегрегирующий
элемент остается на поверхности с более высокими индексами, как видно из
рис. 7.11, а. На рис0 7.11, б показана анизотропия сегрегации на диаграмме
в полярных координатах плоскостей разрушения. Ясно, что на плоскостях
с низкими индексами уровень сегрегаций существенно ниже.7.2.2. Измерения на свободной поверхности
7с2*2.1 „ Нагрев образцаВ случае поверхностных сегрегаций измеряют как сегрегации легирую¬
щих элементов, так и сегрегации примесейо Для металлургов важны обе
группы элементов, тогда как исследователи в области металлических ката¬
лизаторов сосредоточивают внимание на сегрегации легирующих элементов*,
Большую часть измерений поверхностных сегрегаций производят методом
ЭОСо Небольшие образцы материала обычно помещают на нагретый столик
в прибор, очищают измеряемую поверхность ионным травлением и затем
после соответствующей термической обработки, проводимой в сверхвысо¬
ком вакууме прямо в приборе, проводят анализ» В НФЛ для нагрева образ¬
цов небольшого размера до 1000 °С использовали специальные легкие столи¬
ки, при этом потребление энергии составило всего несколько ватт. Такая
установка облегчает дегазацию вакуумной системы и позволяет поддержи-
М.П. Сих293вать высокий вакуум. Это важно, поскольку при загрязнении поверхностей
кислородом или углеродом равновесие между сегрегациями других элемен¬
тов может быть нарушено. Например, поверхность сплавов никель — золото,
которая обычно обеднена никелем, может обогатиться никелем в присут¬
ствии кислорода или водорода. Вторым преимуществом является то, что
под влиянием магнитных полей, связанных с использованием высоких энер¬
гий, в некоторых спектрометрах измерения должны проводиться на выклю¬
ченном из сети приборе. При использовании источников низкой мощности
образец охлаждается за несколько секунд, в частности достаточно быстрое
охлаждение от 850 °С позволяет сохранить сегрегации, существовавшие при
этой температуре»Еще одной проблемой, связанной с измерением поверхностных сегрега¬
ций, является испарение летучих элементов0 Измерения в условиях истинно¬
го равновесия требуют отсутствия испарения с поверхности. Из таблицы за¬
висимости давлений паров от температуры, приведенной Хонигом и Краме¬
ром [ 49], видно, что многие из представляющих интерес для исследователей
элементов, образующих сегрегации, также очень летучио Это, вообще гово¬
ря, ограничивает максимальные температуры измерения, позволяющие полу¬
чить значимые результаты, температурами ниже 850 °С. При более высо¬
ких температурах сегрегирующие элементы испаряются и их содержание в
объеме образца снижается до такой степени, что элемент может быть не об¬
наружен. Испарение можно уменьшить путем частичного заключения поверх¬
ности в оболочку или работы на дне щели [ 50]. Однако эти меры позволяют
увеличить рабочие температуры всего примерно на 50 °С0 Проверку испаре¬
ния легко осуществить, собрав эмитированные атомы на расположенную
поблизости холодную подложку, которая затем может быть проанализирова¬
на в оже-спектрометре.7.2.2.2. Характеристика области сегрегацииПрофили концентраций, как описано выше, показывают, что элементы,
сегрегирующие на поверхности, расположены на наружной атомной плоскос¬
ти. Поверхностным сегрегациям в двойных системах и на плоскостях мо¬
нокристаллов с низкими индексами посвящено гораздо больше работ, чем
зернограничным сегрегациям.» В недавно опубликованной работе Сиха [ 51]
содержится большое количество данных по двойным системам и рассмотрен
широкий круг подложек, включающий серебро, золото, медь, железо, ни¬
кель, палладий, платину и рутений с аналогичными сегрегирующими элемен¬
тами» Гистограммы разброса концентрации сегрегирующих элементов по
плоскости кристалла не опубликованы, так как в большинстве работ отме¬
чено равномерное распределение сегрегирующего элемента по поверхности»
Однако при подробном изучении сегрегации золота в никеле Джонсон и др.
2947. Оже-спектроскопия в металлургиитРис. 7.12. Ориентационная зависимость сегрегации золота на зернах никеля при 700 °С
и относительное расположение зерен на поверхности [52].[ 52] провели анализ сегрегаций и ориентировок поверхностей восемнадца¬
ти зерен, как показано на рис» 7012. Буквы соответствуют зернам, показан¬
ным в левой части рисунка, а цифры примерно пропорциональны уровню сег¬
регаций золота в зернах никеля на полярной диаграмме* Как и ожидалось,
распределение поверхностных сегрегаций, показанное на рис. 7012, являет¬
ся широким. Движущей силой сегрегации служит уменьшение поверхностной
энергии [ 20]. Таким образом, сегрегирующие элементы концентрируются
главным образом на таких плоскостях зерен чистого никеля, поверхностная
энергия которых может быть снижена в наибольшей степени в присутствии
атомов золота, удовлетворяющих требованиям по минимальной локальной
энергии разорванных связей» Такие условия возникают на плоскости (014),
где концентрация легирующего элемента максимальна.В данном разделе описана возможность использования ЭОС для общей
характеристики сегрегации элементов» Ниже рассматривается теория сег¬
регации, проиллюстрированная измерениями с помощью ЭОС»7.3. Теория сегрегацииВ последние десятилетия в связи с увеличением массы данных, полу¬
ченных с помощью ЭОС и доступных для обработки, быстро развиваются
М.П. Сих295теории сегрегации. Теории адсорбции на поверхностях раздела твердых тел,
а также на поверхности твердого тела в вакууме являются либо прямыми
аналогами, либо развитием теорий, хорошо известных в области адсорбции
газов на свободных поверхностях твердых тел» В данном разделе различные
теории будут представлены последовательно по мере усложнений условий
адсорбции. Эти теории описывают равновесное состояние, однако, поскольку
в практических ситуациях процесс лимитируется диффузией, представлена
также кинетика процесса сегрегации.7.3.1. Теория поверхностных и зернограничных сегрегаций
в двойных системах Ленгмюра - МаклинаВ самой первой теории, специально разработанной для границ зерен,
Маклин [ 18] предложил модель, в которой растворенные атомы располага¬
лись по границе зерна и в кристаллической решетке в зависимости от раз-Рис. 7.13. Зависимость Ленгмюра — Маклина для зернограничных сегрегаций кислоро¬
да в молибдене [53]. Т - температура закалки. Общее содержание кислоро¬
да 6 — 9 Ю^6 лежит в пределах растворимости в твердой фазе, а свободная
энергия сегрегации AG равна (37414 + 65,2 Т) Дж/моль.
2967. Оже-спектроскопия в металлургииности энергии AG, являющейся свободной энергией сегрегации. Используя
простую статистическую механику, он обнаружил, что энергия системы ми¬
нимальна при частичном покрытии границ зерен моноатомным слоем сег¬
регирующего элемента Хь, гдеЗдесь X® - доля покрытия границы зерна моноатомным слоем сегрегирую¬
щего элемента при насыщении, Хс - объемная молярная доля растворенно¬
го элемента. Были опубликованы данные о том, что в нескольких системах
со свободными поверхностями результаты измерения сегрегаций соответст¬
вуют уравнению (7.4) [ 17], но наилучшей иллюстрацией теории являются дан¬
ные Кумара и Эйре [ 53] по зернограничной сегрегации кислорода в молиб¬
дене, представленные на рис. 7.13.7.3.2. Изменение свободной энергии зернограничных
сегрегаций дGgfe в двойных системахЗначения ЛGgb были оценены Маклином [18] по энергии упругой дефор¬
мации, высвобождаемой при сегрегации растворенных атомов. В этом
случае для получения корректных значений введен поправочный коэффици¬
ент, равный 2, однако большей точности в оценке AGg$ достигли Сих и
Хондрос [54]. Используя теорию газовой адсорбции Брунауэра, Деминга и
Теллера, известную как усеченная теория БЭТ, они предложили соответству¬
ющее уравнение для твердого состояния [ 54]:где растворимость в твердом состоянии Х°с является важным параметром,
для которого имеется много данных в справочниках по металлургии. При
минимальных значениях растворимости, когда Xg составляет моноатомный
слой, можно записатьгде К = ехр (— ЛG/RT) и $ gb - коэффициент обогащения границ зерен. Урав¬
нение (7.6) очень хорошо описывает совокупность экспериментальных данных,
полученных с помощью ЭОС и других методов, которые показывают, что
М.П. Сих297Рис. 7.14. Кривая расчетных значений коэффициента зернограничного обогащения [40].Pg6 зависит от Xj для растворимостей в интервале от 10~2 до 100%, Из
совокупности этих данных, представленных на рис. 7.14, очевидно, что(7.7)Справа на рис. 7.14 приведены значения $gb, полученные для двойных и бо¬
лее сложных сплавов, для которых значение Х°с неизвестно. Из приведенных
данных можно сделать очень важный вывод, что сегрегации фосфора, олова
и сурьмы в легированных сталях примерно на порядок величины больше, чем
в чистом железе* Этот вопрос будет еще раз рассмотрен ниже.
2987. Оже-спектроскопия в металлургииТаким образом, усеченная теория БЭТ является мощным инструментом
для расчета сегрегаций в двойных системах, и видно, что изменение свободной
энергии зернограничной сегрегации AGg& в интервале от 0 до -100 кДж/моль
соответствует (AGSol-9± 5) кДж/моль. Для двойных систем разрабо¬
тано много других теорий адсорбции [ 54], но усеченная теория БЭТ пред¬
ставляется наилучшей для их расчетов»7.3.3. Изменение свободной энергии сегрегации на
поверхности в двойных системахИзменение свободной энергии сегрегации на поверхности пытались
рассчитать различными способами: по энергии связи [55-57] и по высвобо¬
ждению энергии деформации [ 56 -58]. При использовании первого метода
получаем AG = у$ -у^, где у^ - поверхностная энергия (в килоджоулях
на моль) атомов чистой матрицы В, а у^ - поверхностная энергия чисто¬
го сегрегирующего элемента Л. Для металлов в твердом состоянии поверх¬
ностная энергия связана с температурой плавления, так что AG можно оце¬
нить по разности температур плавления сегрегирующих атомов и атомов
матрицы и .Используя этот подход, Сих [ 51] с помощью регрессионного анализа
сопоставил экспериментальные данные для многих систем и вывел уравне¬
ние для расчета коэффициента поверхностного обогащения объединив
параметры, получаемые из теории поверхностной энергии, и энергии, вы¬
свобождаемой при деформации(7.8)где Q - молярный параметр идеальных растворов двойной системы А В, аа А и ав ~ атомные размеры атомов А и В, рассчитанные, как описано в
гл. 5, в соответствии с уравнением (5.8). Параметр М равен единице для
а а > а в и НУЛЮ ал < а в* Первый член представляет влияние энергии
разрыва связи и является основным членом для систем сегрегирующих
элементов, в которых размеры атомов отличаются менее чем на 10%. Вклад
второго члена, учитывающего химическое взаимодействие Л и В, невелик.
Последний член учитывает высвобождение энергии деформации, и его вклад
существен только тогда, когда размер атомов сегрегирующих элементов
больше, чем атомов матрицы, но не в обратном случае, поскольку тогда
деформация всегда мала [ 59]. Уравнение (7.8) хорошо согласуется с экспе¬
риментом и позволяет рассчитать AGS со стандартным отклонением
9 кДж/моль в интервале изменений свободной энергии сегрегации на поверх¬
ности от -80 до ?0 кДж/моль. На рис. 7.15 показана корреляция расчетных
и экспериментальных величин.
М.П. Сих299Рис. 7.16. Соотношение между расчетными и экспериментальными коэффициентами
обогащения поверхностных сегрегаций L 51].7.3.4. 'Конкуренция мест в кристаллической решетке
в простых тройных системахОпубликовано большое количество данных по измерениям в тройных и
более сложных системах, однако немногие из них в полной мере характери¬
зуют явление конкуренции мест в кристаллической решетке. Первый анализ,
проведенный Сихом и Ли [ 50], показал, что для уровня сегрегаций менее
одного моноатомного слоя может иметь место простая линейная конкурен¬
ция мест в решетке. Элементы с сильно выраженной склонностью к сегрега¬
ции могут смещать элементы со слабо выраженной склонностью к сегрега¬
ции примерно в соотношении атом на атом. Такая конкуренция была впер¬
вые продемонстрирована для олова и серы на железной подложке и позднее
для сурьмы [ 61] и кислорода [ 62] с серой. Во всех этих случаях, если эле¬
мент, смещаемый серой, сильно сегрегирует в отсутствие серы до степени,
3007. Оже-спектроскопия в металлургииРис. 7.16. Конкуренция мест в решетке между кислородом и серой на поверхностижелеза, иллюстрирующая случаи с коэффициентами замещения кислородом
серы в соотношении 1 : 1 и 2 : 1 [ 62].близкой или превышающей один моноатомный слой, начальное смещение
происходит в большем соотношении, чем смещение атом на атом,до образо¬
вания 25% моноатомного слоя серы, после чего процесс идет, как описано
выше. Это проиллюстрировано на рис. 7.16.Конкуренции мест в решетке, в том виде, как она описана выше, не сле¬
дует ожидать в случае сегрегаций по границам зерен, поскольку даже при
сегрегации в виде полного моноатомного слоя возможна дальнейшая сегре¬
гация растворенных атомов, которая снижает энергию деформации и энер¬
гию разорванных связей атомов матрицы просто вследствие наличия атомов
матрицы на обеих поверхностях раздела Эти избыточные места в решетке по
сравнению со свободной поверхностью делают конкуренцию менее вероятной,
и для систем олово - сера и кислород - сера конкуренции мест в решетке
по границам зерен не наблюдали вообще [32, 63]. Конкуренцию мест в решет¬
ке по границам зерен наблюдали между азотом и серой в железе [64], но
не при всех условиях [63]. Более важным является наблюдаемое в [ 65] замеще¬
ние фосфора углеродом. Это очень существенно с точки зрения проблем
разрушения и будет ниже обсуждено более подробно.7.3.5. Более сложные двойные системыВ рассмотренных выше теориях принято, что сегрегирующие атомы не
взаимодействуют между собой. Если допускается, что существует энергия
взаимодействия со между двумя соседними адсорбированными атомами, так
М.П. Сих301что они притягиваются (со отрицательна) или отталкиваются (со положительг
на), то может быть выведено уравнение для твердого состояния аналогично
теории адсорбции Фаулера [ 66]:(7.9)Если со = 0, то эта теория сводится к теории Ленгмюра и Маклина. Однако
по мере увеличения отрицательного значения со темп ускорения сегрегации
с понижением температуры увеличивается до тех пор, пока в конечном ито¬
ге при определенной температуре рост сегрегаций не становится прерывис¬
тым. Это изменение поведения и данные по измерениям сегрегации селена
и теллура в железе методом ЭОС показаны на рис. 7.17 [67]. Для ZlW ниже
-4RT форма кривых становится S-образной, как показано для случая
Z jco= — 6ft Т- В этом случае сегрегация не соответствует S -образному
решению уравнения (7.9), а перемещается с остановками от А до В. Как это
происходит, видно из кривой G в функции от Хъ, где минимум при высокой
степени покрытия имеет в конечном итоге более низкую абсолютную энер¬
гию, чем при низкой степени покрытия [68].Рис. 7.17. Зернограничные изотермы Фаулера и экспериментальные результаты Пи»
чарда и др. [ 67] для Se и Те в железе [54].
3027. Оже-спектроскопия в металлургии7.3.8. Сегрегация в тройных системахВ 1975 г. Гутман [69] распространил теорию Фаулера на случай взаи¬
модействия совместно сегрегирующих элементов в многокомпонентных сис¬
темах. Эта теория, важная для объяснения поведения сегрегаций, приводяще¬
го к межкристаллитному разрушению конструкционных материалов, даетгде Х^и Х£2 - молярные доли моноатомных слоев сегрегаций примесей и
легирующих элементов, объемное содержание которых обозначено Хс 1 и Хс 2
соответственно. Кроме того,где AGJ и AG§ - изменения свободной энергии сегрегации примеси и леги¬
рующих элементов в матрице. Коэффициент взаимодействия а}2 относится
к изменению энергии связи с ближайшим соседом при формировании связи
легирующий элемент - примесь и получается из измерений влияния легирую¬
щих элементов на растворимость примесей [ 70]. Если величина а [2 отрица¬
тельна, сегрегация легирующих элементов способствует сегрегации приме¬
сей. Аналогично результату Фаулера, показанному на рис. 7.17, по мере
того как величина а/2 становится более отрицательной, форма кривой
приближается к S -образной и в конце концов представляет область, в кото¬
рой уровень сегрегации прерывисто увеличивается от низкого до высокого.
Общее влияние взаимодействия Гутмана можно видеть на рис. 7.14. Введе¬
ние легирующих элементов с отрицательной величиной а [ 2 уменьшает раст¬
воримость примесного элемента. Одновременно, согласно уравнениям(7.10) и (7.11), уровень сегрегаций растет. Таким образом, введение леги¬
рующего элемента приводит к тому, что исходная точка двойной системы на
рис. 7.14 перемещается по диагонали вправо вверх, оставаясь, однако, в пре¬
делах общей корреляции [71].В общем эта модель широко принята в настоящее время и позволяет точ¬
но описать некоторые явления, наблюдаемые при охрупчивании. Увеличение
свободной энергии сегрегации элементов одного типа под влиянием элемен¬
тов другого типа, описанное уравнением (7.11), было подтверждено тщатель¬
но проделанными экспериментами Дюмулена и Гутмана [ 72] по сегрегациям
на поверхности в стали, как видно из их данных, приведенных на рис. 7.18.Исходная теория Гутмана была видоизменена Сихом [73], исключившим
конкуренцию мест, не наблюдавшуюся на практике. В уравнении (7.10) сум-(7.10)(7.11)
М.П. Сих303Рис. 7.18. Увеличение свободной энергии поверхностной сегрегации легирующих эле¬
ментов в стали в зависимости от сегрегации углерода и азота L 72J.ма j членов просто заменена *-м членом. Это упрощает расчеты и лучше
описывает экспериментальные результаты. Ряд более сложных теорий под¬
робно описан Гутманом и Маклином [ 71] и Гутманом [ 74].7.3.7. Кинетика сегрегацииВ практических случаях, когда сегрегация играет важную роль, часто
атомы сегрегирующих элементов не имеют достаточно времени, чтобы дос¬
тичь равновесного уровня, определяемого теориями адсорбции. В настоящем
разделе проанализирована кинетика сегрегации.Большинство моделей кинетики основаны на подходе Маклина [18]. При¬
нимается, что растворенные атомы сегрегируют по границе между двумя
бесконечными полукристаллами с равномерным содержанием растворенного
элемента или по поверхности одного бесконечного полукристалла. Диффузия
в кристаллах описывается законами Фика, а отношение содержания раство¬
ренного элемента на границе зерна к содержанию в прилежащем атомном слое
объема дается постоянным коэффициентом обогащения (3, получаемым из
уравнения (7.6). Кинетика сегрегации, таким образом, описывается выраже¬
нием
3047. Оже-спектроскопия в металлургиигде F = 4 для границ зерен и 1 для свободной поверхности, Xb(t ) - содер¬
жание элемента на границе зерна в момент t,D- коэффициент объемной
диффузии растворенного элемента, / - величина, связанная с размерами
атомов растворенного элемента и матрицы b и а соответственно соотноше¬
нием / = а36~2. Для малых времен процесса уравнение (7.12) аппроксими¬
руется выражениемУравнения (7.12) и (7.13) описывают в действительности предельные
случаи общей задачи. На практике (Ь является единственной константой для
разбавленных систем с низким уровнем сегрегаций. По мере протекания сег¬
регации величина р обычно снижается в результате насыщения. Если началь¬
ное значение р велико и быстро снижается при достижении сегрегацией насы¬
щения, уравнение (7.13) применимо вплоть до состояния насыщения [75, 76].
Подробный анализ насыщения в рамках адсорбционной теории Ленгмюра - Мак¬
лина приведен Роуландсом и Вудрафом [ 77]. Их анализ, интерпретация кото¬
рого дана на рис. 7.19, показывает, как временная зависимость сегрегации
изменяется от уравнения (7.12) к уравнению (7.13) по мере приближения рав¬
новесного уровня сегрегации ^ («,) к уровню насыщения Х°у(7.13)Рис. 7.19. Кинетика сегрегации в двойных системах с различной степенью насыщения
при достижении равновесной концентрации [ 77].
М.П. Сих305Более сложная проблема кинетики сегрегации в тройных системах изуче¬
на Тайсоном [78]. Тайсон использовал подход Маклина, включив теорию
Гутмана для определения концентрации по границам зерен. Он обнаружил, что
применение уравнения (7.13) для тройной системы с низкой концентрацией
примеси и высокой концентрацией легирующего элемента часто дает хорошие
результаты, если конечный уровень сегрегаций близок к насыщению, а при¬
менение уравнения (7.12) - при низком конечном уровне сегрегаций. Однако
для некоторых критических случаев, в которых два типа взаимодействующих
элементов имеют аналогичные значения D$~2, на кривых, описывающих сег¬
регации, возникает промежуточное плато.Кинетику рассасывания сегрегаций в прошлом описывали как процесс,
протекающий гораздо быстрее, чем образование сегрегации. Это неверно, и
в таком случае применимы уравнения (7.12) и (7.13), где члены Хъ (0) и
X ь (оо) меняются местами.Все сказанное выше справедливо для полностью закрытых поверхностей
или для границ зерен. При экспериментальном изучении поверхностных сег-
регаций в вакуумной системе методом ЭОС возможно испарение сегрегирую¬
щего элемента. Ли и Сих [75] оценили влияние испарения с поверхности,
сопутствующего сегрегации на поверхности, как показано на рис. 7.20. Вна¬
чале не наблюдается отклонения от результата, полученного Маклином при
решении уравнения (7.12). Однако как только сегрегация завершается, ско¬
рость испарения увеличивается и материал в объеме обедняется растворен¬
ным элементом. Сегрегация достигает максимума и затем необратимоРис. 7.20. Временная зависимость сегрегации на поверхности с различной скоростью
испарения, описываемой параметром Е [75].
3067. Оже-спектроскопия в металлургииуменьшается до низких значений по мере очистки подложки. Цифры у кривых,
соответствующие значениям Е, пропорциональны скорости испарения сег¬
регирующего элемента. Иллюстрацией может служить сегрегация олова на
поверхности сплава Fe - 0,22% Sn при 700 °С, которая представляет собой
случай слабой сегрегации с Е = 0,007, а пик сегрегации достигается через
15 мин [75].7.4. Влияние материала на сегрегациюСегрегации растворенных элементов на поверхности и по границам зерен
в твердых веществах образуют зоны дискретного состава с дискретными
свойствами, определяющими эксплуатационные характеристики всего мате¬
риала в целом аналогично тому, как цемент определяет прочность кирпич¬
ных сооружений. Многие свойства конструкционных материалов существен¬
но зависят от влияния сегрегации на энергию межфазных поверхностей,
сцепление и диффузию. Для специфических случаев разрушения важны и
другие свойства, но Сначала рассмотрим влияние сегрегации на основные
свойства материалов.7.4.1. Сегрегации и основные параметры материалаВлияние сегрегации на поверхностную и межфазную энергии в настоя¬
щее время хорошо изучено [20, 21]. Объединение адсорбционной теории
Гиббса [21] с уравнениями (7.4) и (7.5) дает уравнение [49, 79](7.14)где уъ и - межфазная или поверхностная энергия в материале, имеющем
сегрегации, и в чистом материале соответственно, а - концентрация сег¬
регирующего элемента (в моль/м2) в одном атомном слое. Это соотношение,
многократно подтвержденное экспериментом, показывает, что сегрегация
атомов снижает поверхностную энергию, при этом энергия межфазной поверх¬
ности снижается в тем большей степени, чем более поверхностно активным
является сегрегирующий элемент. Объединяя теорию Борисова, Голикова и
Щербединского [ 80] с уравнением (7.14), Бернардини и др. [81] вывели урав¬
нение для коэффициента самодиффузии Dgb \(7.15)где ®gb ~ коэффициент диффузии на границе с сегрегирующим элементом,
D0 - коэффициент диффузии на чистой границе и Ъ.. - имеющий малуюgoвеличину член, учитывающий влияние растворенного элемента на коэффици-
М.П. Сих307ент объемной диффузии Dv, определяемый из уравнения(7.16)Из уравнения (7.15) видно, что устранение разупорядочения, внесенного
сегрегацией растворенного элемента, уменьшает пути быстрого переноса,
обычно связанные с границами зерен. Эксперименты показывают, что коэф¬
фициент самодиффузии на границах зерен может быть меньше на порядок
величины и что аналогичный, но более сильный эффект наблюдается при диф¬
фузии по границам зерен растворенных элементов. В прошлом измерение
диффузии по границам зерен всегда производили методом меченых атомов,
однако, как показали Бернардини, Ли и Хондрос [82], в настоящее время эти
результаты для всех систем можно получить, измеряя с помощью ЭОС перенос
растворенного элемента через тонкие фольги.Влияние легирующих элементов на сцепление между зернами было пред¬
метом многих исследований и противоречивых суждений, здесь не рассматри¬
ваемых. В основном мнения расходятся относительно утверждения, с одной
стороны, что сегрегирующие атомы занимают дыры в границах, запирая элект¬
роны металлической связи и, следовательно, ослабляя ранее существовав¬
шую связь металл - металл [ 83], с другой стороны, что сегрегирующий атом
замещает атом матрицы, заменяя связь металл - металл более слабой
связью металл - сегрегирующий атом [ 84]. В приближении идеальных раст¬
воров этот последний подход согласуется с термодинамической теорией
[ 85] и позволяет сделать вывод, что именно энергия связи атомов на едини¬
цу площади определяет способность сегрегирующих атомов ослаблять или
упрочнять границы зерен. Все эти члены могут быть рассчитаны по данным
Халтгрена и др. [ 86]. На рис* 7.21, а показано рассчитанное таким образом
относительное влияние различных элементов, сегрегирующих в железе.По ординате отложены относительные значения, но они примерно в 800 раз
выше влияния моноатомных слоев сегрегированных элементов на идеальную
работу разрушения, отнесенную к проценту моноатомного слоя. Из рис. 7.21, а
ясно, что фосфор, олово, сурьма и мышьяк способствуют охрупчиванию ста¬
ли, тогда как углерод и молибден уменьшают это воздействие. Имеющиеся
экспериментальные результаты [32, 64, 87 - 90] о влиянии сегрегирующих
элементов по отношению к фосфору проанализированы Сихом [91], как по¬
казано на рис. 7.21, б» Эти результаты очень хорошо подтверждают первоначальные
расчеты, за исключением влияния углерода, который действительно ослаб¬
ляет охрупчивающее действие, но в меньшей степени, чем предполагали
раньше.Расчеты для элементов, сегрегирующих в железе, могут быть обобще¬
ны для всех матриц в приближении идеального раствора. На диаграмме, по¬
казанной на рис. 7.22, по оси ординат отложена плотность энергии связи.
Элементы, находящиеся на диаграмме ниже каких-либо других элементов,
3087. Оже-спектроскопия в металлургииРис. 7.21. Относительное влияние эквивалентных концентраций элементов, сегреги¬
рующих в железе, на повышение вязкости и охрупчивание: а - расчетные
значения [73], б — результаты измерений относительно фосфора [ 91].С — L65J; Си - [87]; Мо - [ 8в] (Сих, не опубликовано); S - [ 32],
Si, Sn — [90]; Sb,Sn - [89].в случае сегрегации снижают сцепление по границам зерен этих элементов
приблизительно пропорционально вертикальному расстоянию между положе¬
нием этих элементов на рисунке. Таким образом, с этими данными хорошо
согласуется наблюдаемое охрупчивание меди висмутом [ 92], вольфрама
калием [93] и никеля серой [94]. Эта диаграмма, в частности, предсказывает
улучшение пластичности тройные сплавов платины с родием и вольфрамом при
добавке бора [95].
М.П. Сих309Рис. 7.22. Общая диаграмма охрупчивание - вязкость для матрицы и сегрегирующих
элементов в приближении идеального раствора 173J- Например, при исполь¬
зовании железа в качестве матрицы все точки, расположенные выше пунк¬
тирной линии, повышают вязкость, а расположенные ниже — охрулчивают,
степень развития эффекта зависит от расстояния от точек до линии.7.4.2. Отпускная хрупкостьАнализ сцепления межфазных поверхностей, рассмотренный выше, явля¬
ется Центральным при решении проблемы межкристаллитного низкотемпераг
турного разрушения. Наиболее ярким примером этого явления на практике
служит охрупчивание низколегированных сталей при их нагреве в интервале
температур 450-550 °С, что привело к появлению термина "отпускная хруп¬
кость”. Анализ причин многочисленных поломок в промышленности показал,
что фосфор, содержание которого в промышленных сталях обычно составляет
0,008 - 0,016%, вызывает разрушение хромомолиоденовых сталей, а комби¬
нация фосфора с оловом - разрушение никель-хромомолибденовых сталей
[96].При испытании сталей на ударный изгиб в интервале температур от
-100 до + 100 °С обычно обнаруживают, что при высоких температурах
стали являются вязкими, а при низких - хрупкими. Температура перехода,
которая должна быть возможно более низкой, увеличивается приблизительно
линейно при образовании сегрегаций, если это приводит к межкристаллит-
ному разрушению. Таким образом, измеренные температуры вязкохрупко-
го перехода для стали S АЕ 3140, содержащей 0,015% фосфора, показанные
на рис. 7.23, отражают степень сегрегации фосфора. Расчеты сегрегации
3107. Оже-спектроскопия в металлургииРис. 7.23. Диаграмма время — температура, показывающая охрупчивание сталиSAE 3140, содержащей 0,015% фосфора [97]. Скорректированная темпе¬
ратура вязкохрупкого перехода, ° С:1) -60; 2) -55; 3) -50; 4) -45; 5) -40; б) -35; 7) -30; 8) “25;9) —20; 10) -15; 11) -10; 12) -5; 13) -0; 14)+ 5; 15) +10; 16) +15;17) +20; 18) +25; 79) +30.фосфора в этой стали с использованием теории сегрегации в тройных систе¬
мах Гутмана и кинетического уравнения Маклина показали очень хорошее
согласие между кривыми с постоянным уровнем сегрегации и кривыми с
постоянной температурой перехода. Поэтому возможно распространение
полученных данных на периоды до 30 лет, как показано на рис, 7.24 [73]. На
практике при таком распространении необходима некоторая осторожность,
так как по истечении длительного времени твердость стали обычно снижа¬
ется и пластичность повышается. Изменяется также перераспределение ле¬
гирующих элементов между карбидами и твердым раствором в матрице; при
этом сегрегация и охрупчивание усиливаются или ослабляются по сравнению
с рассчитанными простыми методами [ 88, 98]0Теория Гутмана была подтверждена недавно опубликованным обзором
его работ, охватывающих широкий круг проблем [ 74 ], и работой Макмаго-
на и сотр., в которой показано, что никель и хром усиливают сегрегацию
фосфора [99], сурьмы [ 100] и олова [ 101]. Однако в работах Эрхарта и
Грабке [ 65] и Брайанта [ 102] высказывается совершенно противоположная
точка зрения. При изучении серии сплавов железа с углеродом, фосфором
и хромом, введенными по отдельности и в комбинации, Эрхарт и Грабке по¬
казали, что добавка хрома к сплаву железа с фосфором не влияет на сег-
М.П. Сих311Рис. 7.24. Диаграмма сегрегации, соответствующей рис. 7.23 [ 73].регацию фосфора, тогда как добавка углерода ослабляет сегрегацию фосфо¬
ра почти в пять раз. Это обусловлено непосредственной конкуренцией мест
в решетке, при которой сумма долей моноатомных слоев сегрегирующих
фосфора и углерода остается постоянной» Добавка хрома в сплав железо -
углерод - фосфор запирает углерод в карбидных выделениях, так что сег¬
регация углерода подавляется, а сегрегация фосфора достигает более вы¬
сокого уровня, близкого к уровню в сплаве железо - фосфор. Таким обра¬
зом, добавка хрома усиливает сегрегацию фосфора не за счет формирова¬
ния прямых связей, а вследствие конкуренции между углеродом и фосфором
за занятие мест в решетке. Сегрегации, аналогичные наблюдаемым в спла¬
ве железо - хром - углерод - фосфор, были обнаружены Брайантом [102]
в хромоникелевой стали. Кроме того, Брайант нашел, что увеличение содер¬
жания хрома, марганца и никеля не влияет на сегрегацию фосфора. Очевид¬
но, что необходимо продолжение работ в данной области для выяснения этих
противоречий и понимания проблемы отпускной хрупкости. С практической
точки зрения эта проблема до некоторой степени разрешена за счет исполь¬
зования более совершенных процессов выплавки стали и более чистых руд,
позволяющих удалить фосфор и олово, или применения активных добавок,
которые, как показали Сих, Спенсер и Хондрос [ 103], полностью нейтрализу¬
ют влияние примесей.7.4.3. Коррозионное растрескивание под напряжениемМежкристаллитное разрушение под действием коррозии под напряжени¬
ем является не таким простым явлением, как непосредственное нарушение
сцепления зерен при развитии отпускной хрупкости. В данном случае воз¬
можно образование сегрегаций, которые не влияют на ударную вязкость
или пластичность, однако вызывают разрушение в процессе эксплуатации
в некоторых электролитах. Ли и Хондрос [104, 105], например, показали,
что определенные сегрегирующие элементы снижают время до разрушения
низкоуглеродистых сталей, работающих под нагрузкой в нитрате аммония,
3127. Оже-спвктроскопия в металлургиии не влияют в случае работы в парафине. Относительное влияние сегрегирую¬
щих элементов пропорционально разности между табулированными значения¬
ми равновесного электродного потенциала каждого сегрегирующего элемен¬
та и железа. Считают, что механизмом разрушения является анодное раст¬
ворение у вершины трещины, которому способствует образование гальва¬
нического элемента между атомами сегрегирующего элемента и матрицы.
Одни атомы способствуют растворению других в зависимости от электрод¬
ного потенциала относительно железа. Обнаружено, что вредными элемен¬
тами являются расположенные в порядке убывания их активности кальций,
фосфор, сера, алюминий, медь, никель, олово, сурьма, мышьяк и цинк.7.4.4. Растрескивание при отжиге для снятия напряжений
и охрупчивание при ползучестиТакого рода разрушение наблюдается в ползучестойких сталях в усло¬
виях нагружения при рабочих температурах. Охрупчивание при ползучести
происходит при низких напряжениях в процессе эксплуатации, а при отжиге
для снятия сварочных напряжений в зоне термического влияния сварных
соединений могут образоваться трещины. Межкристаллитное разрушение
часто вызвано образованием пор по границам зерен, которые зарождаются
и растут с течением времени в условиях воздействия повышенных темпе¬
ратур и напряжений до тех пор, пока оставшееся сечение материала не
уменьшится до такой степени, что умеренные нагрузки вызовут разрушение.На основе теории Скелтона [ 106], показавшего, что деформация при
разрушении е t в условиях образования пор описывается уравнениемгде N -скорость зарождения поры, Сих [23] предположил, что примеси могут
быть ответственными за охрупчивание, действуя в двух не связанных между
собой направлениях: а) путем сегрегации на поверхности в направлении к
поверхности зародышевой поры, уменьшая поверхностную энергию и увели¬
чивая N, и б) путем сегрегации по границам зерен, уменьшая Dgb, как по¬
казано в уравнении (7.15). В обоих случаях деформация при разрушении
уменьшается и соответственно усиливается охрупчивание. Проблема зарож¬
дения пор была недавно изучена на этой основе более подробно [107] и по¬
лучены новые доказательства влияния поверхностных и зернограничных сегре¬
гаций фосфора и олова на разрушение низколегированных сталей в резуль¬
тате образования пористости по границам зерен [ 108, 109],
М.П. Сих3137.5. ЗаключениеКак видно из рис. 1.2 гл. 1,ЭОС оказывает сильное влияние на решение
многих технологических проблем. Так, в металлургии решаются давно на¬
зревшие задачи, разрабатываются новые материалы и процессы. В этой главе
проанализирована классическая проблема адсорбции и определено ее влияние
на широкий круг вопросов, связанных с разрушением материалов. Использо¬
вание сканирующей ЭОС привело к пониманию атомных механизмов процессов
разрушения [ 110]: будь то влияние сегрегирующих элементов на прочность
связи, электрохимию, энергию поверхностей раздела или перенос материа¬
ла. Это понимание позволило производителям и потребителям металла вы¬
работать эффективные предупредительные меры, например вводить добавки
[ 103, 111], как описано выше, или изменять содержание легирующих элемен¬
тов в сплаве.Литература1. Stout D.A., Gavelli G.f Lumsden Staehle R,W.9 Appl. Surf. Anal.ASTM STP, 699, 42 (1980).2. Conner G.R. Appl. Surf. AnaL, ASTM STP, 699, 54 (1980).3. Mathieu J*B., Mathieu H*J; Landolt D-, Je Electrochem. Soc., 125, 1039
(1978),4. Lea C„ Met, Sci,, 13, 301 (1979),5. Bullock Eo, Lea G, McLean M<, Met Sci., 13, 373 (1979),6. Grabke Paulitschke W*t Tauber G-, Viefhaus H. Surf. Sci., 63, 377
(1977).7. Grabke H»J* Mater. Sci. Eng., 42, 91 (1980).8. Lea C„ Proc. Eighth Int. Vac. Congr*, Vol. II, Vacuum Technology and
Vacuum Metallurgy, p* 468, Cannes (1980); Le Vide les Couches Minces,
1201, Suppl., 468 (1980).9. Edmonds D<V<, Jones P*N*, Met* Trans,, 10A, 289 (1979).10. Waters R.E., Charles JtA., Lea C., Met. Technol., 8, 194 (1981).11. Stoddart C*T.H., Lea C*, Dench W.A., Green P., Pettit H.R., Met. Technol.,
6, 176 (1979).12. Mathieu H*J; Landolt D., Schumacher ft., Wear, 66, 87 (1981).13. Schumacher ft., Gegner £., Schmidt Л., Mathieu H*]*t Landolt D., Tribology,
13, 311 (1980).14. Lea C., Roebuck ВMet. Sci., 15, 262 (1981).15. Kalderon D., Proc. Inst. Mech. Eng., 186, 341 (1972).16. Seah M.P., Surf. Sci., 80, 8 (1979).17. Hondros E*D*, Seah M*P*, Int. Met. Revs,,22, 262 (1977).18. McLean D., Grain Boundaries in Metals, Oxford University Press, London,
1957.
3147. Оже-спектроскопия в металлургии19. Arnold J*0*, J. Iron Steel Inst., 45, 107 (1894).20. Hondros E.D*, McLean D%, Monograph 28, Society of Chemical Industry, Lon¬
don, 1968*21. Hondros E*D, in: Proc. Melbourne Conf. on Interfaces (ed. R.C. Gifkins),
Butterworths, 1969.22. Beevers C*J*, Met. Sci., 14, 418 (1980).23. Seah M*P*, Phil. Trans. Roy. Soc., A295, 265 (1980).24. Nicholas M*G., Old C*F*, J. Mater. Sci., 14, 1 (1979).25. Woodward Burstein G*T*, Met. Sci., 14, 529 (1980).26. Lea C*, Seah M*P*, Scripta Metall., 9, 583 (1975).27. Suzuki H*G*, Ono M*, Trans. Iron and Steel Institute of Japan, 12, 251 (1972).28. Seah M.P*, Surf. Sci., 53, 168 (1975).29. Davis L.E., MacDonald NX*, Palmberg P.W., Riach G.E., Weber R*E*,
Handbook of Auger Electron Spectroscopy, 2nd ed., Physical Electronics
Industries Inc., Minnesota, 1976.30. Seah M*P*, Dench W*A. Surf. Interface Anal., 1, 2 (1979).31. Ichimura S., Shimizu R. Surf. Sci., 112, 386 (1981).32. Seah M.P*, Hondros E.D., Proc. Roy. Soc. London, A335, 191 (1973).33. Walsh J.M*, Gumz K*P., Anderson N.P., Quantitative Surface Analysis of
Materials, ASTM STP, 643. 72 (1978).34. Wei R.P., Simmons G.W*, Scripta Metall., 10, 153 (1976).35. Smialowski M*, Hydrogen in Steel, Pergamon, New York (1962).36. Menyhard M*, Surf. Interface Anal., 1, 175 (1979).37. Seah M.P*, Sanz J*M*, Hofmann S*, Thin Solid Films, 81, 239 (1981).38. Powell B.D %, Mykura H*, Acta Metall., 21, 1151 (1973).39. Johnson WX*, Smartt H.BMet. Technol., 6, 41 (1979).40. Seah M*P., J. Vac. Sci. Technol.,17, 16 (1980).41. Shimizu H<, Ono M., Nakayama K*, Surf. Sci., 36* 817 (1973).42. Ho P*S*, Lewis J.E., Howard J*K*, J. Vac. Sci. Technol., 14, 322 (1977).43. Kelly R*, Surf. Sci., 100, 85 (1980).44. Edwards BX*, Eyre B.L., Gage G., Acta Metall., 28, 335 (1980).45. Powell B.D*, Woodruff D*P., Philos. Mag., 34, 169 (1976).46. Kameda ]., McMahon C.J*, Met. Trans., 11 A, 91 (1980).47. Watanabe T*, Kitamura S., Karashima S., Acta Metall., 28, 455 (1980).48. Suzuki S*, Abiko K*, Kimura H*, Scripta Metall., 15, 1139 (1981).49. Honig R*E*f Kramer D.A., RCA Review, 30, 285 (1969).50. Seah M*P., Lea С*, Philos. Mag., 31, 627 (1975).51. Seah M*P*, J. Catal., 57, 450 (1979).52. Johnson WX*, Charka N.G., Ku R., Bomback J.L., Wynblatt P*P*, J. Vac.
Sci. Technol., 15, 467 (1978).53. Kumar A., Eyre B.L., Proc. Roy. Soc. London, A370, 431 (1980).54. Hondros E.D., Seah M.P*, Met. Trans., 8A, 1363 (1977).55. Williams F.L., Nason D., Surf. Sci., 45, 377 (1974).
М.П. Сих31556. Wynblatt P., Ки R.C., in: Proc, ASM Materials Science Seminar ’ Interfacial
Segregation’ (eds, W.C. Johnson, J.M. Blakely), p. 115, ASM, Metals Park,
1979.57. Burton J.J., Machlin E.S., Phys. Rev. Lett., 37, 1433 (1976).58. Miedema A.R., Zeits. f. Metallkunde, 69, 455 (1978).59. Tsai N.H., Pound G.M., Abraham F.F., J. Catal., 50, 200 (1977).60. Lea С», Seah M.P*, Surf, Sci., 53, 272 (1975).61. Burstein G.T., Clayton J.Q., Scripta Metall., 13, 1099 (1979).62. Thomas M.T., Baer D.R., Jones R.H., Bruemmer S.M., J. Vac. Sci. Technol.,
17, 25 (1980),63. Jones R.H., Bruemmer S.М., Thomas M.T., Baer D.R., Met. Trans., 12A,1621 (1981).64. Tauber G., Grabke H.J., Ber. Bunsenges. Phys. Chem., 82, 298 (1978).65. Erhart H; Grabke H.J., Metal Sci., 15, 401 (1981).66. Fowler R.H., Guggenheim E.A., Statistical Thermodynamics, Cambridge
University Press, 1939.67. Pichard С., Guttmann М., Rieu J., Goux C., J. de Phys., 36, C4-151 (1975).68. Seah M.P., J. Phys. F: Metal Phys., 10, 1043 (1980).69. Guttmann М., Surf. Sci., 53, 213 (1975).70. Guttmann М., Met, Sci., 10, 337 (1976).71. Guttmann M., McLean D., in: Proc. ASM Materials Science Seminar ’Interfa¬
cial Segregation' (eds. W.C. Johnson, Blakely J.M.), ASM, Metals Park,1979, p. 261.72. Dumoulin Ph., Guttmann М., Mat. Sci. Eng., 42, 249 (1980).73. Seah M.P. Acta Metall., 25, 345 (1977).74. Guttmann М., Phil. Trans. Roy* Soc., A295, 169 (1980).75. Lea C., Seah M.P., Philos. Mag., 35, 213 (1977).76. Hofmann S.t Erlewein , J. Surf. Sci., 77, 591 (1978).77. Rowlands G., Woodruff D.P*, Philos. Mag., 40, 459 (1979).78. Tyson W.R., Acta Metall., 26, 1471 (1978).79. Seah M.P., Proc. Roy. Soc., A349, 535 (1976).80. Borisov V.T., Golikov V.M., Scherbedinskiy G.V., Physics Metals Metallogr.,
17, 80 (1964).81. Bemardini J., Gas P., Hondros E.D., Seah M.P., Proc. R. Soc. London,A379, 159 (1982).82. Bemardini J., Lea C., Hondros E.D., Scripta Metall., 15, 649 (1981).83. Briant C.L., Messmer R.P., Philos. Mag., 842, 569 (1980).84. Seah M.P., Acta Metall., 28, 955 (1980),85. Hirth J. P., Rice J.R., Met. Trans., 11 A, 1501 (1980).86. Hultgren R., Desai P.A., Hawkins D.T., Gleiser М., Kelly K.K., Selected
Values of the Thermodynamic Properties of Elements, ASM, Metals Park
(1973); Selected Values of the Thermodynamic Properties of Binary Alloys,
ASM, Metals Park, 1973.
3167. Оже-спектроскопия в металлургии87. Hasegawa М., Nakajima N; Kusunoki N., Suzuki КTrans JIM, 16, 641(1975).88. Dumoulin Ph., Guttmann MFoucoult MPalmier М., Wayman М., Biscondi М.,
Met. Sci., 14, 1 (1980).89. Takayama SOgwa Т., Fu Shin^Cheng, McMahon C./., Met. Trans., 11A,1513 (1980).90. Kameda J., McMahon C.J., Met. Trans., 12A, 31 (1981).91. Seah M*P. Materials Science Club Bulletin, 64, 2 (1981).92. Hondros E.D., Mclean D., Philos. Mag., 29, 771 (1974).93. Simpson R*P., Dooley G.J., Haas T.W., Met. Trans., 5, 585 (1974).94. Johnson W.C., Doherty JtE., Kear B.H., Giamei A.F., Scripta Metall.,S0 971 (1974).95. White C.L., Keiser J.R., Braski D.N., Met. Trans., 12A, 1485 (1981).96. Hondros E.D., Seah M.P*, Lea С., Met. Mater., January 1976, 26 (1976).97. Carr F.L., Goldman M., Jaffee L.D., Buffum D.C., Trans. AIME, 197,998 (1953).98. Edwards B.L., Eyre B.L., Gage G., Acta Metall., 28, 335 (1980).99. Mulford R.A., McMahon C.J. Pope D.P., Feng H. C., Met. Trans., 7A,1183 (1976).100. Mulford R.A., McMahon C.J., Pope D.P., FengH.CMet. Trans., 7A, 1269(1976).101. Cianelli A.K., FengHX., Ucisik A.H., McMahon C.J*, Met. Trans., 8A,1059 (1977).102. Briant C.L., Scripta Metall., 15, 1013 (1981).103. Seah M.P., Spencer P.J., Hondros E.D., Met. Sci., 13, 307 (1979).104. Lea C., Hondros E.D., Proc. Roy. Soc. London, A377, 477 (1981).105. Hondros E.D., Lea С., Nature, 289, 663 (1981).106. Skelton R.P., Met. Sci., 9, 192 (1975).107. McLean D«,Metals Forum, 4, 44 (1981).108. Hippsley C.A., Knott J.F., Edwards B.C., Acta Metall., 30, 641 (1982).109. Hartweck W., Grabke H.J., Scripta Metall.* 15„ 653 (1981).110. Seah M.P., Hondros E.D. Proc. NATO Advanced Research Institute ’Atomis-
tics of Fracture’, Calcatoggio, Corscia, NATO Conf. Ser., VI, Materials
Science v. 5, Plenum, New York, 1981, p. 855.111. Middleton C.J., Met. Sci., 14, 107 (1981).112. Kalderon D., Proc. Inst. Mech. Eng., 186, 341 (1972).Литература, добавленная переводчиком1. Aitchison I., Davidson R.D., J. Vac., Sci. and Technol., A20 775 (1984).2* Bermudez V.M., Appl. Surf. Sci., 17, 12 (1983).3? Biggiero G., Luzzi J., Appl. Phys., 54, 4241 (1983).4? Biggiero G., Luzzi J., Vacuum, 33, 723 (1983).
М.П. Сих3175* Berghaus ThNeddemeyer H., Radlik WRogge VPhys. Ser., 74, 194(1983).6.*Elliott J.R., Williams R.S., Keller R.L., Taylor R., J. Nucl. Mater., 123,
1151 (1984).ж7. Ge-Lun-Long, Hu-Zkuang-Qi, Shi-Chang~Hsu, Pract. Metallogr., 21, 42(1984).8* Hofmann H., Hofmann S*, Ser. Met., 18, 77 (1984).9* Jardin С., Michel P., J. microsc. et spectrosc. electron., 9, 439 (1984).
10? Hegde M.S., Kumar Sampath T.S., Mallyo R.M., Appl. Surf. Sci., 17, 97
^ (1983).11. Brytov I. A., Wander Neshpor V.S., J. Phys. (Fz.), 45, Suppl. 353
(1984).12? LellerM.V., Kargol J.A., Appl. Surf. Sci., 8. 63 (1984).13. Lergely G., 8th Congress Mater. Test, Budapest, 28 Sept. — 1 Oct. 1982
Lectures, Vol. 3, Budapest, 1982.14* Nemoshkalenko V.V., Gorsky V.V., Ivanova E.K., Gonchorenko A.B.,J. Phys. (Fz.), 45, Suppl. 643 (1984).15. Okado Katsuzo, Takahashi Noboru et al. Bull. Jap. Soc. Precis. Eng.,
18, 33 (1984).l6?Pulkinen R.E., Lahdeniemi E., J. Mater. Sci., 18, 3421 (1983).
ntOciepa L.G., Thin Solid Films, 120, 123 (1984).18* Rachocki K.D., Broun D.R., Springer R,W,, Arendt P.N., Appl. Surf. Sci.,
18, 165 (1984).19* Sawchyn JDraper C.W., Appl. Surf. Sci., 18, 86 (1984).20*Seah M.P., J. microsc. et spectrosc. electron, 8, 177 (1983).21? Smith J.F., Southworth H.M., Surf. Sci., 122, L619 (1982).22* Vardiman R.G., Singer I.I., Material Lett., 2, 150 (1983).23*King T.S., Donnelly R.G., Surf. Sci., 151, 374 (1985).24*Waolsworth J., Chellmann D.J., Ser. Met., 19, 181 (1985).25tSiuda R., Surf. Sci., 140, 472 (1984).26* Агеев B.H., Рутъков E.B., Тонтегоде А.Я., Холина Н*А., ФТТ, 23, № 8,
2248 (1981).27* Гомоюнов М.В., Пронин И.И., Поверхность: Физ., химия , мех., 7, 44(1982).28? Ибрагимов В.Я, Ильин А.М., Зашквара В.В., Поверхность: Физ., химия,
мех., 1, 66 (1984).29. Косячков А.А., Черепин В.Т., Металлофизика, 2, 57 (1982)*30.* Ашхопов О.Г., Шебзухов А.А,, Поверхность: Физ0, химия, мех., 4, 60(1983).
Глава 8
ПРИМЕНЕНИЕ
ЭЛЕКТРОННОЙ СПЕКТРОСКОПИИ
В ГЕТЕРОГЕННОМ КАТАЛИЗЕТ. Л. Баррv8.1. ВведениеОсновным содержанием этой главы является использование методов
электронной спектроскопии (и в первую очередь рентгеновской фотоэлектрон¬
ной спектроскопии, РФЭС, или ЭСХА) в гетерогенном катализе* Мы намере¬
ны всесторонне рассмотреть этот вопрос. Несмотря на то что в этой главе
будет дан обстоятельный (а иногда и детальный) обзор имеющейся литера¬
туры, поставленная цель заключается не в исчерпывающем обзоре вопроса,
а в охвате наиболее важных достижений.Катализ являлся одной из областей приложения ЭСХА, на которую осо¬
бо обратил внимание Зигбан в своих пионерских работах (см., например,[ 1]), и частично благодаря этому был опубликован ряд как фундаментальных,
так и прикладных работ в области катализа в течение первых нескольких
лет коммерческого использования ЭСХА-спектрометров (1969-1974 гг.).
Многие из этих попыток оказались не очень плодотворными, и в ряде случа¬
ев незрелость метода приводила к недоразумениям и даже ошибкам. Тем
не менее был достигнут, вообще говоря, заметный успех, особенно в фунда¬
ментальных областях, что содействовало развитию во второй, более продук¬
тивной фазе (с 1975 г. до настоящего времени). Автор полагает, что эта
вторая фаза закончилась, и мы находимся теперь в начале значительно
более продуктивного периода» Поэтому в данной главе мы сконцентрируем
свое внимание на развитии методов и приложений электронной спектроскопии,
имевшем место в течение последнего периода» В последние годы были ис¬
следованы многие области катализа. Здесь для краткости рассматриваются
только три из них, тем не менее этого будет достаточно, чтобы оценить сам
предмет и размер того влияния, которое в настоящее время оказывает элек¬
тронная спектроскопия на понимание катализа»Исследованиям в области катализа методами электронной спектроско¬
пии до некоторой степени способствует их близость к большой "фундамен¬
тальной" области изучения контролируемых реакций на поверхностях моно¬
кристаллов металлов и сплавов. Несмотря на то что эту область можно рас-^ ToL„ ВатГр Corporate Research Center, UOP Inc0 Ten UOP Plaza, Des Plaines,
Illinois 60016, USA0
Т.П. Барр319сматривать как важное звено изучения поверхности в гетерогенном катализе,
количество точек тесного соприкосновения между этими двумя областями,
как мы это увидим, весьма ограниченно. В этой связи очень полезно озна¬
комиться с тремя обзорами Иейтса [ 2], Иейтса и Мейди [ 3] и Фишера [ 4],
написанными примерно в 1975 г., в которых подчеркивается значение взаимо¬
связи между фундаментальной наукой о поверхности и теоретическим, а
также прикладным катализом. Авторы этих обзоров были едины в предска*
зании того, что упомянутая выше взаимосвязь будет главным моментом при
изучении поверхности в следующие за 1975 годы. Все эти обзоры содержат
детальные предсказания будущего (уже наступившего)» Интересно сравнить
великолепные предсказания этих авторов с последними достижениями, ко¬
торые суммированы в конце данной главы.Подход, основанный на использовании многих методов, по-видимому,
играет главную роль в упомянутых фундаментальных исследованиях. На са¬
мом деле очень часто РФЭС и ЭОС выступают в таких исследованиях на вто¬
ром плане. С другой стороны, из дальнейшего станет ясно, что, хотя ион¬
ные методы и методы структурного анализа, как принято считать, имеют
все возрастающее значение в прикладном катализе, основным источником
информации все еще являются методы электронной спектроскопии, и особен¬
но РФЭС.Недавно были опубликованы многочисленные очень хорошие обзоры,
посвященные изучению природы хемосорбции, поэтому в данной главе мы
сконцентрируем свое внимание на менее обстоятельно проанализированных
"прикладных” областях. Хотя подробный анализ работ первой из упомяну¬
тых областей не вписывается в тематику настоящей главы, некоторое пред¬
ставление о взаимосвязи между прикладной и фундаментальной областями
можно получить из отдельных примеров, относящихся к последней области,
особенно в разделе, посвященном катализу на платиновых металлах.Перед началом этого изложения уместно сделать несколько замечаний
относительно терминологии. Во-первых, термин фундаментальные будет
использоваться в этой главе для обозначения тех работ, которые проводи¬
лись в значительной степени с целью улучшения или расширения возможнос¬
тей электронной спектроскопии. Большая часть этих вопросов достаточно
подробно обсуждается в других разделах книги. По этой причине мы будем
затрагивать здесь эти вопросы только в том случае, если они представля¬
ют интерес для катализа. В других случаях читатель отсылается к соответ¬
ствующим разделам данной книги. Выражение "фундаментальные исследо¬
вания в катализе", с другой стороньцбудет использоваться для обозначения
тех работ, которые ставились с целью моделирования каталитических свойств
путем исследования материалов, специально изготовленных таким образом,
чтобы они проявляли только требуемые свойства. Подавляющее большинство
работ таких исследователей, как Сомораи, Мейдикс и Уайт (и фактически
вся область хемосорбции) попадает в эту категорию исследований. В настоя-
3208. Применение электронной спектроскопии в гетерогенном катализещей главе рассматриваются только основные моменты этих исследований.
Читателям, заинтересованным в более детальном рассмотрении, следует
обратиться к многочисленным недавно опубликованным обзорам. Основное
внимание в данной главе уделяется прикладной области, которая характери¬
зуется приложением электронной спектроскопии к промышленным или моде¬
лирующим промышленные каталитическим системам. Такие системы сос¬
тоят из металлов, окислов, сульфидов и других веществ, называемых ак¬
тивными присадками, которые обычно наносятся на разнообразные окис¬
лы с широкой запрещенной зоной, называемые носителями.Все рассматриваемые в этой главе материалы исследовались в твердом
состоянии. Хотя некоторые из катализаторов изучались без носителей (см. ниже),
все они рассматриваются как гетерогенные катализаторы (по крайней мере для
целей настоящей главы). Это важный момент, так как он предполагает, что боль¬
шинство (если не все) изучаемых процессов по крайней мере инициируются
на поверхности этих каталитических материалов, и потому если можно разра¬
ботать метода анализа их поверхностей до, во время и после протекания исследуемой
реакции, то это может дать весьма полезную информацию. Не рассмотренным
с точки зрения такого подхода в этой главе остается очень важный вопрос
(на который до сих пор ответа нет): из чего состоит поверхность катализато¬
ра? Этот момент особенно неопределен в случае промышленных катализаторов,
когда используемые вещества часто приготавливаются таким образом, что
большая часть их ( кажущейся) активной поверхности изолирована внутри
пор или внутри сферических зерен или выступов. Большая часть активной
поверхности катализатора, будучи доступной для реагентов (под давлением),
может быть фактически недоступной для традиционных инструментов ана¬
лиза поверхности, например для электронного спектрометра. На практике
непостоянство и часто неопределенность контакта между поверхностью ве¬
щества и анализирующим зондом остаются основной заботой эксперимента¬
торов-Все это означает, что от ученого, работающего в прикладных областях
науки о поверхности, требуется большое искусство, чтобы таким способом
приготовить материалы, при котором изучаемые поверхности в максималь¬
ной степени "видны” спектрометру. Независимо от того, насколько искус¬
ным является экспериментатор, в этом вопросе имеется некоторая неопре¬
деленность- Еще более важным является связанное с этим моментом пред¬
положение, к которому часто прибегают в электронной спектроскопии (осо¬
бенно в ЭСХА), состоящее в том, что спектр отражает состояние исследуе¬
мого материала статистически, т« е. этот спектр можно использовать для
описания разумного среднего состояния поверхности катализатора. В этой
главе не будут рассматриваться подробно работы, в которых делаются по¬
пытки отстоять это предположение. (Фактически число работ, в которых пы¬
тались решить эту задачу, весьма незначительно-) Читатель должен выра¬
ботать свое собственное мнение в этом спорном вопросе; тем не менее еле-
Т.Л. Барр321дует помнить, что были опубликованы сотни работ, в которых это предполо¬
жение считалось выполненным, и в подавляющем большинстве этих работ
было показано, что данные, полученные ЭСХА, непосредственно коррелиру¬
ют с химическими и физическими изменениями.8.1.1. «Краткий обзор исследований катализа методамиэлектронной спектроскопии, проведенных до 1975 г.Состояние исследований катализа с использованием электронной спек¬
троскопии в течение первой половины 70>х годов обсуждалось также в обзо¬
рах Мак-Кэррола [ 5] и Томпкинса [ 6].Томпкинс в основном уделил внимание обзору состояния "физики по¬
верхности1', или, как мы это назвали, фундаментальным исследованиям.
Прежде всего его интересовали адсорбция окиси углерода и реакции с ее
участием на поверхностях монокристаллов, а также спектры валентных зон,
получаемые методами УФЭС и РФЭС» Мак-Кэррол, с другой стороны, отме¬
чал в начале своего обзора, что "гетерогенный катализ в практическом
смысле мало выиграл” от прогресса в физике поверхности. Он также отме¬
тил, что "практическое значение для человечества . . . полностью разрабо¬
танной научной основы катализа было бы огромно” [ 5]„Мак-Кэррол описал существенные достижения в некоторых фундаменталь¬
ных и прикладных областях, а также привел доводы, подтверждающие спо¬
собность физики поверхности решать задачи промышленного катализа, не¬
смотря на очевидное различие непредсказуемого поведения поверхностей в
этих двух областях- Он показал, как исследования методами ДМЭ и ДБЭ
поверхностей монокристаллов, свойства которых изменены благодаря по¬
крытию адсорбентами, могут иметь отношение к прикладным задачам, если
используемые газы аналогичны тем, которые осаждаются на металлах в
промышленных катализаторах. Например, "переориентация” поверхности
Ni (111) в присутствии серы, как оказалось, вызывается наружным слоем
Ni (100) (2 х 2) - S [7]. Мак-Кэррол также отметил, что вследствие этой
переориентации катализатор может вести себя по отношению к молекулам
нефти как катализатор, имеющий новую поверхность с существенно иными
активностью и селективностью.После краткого обсуждения дегидроциклизации на платине (аналогичный
процесс изучался Сомораи, см. ниже) Мак- Кэррол рассматривает некоторые
ранние приложения электронной спектроскопии к катализу. РФЭС-исследова-
ния Карберри, Кучинского и Мартинеса [ 8] показали, что у-облучение инду¬
цирует миграцию щелочных металлов (например, цезия) к поверхности на¬
несенных серебряных катализаторов, усиливая таким образом способность
этого катализатора окислять этилен.Рассматривались также предварительные исследования Ван Сантена и
Захтлера [ 9], а также других исследователей, посвященные платиновым
3228. Применение электронной спектроскопии в гетерогенном катализесплавам, которые, как предполагалось, являются исходным материалом
биметаллических восстановительных катализаторов для реакций дегидро¬
циклизации . Мак-Кэррол сомневается в уместности этой работы. Однако
последующее развитие, по-видимому, подтвердило правильность этих ран¬
них попыток (см. ниже).Мак-Кэррол упоминал также о предварительных исследованиях методом
РФЭС систем Со -Mo -S -А 1203, выполненных Стивенсом и Эдмондсом
[ Ю].Он подчеркнул, что эта работа служит примером " более корректного
использования метода РФЭС", не подозревая, конечно, о многочисленных
противоположных случаях, которые вскоре появятся в растущем потоке ли¬
тературы по этим системам (см. ниже). Наблюдение Стивенса и Эдмондса
[ 10], что MoIV в форме Mo S2 оказывает решающее влияние на характерис¬
тики этой каталитической системы, оказалось довольно пророческим.Были рассмотрены также исследования Бхасина [11], выполненные ме¬
тодом ЭОС, В этой работе суждения об ожидаемых характеристиках катали¬
затора Cu/Si02-A^Q при наличии свинца и магния основывались на изме¬
рениях отношений поверхностной концентрации к объемной концентрации
свинца, магния и кремния. Ввиду предполагаемых эффектов разрушения
под действием электронного пучка некоторые из этих выводов могут вы¬
зывать вопросы, тем не менее сами результаты указывают на потенциаль¬
ную полезность ЭОС.Имеется несколько прикладных РФЭС-работ по катализу, опубликован¬
ных в самом начале 70-х годов, которые не были отражены в обзоре Мак-
Кэррола. Большинство из них были выполнены, по-видимому, под впечатле¬
нием от пионерской работы Дельгаса, Хьюза и Фэдли [ 12]. В этих рабо¬
тах описано также исследование Брайнена и Мелера [ 13] адсорбции родия
на угле. В работе [ 13] указывается на потенциальные возможности РФЭС
как метода некоторых относительных количественных измерений, являю¬
щихся очень важным моментом для оценок в области катализа, особенно
в тех случаях, когда приняты меры для правильного учета параметров
носителя (размер пор и т- п.) (количественные характеристики можно най¬
ти в соответствующем разделе этого обзора). Брайнен использовал метод
РФЭС также для исследования отравления катализаторов [14]. В более ран¬
ней работе Матьенцо и др. [ 15] методом РФЭС получили энергии связи, ко¬
торые крайне необходимы для дальнейшего изучения никелевых катализа¬
торов-Работа Линдсея и др. [ 16] наряду с многими другими посвящена изуче¬
нию носителей. Эти авторы представили результаты по сдвигам энергий свя¬
зи различных систем, основанных на окиси алюминия- Развивая количест¬
венные соображения Дельгаса-, Хьюза и Фэдди [ 12], а также Брайнена и Ме¬
лера [ 13], Темпре, Делафос и Контур [ 17] использовали РФЭС для изучения
цеолитов с различными отношениями концентраций кремния и алюминия
(цеолиты являются материалами, которые потенциально можно использовать
Т.Л. Барр323в качестве как носителей, так и катализаторов). Было доказано, что по ме¬
ре увеличения отношения объемной концентрации кремния к объемной кон¬
центрации алюминия наблюдается интенсивная деалюминизация поверхности,,
Путем наблюдения за изменениями энергии связи Се Ы были также заре¬
гистрированы изменения состояния окисления цериевого катиона.Дополнительные данные по каталитическим системам Со -Mo -S -
А120^ были представлены Армуром и др. [18], которые обнаружили, что
сульфидирование уменьшает энергии связи молибдена и кобальта. Эта же
система была исследована Каннесоном и Гранжем [ 19], которые, используя
данные РФЭС, впервые высказали предположение о присутствии как MoS2,
так и Моб+.Хотя каждое из упомянутых выше исследований катализаторов методом
РФЭС, а также другие исследования, опубликованные между 1970 и 1975 гг.,
были по отде чьности весьма информативны, все вместе они лишь подтвер¬
дили вывод Иейтса и Мейди[ 2, 3] (повторенный Мак-Кэрролом [ 5]) о том,
что к 1975 г. возлагавшиеся на РФЭС надежды еще не были реализованы.8.2. Основы электронной спектроскопии применительнок изучению катализа8.2.1. введениеБольшинство исследований методом РФЭС начинаются с качественно¬
го наблюдения обзорных спектров при последующем быстром увеличении
масштаба с целью выделения наиболее интенсивных линий остовных уров¬
ней. Часто эти линии в изобилии содержат количественную и химическую
информацию, и многие исследования методом РФЭС никогда не выходят
за эту фазу. Очень часто, однако, весьма малые изменения в составе,
структуре или химическом состоянии системы представляют первостепен¬
ный интерес, и эти изменения могут не "отражаться” в "традиционных”
частях остовных спектров. По этой причине другие, редко используемые и
обычно малоизученные особенности РФЭС были проанализированы и ис¬
пользованы в экспериментах, среди которых недавно появились и экспери¬
менты с материалами, обладающими каталитическими свойствами- Наибо¬
лее обещающими среди таких особенностей являются индуцированные
РФЭС-спектры потерь энергии, оже-спектры и спектры валентных зон. Все
они были рассмотрены в других главах этой книги; поэтому здесь мы их
рассмотрели только потому, что они являются новой попыткой использования
РФЭС для изучения катализаторов и материалов, имеющих отношение к ка¬
тализу. Также (и по той же причине) в настоящей главе кратко излагаются
последние работы с использованием СХПЭЭ, синхротронного излучения и
преадсорбции.Однако прежде чем обсуждать эти более "тонкие" моменты будет по¬
учительно (и в некоторой степени отрезвляюще) ознакомиться с двумя послед-
3248. Применение электронной спектроскопии в гетерогенном катализеними обзорами [20, 21], в которых можно найти ответы на вопросы о кор¬
ректности и точности количественных данных и значений энергии связи как
в РФЭС, так и в ЭОС.8.2.2. Обзоры по энергиям связи и
интенсивностям линийОчень важный обзор, имеющий отношение к катализу, был опубликован
в 1977 г. Мейди, Вагнером и Джоши [20]о В нем содержится информация о
работах большого числа промышленных и академических лабораторий, вклю¬
чая и лабораторию автора, использующих методы РФЭС и ЭОС. Этот обзор
отличается тем, что в нем собраны и проанализированы результаты измере¬
ний положений линий и их интенсивности для " стандартного" силикагеля,
окиси алюминия и веществ из Na А, Для всех этих данных был обнаружен
значительный разброс (рисе 8.1) не только в том случае, когда сравнивались
их абсолютные величины, но и в случае использования некоторых тонких
относительных (с целью устранения случайных факторов) методов их пред¬
ставления. Значения разброса во всех указанных работах показывают, что в
этих работах все еще не был получен ответ на многие вопросы, связанные
с подготовкой образцов, зарядкой (и ее устранением), количественными из¬
мерениями и даже калибровкой энергетической шкалы. Особенно это наблю¬
дается при анализе образцов из непроводящих материалов»Из-за отсутствия согласованности этих результатов было решено "вер¬
нуться к первоосновам" и проанализировать три чистые поликристалличес-
кие фольги из проводящих металлов: никеля, меди и золота. Разброс данных,
полученных в этом исследовании [21], был значительно меньше результатов
первых работ; тем не менее колебания как интенсивности, так и энергии свя¬
зи оказались значительно больше, чем предполагалось. Во многих отноше¬
ниях это второе исследование было даже более обескураживающим,чем пер¬
вое, поскольку указанные металлы обычно использовались для калибровки
электронных спектрометров. Эти исследования, если их рассматривать объек¬
тивно, имели огромное значение для электронных спектроскопистов (особен¬
но для тех, кто имеет дело с непроводящими материалами), так как они спо¬
собствовали переходу к периоду критического анализа и учета на практике
вопросов, связанных с поверкой и калибровкой. Благодаря этим исследоваг
ниям большинство установок работает теперь с более высокой воспроизво¬
димостью (и точностью). Тем не менее полагаться на интерпретацию, ос¬
нованную на единичных измерениях, следует с большой осторожностью, осо¬
бенно если ключевые моменты этой интерпретации связаны с малыми вариа¬
циями энергии и/или интенсивности линий.
ЦеолитРиг 8.1. а - сравнение энергии связи А1 2р в цеолите с энергией связи А1 2р в
окиси алюминия. Светлые кружки - с использованием электронной ком¬
пенсационной пушки; темные кружки — относительно С 1* . б — сравнение
энергии связи Si 2р в двуокиси кремния с энергией связи Si 2р в цеолите.
Светлые кружки - с использованием электронной компенсационной пушки;
темные кружки — относительно С 1s. в - разности энергий РФЭС-линий;1) сравнение А! 2р -Al 2 s для цеолита с А1 2р -А1 2 s для окиси алюми¬
ния, 2) сравнение Si 2р - Si2s для цеолита с Si 2р -Si 2s для двуокиси
кремния; 3) сравнение Al 2р - 01s для цеолита с А1 2р -01s для окиси
алюминия; 4) сравнение Si 2Р - 01s для цеолита с Si 2р -01s для двуоки¬
си кремния [ 20].
3268. Применение электронной спектроскопии в гетерогенном катализе8.2.3. Новые методы и катализ
8.2.3J. РФЭС-спектры потерьСпектры потерь, начиная с их открытия Зигбаном более чем два деся¬
тилетия назад, считались странным и неприятным явлением в ЭСХА. Вна¬
чале причины появления этих различающихся по размерам "сателлитов” в
высокоэнергетичной части остовных, оже- и валентных пиков были непо¬
нятны. Однако тщательные (и иногда противоречивые) экспериментальные
и теоретические исследования через некоторое время показали, что эти
вторичные пики, связанные с большим числом неупругих столкновений са¬
мой различной природы, в равной степени зависят как от скорости фотоэлек¬
трона, так и от вероятности вторичных переходов. Во всяком случае, как
это обсуждалось в гл. 3, вклад в эти сателлитные пики дает сложная смесь
встряски (shake-up), (стряхивания) (shake-off) мультиплетных, плазмонных,
межзонных и других переходов.Подробный анализ физических причин му льтип летных переходов и явле¬
ния встряски был опубликован в работах Фэдли [22] и других авторов [23]*
Из всех упомянутых выше механизмов потерь энергии эти два механизма,
вероятно, изучены лучше всего и определенно чаще всего используются
на практике. Они приводят к заметным особенностям в РФЭС-спектрах мно¬
гих систем, содержащих переходные металлы» Сателлиты встряски исполь¬
зуются при анализе каталитических систем, особенно тех, у которых в ка¬
честве активных добавок применяются переходные металлы. Типичный при¬
мер такого подхода содержится в работе Нг и Эркюля [24], которые иссле¬
довали катализатор никель - вольфрам - глинозем» В этой работе появление
(или исчезновение) мультиплетных сателлитов и сателлитов встряски нике¬
ля являлось основной информацией для анализа изменений состояния окис¬
ления никеля при работе катализатора. Однако такой анализ не является
столь простым, как это может показаться. Например, попытки его исполь¬
зования для описания различных стадий обработки каталитической системы
Со -Mo -S -А120^ привели к интересной полемике [25, 26], так как
оказалось, что в некоторых случаях халькогениды ряда переходных метал¬
лов могут вовсе не иметь сателлитов (или химических сдвигов), даже если
соответствующие окислы той же степени окисления имеют очень заметные
сателлиты и сдвиги [ 27].Использование РФЭС-сателлитов для химического анализа автор нас¬
тоящей главы распространил на относительно слабые линии потерь ряда
"керамических” (типичных для носителей) окислов, например Al203,Si02
[28] и других материалов, которые можно в широком смысле рассматривать
как широкозонные полупроводники.Было показано, что [31], начиная с ряда соединений индия [29, 30],
а) заметные (хотя и слабые) воспроизводимые линии потерь могут возни¬
кать для большинства систем с ненулевой запрещенной зоной (рис. 8.2);
Т.П. Барр327Рис. 8.2. Спектры потерь 1пЗd для: In0 (A), InN(B) и Ii^O^c) (а) и InSb (б) [31].б) для серии соединений с общим катионом (например, индий) или анионом
(например, сурьма) эти линии потерь сдвинуты относительно друг друга на
величину, которая в общем превышает величину соответствующих химичес¬
ких сдвигов (Зигбана) вплоть до нескольких электронвольт (табл. 8.1);в) несмотря на их малую интенсивность, эти линии теперь часто можно де¬
тектировать для сложных систем; г) квантовомеханический анализ причин
возникновения этих линий показывает, что они прежде всего возникают
из-за внутреннего (дырка) или внешнего (фотоэлектрон) возбуждения окру¬
жающей плазмы валентной зоны с некоторыми (часто существенными) воз¬
мущениями со стороны межзонных и других неколлективных эффектов;д) несмотря на то что источником этих линий потерь является конечное
3288. Применение электронной спектроскопии в гетерогенном катализеТаблица 8.1. Идентифицированные энергии линий
потерь (относительно соответствующих основных РФЭС-
пиков) для некоторых индиевых систем. Значения
энергии приведены в электронвольтахIn0InSbInNIn203ln(3d5/2)i443,9444,1444,3444,851п(3^/2)2451,3451,5451,7452,3s,——11,113,2В,11,513,315,518,5S28*2—11,413,5В211,813,315,818,8х Si20,1—26,531,92 х В!23,126,430,937,22 х В223,226,231,237,53 х В,—40,446,3—3 х В235,2———S-вероятный поверхностный плазмон;В — вероятный объемный плазмонРис. 8.3. Типичные РФОС-спектры потерь Al 2s: A^Og на Al^^a); смешанная система
А1°(б) и А1°,покрытый менее чем одним моноспоем А1203 (в) [2в].
Т.Л. Барр 329Таблица 8.2. Идентификация возможных линий потерь в системе А1203 — А1°Источник фото¬МестоТип переходаРасстояниеРасстояниеэлектроновпереходас потерейот линий AIот линии AIс потерейэнергиив А1203в А1°энергииРхА12р 0,0 (75,5)0,0 (72,9)Р 2A12s——0,0 (120,4)0,0(118,0)0А1°А1° поверхностьS9Д11.51А1АА1°А1° поверхностьSпа13,515г92А1°в13,516.03А!2ОзА1°в18,44А1°ai2o3в21,924,224т35Al?OiАЬ»03А1°/ гр. разд.в26,56о<1> ГО
<<В х S/S х SШ7А1°2 х В2241U8А12о3А1°2 х В31,934,3S — поверхностная эмиссия; Расщепление вследствие потерь (основной плазмон)в —объемная эмиссия Объем Поверхность1*гр. разд. — граница раздела А1° 15,8 11,3А1203 24,4 17,4(дырка) состояние системы, теоретически и экспериментально [ 32] было
показано, что сдвиг этих линий потерь от главных пиков (без потерь) не
зависит от уровня Ферми (зарядки) материала и в первом приближении не
зависит также от эффектов релаксации* Эти расщепления линий, связанные
с потерями, фактически определяются довольно простыми параметрами
эмитирующих систем (например, электронной плотностью состояний валент¬
ной зоны и То п.). Поэтому разности между расщеплением ряда линий потерь
(сдвиги потерь) часто представляют собой химические сдвиги, которые мож¬
но использовать для дифференциации систем точно так же, как и обычные
химические сдвиги (Зигбана). Такой подход использовался, например, для
описания физических изменений, замеченных при росте А1208 после окисле¬
ния поликристаллического алюминия при комнатной температуре [ 32]. Ана¬
логичные свойства были успешно зарегистрированы в процессе изучения по¬
верхности раздела Si02 -SiQ [28]. Основные линии потерь, появляющиеся
в этих случаях, имели внешнюю природу (связанную только с электроном), что
указывало на то, что фотоэлектрон проходит из внутреннего слоя через тон¬
кий наружный слой. Связанные с потерями переходы, которые появляются в
этом случае, позволяют определить методом РФЭС морфологию слоев (нап¬
ример А1203 расположен на А1° или наоборот; рис- 8.3, табл. 8.2) способом,
который ранее был невозможен [28, 32L Такое определение морфологии мо¬
жет быть очень полезно для последующего комбинированного химико- морфо¬
логического изучения сложных слоистых систем.
3308. Применение электронной спектроскопии в гетерогенном катализе8.2.3.2. Оже-спектры при рентгеновском возбужденииКак упоминалось выше, оже-спектры, возбуждаемые рентгеновским из¬
лучением, можно также использовать для изучения каталитических систем.В прошлом имелись немногочисленные, но важные примеры практического
использования этих эффектов релаксации конечного состояния (см. обсуж¬
дение в гл. 3). Тот факт, что такие оже-линии очень сильно меняются с из¬
менением химического состояния и эти изменения часто значительно сильнее
выражены, чем изменения в соответствующих фотоэлектронных пиках, ис¬
пользовался для анализа неорганических материалов [ 33] и побочных продук¬
тов коррозии [34-36] . Аналогичный анализ методом РФЭС изменений в
соединениях, родственных используемым в катализаторах активным добав¬
кам, еще не применялся. Потенциальные возможности использования оже-па-
раметров (см. также приложение 4) для изучения носителей катализаторов,
Цеолитов и связанных с ними материалов были недавно подробно проанализи¬
рованы в работе Вагнера и др» [ 37, 38]. Эта методика может оказаться
эффективным инструментом анализа типичных каталитических систем, так
как оже-параметры не зависят (в первом приближении) от зарядки (и таким
образом аналогичны, в этом смысле, сдвигу линий вследствие потерь)0 Так
как оже-пики, как правило, довольно интенсивны для веществ, обычно исполь¬
зуемых в качестве носителей катализаторов, оже-параметры могут оказать¬
ся чувствительным индикатором таких трудно обнаружимых эффектов, как
взаимодействие между металлами и носителем, спекание носителей и т* п.8.2.3.3. РФЭС- (и УФЭС-) спектры валентных зон и спектроскопия
характеристических потерь энергии электронов (СХПЗЭ )Валентная область часто и достаточно произвольно определяется как
первые 20 эВ энергии связи. В этой области главный вклад в фотоэлектрон¬
ный спектр дают уровни, которые в значительной степени подвержены мо¬
лекулярному (орбитальному) или твердотельному (зонному) влиянию. (Неко¬
торые исследователи, возможно, для удобства расширяют эту область до
50 эВ,)РФЭС-спектры валентных зон очень редко использовались для изучения
катализаторов и связанных с ними систем» С другой стороны, был выполнен
ряд работ с использованием УФ- и синхротронных источников излучения, на¬
правленных на решение некоторых фундаментальных проблем катализа.В качестве примера рассмотрим работу Глэнда, Секстона и Фишера [39],
в которой УФЭС использовалась совместно с СХПЭЭ и термодесорбцией
для изучения взаимодействия 02 с поверхностью Pt (111). Было зарегист¬
рировано три зависящих от температуры состояния: а) молекулярный кисло¬
род (ниже 120 К); б) атомарный кислород (150-500 К) и в) подповерхност¬
ный кислород (1000-1200 К). Данные УФЭС (спектры валентных зон) пред-
Т.Л. Барр331ставляли собой составную часть окончательного анализа результатов этой
работы, они давали информацию о заполнении и обеднении молекулярных
орбиталей, однако для правильного определения конфигурации была необ¬
ходима также информация о колебательных спектрах.В связанной с этими исследованиями работе Киши и Робертса [ 40]
использовались УФЭС и РФЭС для изучения зависящей от температуры ад¬
сорбции N0 на поверхности железа. В работе сообщалось как о Не11 -, так
и о Не1-спектрах валентных зон, которые сопоставлялись с одновременно
возбуждаемыми (РФЭС) линиями остовных уровней О Is и N Is . Этим спо¬
собом были зарегистрированы не только характеристики адсорбата, но так¬
же и процессы десорбции и диссоциации* Молекулярная окись азота N0 явно
адсорбировалась при низких температурах (~ 85 К), в то время как она пол¬
ностью десорбировалась при температурах, близких к комнатной. Остается
неопределенность относительно связи N0 с железом, причем предполагает¬
ся, что одновременное исследование методом СХПЭЭ (аналогичное проведен¬
ному в работе Глэнда, Секстона и Фишера [ 39]) будет, вероятно, полезным.
Эти результаты продемонстрировали важность использования Р£ЭС в подоб¬
ных исследованиях. Таким образом, путем наблюдения за недавно обнару¬
женным пиком N Is при ^ 397 эВ, свидетельствующим об образовании гругг-Рис. 8.4. Типичные спектры валентных зон (0 - 20 эВ) [42].
3328. Применение электронной спектроскопии в гетерогенном катализепы Fe -N, была обнаружена диссоциация N0 даже при 85 Ко Имеются пуб¬
ликации по разнообразным другим направлениям использования УФЭС. Неко¬
торые из них описаны в следующем разделе»Метод СХПЭЭ еще нельзя использовать для изучения промышленных или
модельных катализаторов, так как его применение ограничено поверхностями
монокристаллов* Тем не менее он использовался в ряде работ, аналогичных
описанной выше работе Глэнда, Секстона и Фишера. Например, Томас и
Вайнберг [41] применяли СХПЭЭ и термические методы для изучения адсорб¬
ции N0 на Ru (001). В работе сообщается как о мостиковой, так и о линей¬
ной формах адсорбированной молекулы N0, а также о степени ее диссоциа¬
ции. Полезные данные, в частности для неплоских поверхностей можно по¬
лучить, используя другие методы электронной спектроскопии.Использование РФЭС-спектров валентных зон для изучения имеющих
отношение к катализу материалов было продемонстрировано автором настоя¬
щей главы при изучении свойств цеолитов и глин [ 28, 42]. Были обнаружены
различимые изменения зонной структуры (рис. 8о4), и при помощи этих спект¬
ров (табло 8.3) была зарегистрирована селективная деградация цеолитов, что
позволяло дифференцировать эти материалы.Таблица 8.3.а. Ширины1* некоторых зон в электрон вольтах (±0,1 эВ)КомпонентаШирина зонысг-А120,9,1NaA9ДСаА9,2NaX9,8СаХ9,8NaY10,4CaY10,1Si0210,4^Ширины на полувысоте зоныб. Уменьшение ширины зоны некоторых металлов в элек¬
трон вольтах (±0,1 эВ) после травления ионным распыле¬
ниемКомпонентаИзменение ширины зоныSi020,1NaY0,3CaY0,3Бентонит0,3Каолинит0,7Катапал0,7У-А12030,8NaA0,7
Т.П. Барр333до2.3.4. Фотоэлектронные спектры, возбуждаемые
синхротронным излучением
Источники синхротронного излучения создают ряд преимуществ в фото¬
электронной спектроскопии, которые могут оказаться полезными при ис¬
следовании катализаторов. Например, Миллер и др.[43] для локализации
2р» и 2 s-уровней кислорода, хемосорбированного на поверхности кристалла
Pt 6 (111) х (100), использовали изменяемый в широких пределах диапазон
энергиио Было продемонстрировано увеличение степени покрытия с ростом
температуры, несмэтря на отсутствие образования "реального" окисла.Эти результаты полезны также при изучении адсорбции воды и окиси угле¬
рода как первой стадии их окисления (см. ниже) о8.2.3.5. ПреадсорбцияПредварительная адсорбция, особенно активных атмосферных газов,
может заметно изменять данные, которые связываются с металлами. Пла¬
тина, например, как принято считать, относительно легко восстанавливает¬
ся до Pt0 (в потоке Н2), что может способствовать протеканию каталитичес¬
ких процессов на поверхности дисперсной фазы этого элемента. Фактически
это может и не иметь места, так как оказывается, что все поверхности да¬
же в довольно чистой атмосфере адсорбируют некоторое количество 02 и та¬
ким образом участие кислорода может, иметь место даже тогда, когда окис¬
ление не предполагается» Мак-Кейб и Шмидт [ 44] продемонстрировали, что
покрытые кислородом грани Pt (110) явно в большей степени катализируют
процесс Н2 + СО, чем Pt°. Для обнаружения окисных слоев и измерения их
плотности в этих исследованиях использовался метод ЭОС.8о2.4. Хемосорбция и фундаментальные исследования катализаКак отмечалось во введении, знакомство с развитием этой области очень
полезно для понимания процессов, происходящих на поверхностях промыш¬
ленных катализаторов. Уже опубликован ряд подробных обзоров, посвящен¬
ных приложению электронной спектроскопии к исследованию хемосорбции
и фундаментальному катализу. Эти обзоры широко освещают эти вопросы,
и мы не будем повторять их содержание в этой главе. Чтобы оценить высо¬
кий уровень достижений в этой области,мы рекомендуем заинтересованно¬
му читателю ознакомиться со статьями Брандла и Тодда [45], Гомера
[ 46], Робертса и Мак-Ки [ 47], Сомораи [ 48] (и с библиографией в этих ра¬
ботах)» Перечислим некоторые современные направления фундаментальных
исследований, которые, по мнению автора, представляют особый интерес
для прикладного катализа.
334 8. Применение электронной спектроскопии в гетерогенном катализеРис. 8.5. Диаграмма реакционной способности окиси углерода при 290 °К; диссоциатив¬
ная хемосорбция имеет место для металлов, расположенных слева от жир¬
ной линии. Для железа степень диссоциации при этой температуре относи¬
тельно мала, и поэтому она соответствует промежуточному случаю [55].а. Исследования Мейдикса [49-52] традиционного реагента - муравьи¬
ной кислоты, адсорбированной на поверхностях различных металлов. В этих
исследованиях детектировались промежуточные продукты и были предло¬
жены механизмы изучаемых процессов. Мейдикс также подверг критике пред¬
ложенный другими авторами термин "перепад давления".б. Робертс и др. [53-55] исследовали способность различных перехо¬
дных металлов адсорбировать и диссоциировать N -О. Данные, полученные
методом РФЭС, являлись ключевым моментом в построении диаграмм, ко¬
торые позволили предсказать периодический характер поведения переход¬
ных металлов в зависимости от их электронных свойств (рис. 8.5).в. Уайт и др. [56-64] при исследовании реакций СО + 02 на платинеи палладии в качестве одного из методов измерений использовали оже-спек-
троскопию.г. Бернс и др. [65-69] также провели аналогичные исследования ме¬
тодом ЭОС и разработали механизм реакции S 02 + СО на платине.д. Эртль и др0 [ 70] опубликовали детальный анализ механизма обра¬
зования NH3 из N2 и Н2 на железе. Для процесса, определяющего скорость
реакции, были предложены кинетические уравнения Ленгмюра и Хеншеля
на основе данных об адсорбированных соединениях (N + Н) •8.3. Электронная спектроскопия и прикладной катализ
8.3.1. Современные обзорыИнтересный обзор по прикладному гетерогенному катализу и его связи
с наукой о поверхности был опубликован в конце 1975 г» Будартом [71]* По
объему рассмотренного материала обзор Бударта аналогичен обзору Фишера
[ 4] (см. выше), однако во многих отношениях первый обзор превосходит
второй.
Т.П. Барр335Недавно было опубликовано несколько информативных обзоров, специ¬
ально посвященных приложению электронной спектроскопии к катализу. Сре¬
ди них выделяются обзоры Брайнена [25] и Бриггса [72]. Оба они прежде
всего посвящены практическим приложениям прикладного катализа и пред¬
ставляют особый интерес благодаря оригинальным взглядам авторов и выб¬
ранным для иллюстрации примерам.Все примеры в обзоре Брайнена [25] относятся к нанесенным катализа¬
торам. Определяющим моментом в этом случае является природа результирую¬
щего носителя, что было продемонстрировано во время уже упоминавшегося
исследования родия (относительно неактивного металла) на углероде; угле¬
род был выбран, очевидно, потому, что многие его физические свойства
можно изменять контролируемым образом [13]. Обзор Брайнена [25] также
содержит подробный анализ катализатора Со -Mo -S -А1203, который яв¬
ляется каталитической системой, чаще всего встречающейся в литературе
по РФЭС. Для этой системы, по-видимому, легко регистрировать изменения
ее количественных характеристик, структуры слоев и состояния окисления
(см. раздо По 8.3.5.3), однако трудно их интерпретировать.С другой стороны, Бриггс [72] начал свою статью с описания того, что
он называет тремя "существенными" моментами изучения катализаторов ме¬
тодом РФЭС: а) дезактивация, б) количественные измерения и в) моделиро¬
вание катализаторов (т. е. выявление связей между структурой и свойства¬
ми). Приведено три примера для каждого из этих моментов, однако только
один из них относился к нанесенным катализаторам» Из всех перечисленных
моментов Бриггс считает самым важным моделирование катализаторов,
однако, как он отмечает, в настоящее время этот вопрос менее всего ясен.Еще один ббзор, опубликованный Келли [73], посвящен не только ката¬
лизу, тем не менее он содержит жизненно важные для катализа выводы, так
как в нем подробно рассмотрены вопросы, связанные с отношением поверх¬
ностной концентрации к объемной концентрации и сегрегации на поверхности.
Многие промышленные катализаторы представляют собой двойные и трой¬
ные металлические системы, в которых один из металлов (или его окисел),
как правило, является первичным каталитическим агентом (для гидрирова¬
ния, дегидрогенезации, окисления и т, д»), в то время как другие металлы
(или их окислы, сульфиды и т. д.) действуют прежде всего как промоторы,
замедлители или же каким-либо другим способом влияют на активность
основного металла. Многие исследователи предполагают, что эти полиме¬
таллические системы существуют на носителе работающего катализатора в
виде сплава [74] или кластера. Келли в своей работе обращается в собира¬
тельном смысле как к эмпирическим, так и к экспериментальным методам
определения (и непрерывного контроля) степени перемешивания различных
соединений на поверхности. В обзоре описаны ТЭМ, методы ионной спектро¬
скопии, а также РФЭС и ЭОС. В дополнение к этому Келли недавно сообщил
также о приложении метода РФЭС к этим проблемам [ 75].
3368. Применение электронной спектроскопии в гетерогенном катализе8.3.2. Проблемы и методы использования электронной
спектроскопии в катализе
8.3.2.1. ВведениеОпределения химического состава и концентрации являются двумя ос¬
новными источниками погрешностей при использовании электронной спектро¬
скопии для изучения каталитических систем с носителями» Интересно от¬
метить, что указанные проблемы, как это кажется, достаточно успешно
преодолены в каждой отдельной лаборатории, однако до сих пор не сущест¬
вует общепринятых процедур измерений, особенно в случае определения сдви¬
гов энергий связи, являющихся индикатором изменения химического соста¬
ва (см. обзор результатов, обсуждавшихся в п. 8.2.2). Основные причины
отсутствия согласия в этих областях измерений, как мы увидим, заключа¬
ются в отсутствии возможности зафиксировать нуль энергии связи (пробле¬
ма зарядки) и в неоднородности, которая присуща многим катализаторам
и которая часто нарушает описанную ранее статистическую природу электрон¬
ной спектроскопии- Для краткости дальнейшее обсуждение мы сконцентри¬
руем на влиянии этих моментов на результаты измерений в РФЭС, хотя мно¬
гие выводы в равной степени справедливы и в ЭОС.8.3.2.2. Проблема зарядки и отсчета энергииХимия поверхности как носителя, так и активных добавок катализатора
в основном изучалась методом РФЭС. Как упоминалось выше при обсужде¬
нии особенностей электронной спектроскопии, существуют две принципиаль¬
ные проблемы при использовании этого вида анализа в случае катализаторов:
а) концентрация активного вещества часто настолько мала, что обнаружить
какой-либо химический сдвиг очень трудно (подробнее этот вопрос обсужда¬
ется в соответствующих разделах, посвященных химии активных добавок),
и б) энергию связи носителей, которые в основном являются непроводящими
материалами, трудно зафиксировать из-за их сдвига вследствие зарядки
(см. приложение 2 и рис. 8.6, а и 8.6, б). В других главах этой книги показа¬
но, что величину результирующего сдвига вследствие зарядки определяет
морфология, а также состав каталитических систем. Дополнительные слож¬
ности могут возникать в связи с избирательной зарядкой. С этой проблемой
трудно бороться, особенно если используется обычный прием, заключающий¬
ся в "привязке" шкалы энергий связи к характерному значению для одного
из атомов носителя с последующим отсчетом энергий связи для всех актив¬
ных веществ относительно этого значения- Ни одна из этих проблем не бы¬
ла полностью преодолена или устранена, однако последующее изложение,
возможно, даст по крайней мере полезное представление об этих проблемах
и укажет пути их преодоления-
Т.П. Барр 337Рис. 8.6. Примеры зависимости зарядового сдвига РФЭС-линии 01 s от тока компен¬
сирующей заряд электронной пушки, а - Si02:: наблюдается значительный
сдвиг при выключении компенсирующей пушки, б - NaA:: зарядовый сдвиг
незначителен. Оптимальный ток равен 0(3 мА. 1 - компенсирующая пушка
выключения.Льюис и Келли [ 76] опубликовали обзор, в котором с общих позиций
рассмотрена проблема сдвига вследствие зарядки. Вагнер [ 77], Виндэви и
Вагнер [78] также рассмотрели зарядку и продемонстрировали, как на нее
влияет однородность материалов. Виндэви [79] рассмотрел некоторые воп¬
росы, связанные с тем, в какой мере отсутствие однородности может при¬
водить к неоднородной зарядке [ 14].
3388. Применение электронной спектроскопии в гетерогенном катализеЭти идеи недавно были обобщены в работе автора настоящей главы
[ 80], в которой были определены типы неоднородной зарядки и предложены
модели, основанные на типах морфологических элементов, построенных из
двух веществ с различными параметрами зарядки. В этой работе было показано, что,
даже если активное вещество однородно распределено (диспергировано) на
носителе, оно может проявлять некоторые виды неоднородной зарядки.В работе [80] ив работе Льюиса и Келли [ 76] также был продемонстриро¬
ван тот факт, что зарядовый сдвиг является только частью проблемы от¬
счета энергии. Вторая часть обусловлена "плаванием” (отсутствием свя¬
зи) уровня Ферми многих материалов относительно уровня Ферми спектро¬
метра, что связано с отсутствием термического равновесия. Эта особен*
ность имеет место для непроводящих образцов и для проводящих материа¬
лов, электрически изолированных от спектрометра. Если это имеет место,
то энергии связи в методе РФЭС, регистрируемые относительно нуля (уров¬
ня Ферми) спектрометра, неверны. Таким образом, энергии связи для актив¬
ного вещества катализаторов не только сдвинуты вследствие зарядки (что
естественно, например, для "химии" окислов носителя), но они также в боль¬
шей или меньшей степени сдвинуты из-за этого "плавающего уровня Ферми".
Все эти эффекты зависят от морфологии изучаемой системы» Некоторые кон¬
кретные примеры и модели представлены на рис. 8.6 и 8„7.Эти проблемы привели к анализу и дополнительному использованию раз¬
нообразных операций, направленных на устранение зарядовых сдвигов и
получение истинных энергий связи. К сожалению, ни одна из этих операций
не является универсальной. По-видимому, наиболее часто используются
следующие методы:а. Как упоминалось выше, энергии связи некоторых пиков ключевых
элементов носителя, например А12р в А1203 , как принято считать, имеют
фиксированное значение независимо от состояния системы» Иногда такое
приближение приемлемо; однако существуют также случаи, когда этот под¬
ход не позволяет определить такие важные характеристики, как взаимодей¬
ствие между металлами и носителем. В целом автор настоящей главы счи¬
тает это приближение грубым упрощением, которое часто является некор¬
ректным [28]„б. Чтобы обойти упомянутую трудность, многие авторы полагали, что
пику С Is, связанному с адсорбированными случайными примесями углеро¬
да, можно приписать фиксированное значение энергии, например 285 эВ, и
все другие энергии связи отсчитывать от этого значения. Применимость, а
также слабые места такого подхода недавно были проанализированы в обзо¬
ре Свифта [ 82], который советует соблюдать крайнюю осторожность при ис¬
пользовании этого метода. Метод был проанализирован также в лаборатории
автора [ 39, 80], в результате был сделан-вывод, что многие практики не
понимают важности этих двух независимых, но взаимосвязанных проблем,
т. е. зарядового сдвига и отсутствия связи между уровнями Ферми спектро•
Рис. 8.7. Схемы энергетических уровней для РФЭС-экспериментов. а - проводящий
образец по уровню Ферми (F) находится в контакте со спектрометром. Экс¬
перимент Зигбана. б- образец 1 из полупроводника или диэлектрика. Пока¬
зана зарядка у и ее устранение. Имеет место отсутствие связи уровней
Ферми; фп - энергия, необходимая для компенсации Ч^1 (внешний потен-
циел или зарядка) и перехода электрона с уровня вакуума (Вак.) образца
на уровень вакуума спектрометра. Для упрощения эта энергия, как принято
считать, не влияет на ф5 ^ или . 3) - ф5 - (<рсп - Ф5) -~£кин»£в = *v - Фсп - Екин; Ев = Ев вследствие теплового равновесия (вы-
М AIравнивания уровней Ферми), следовательно, Еп- hv - ф - е причем** СП кин/iv - фсп — величина, зависящая от спектрометра, а Екин определяетсяизмерением. ^ = hv - £|<ин _ 9cnJ = hv - Екин - ^ - v
^контакт + *еа
3408. Применение электронной спектроскопии в гетерогенном катализеметра и образца. Вследствие сложной взаимосвязи этих эффектов при ис¬
следовании носителей, активных добавок и случайных примесей было выска¬
зано предположение о возможной необходимости устранения независимым
способом сдвига, обусловленного зарядкой (возможно, при малых токах
электронов, см. ниже), прежде чем пытаться разрешить проблему уровней
Ферми, а шкалу энергий связи калибровать, например, по линии С Is о Одна¬
ко необходимо отметить, что для практической пригодности даже этой пос¬
ледней методики все анализируемые соединения должны находиться в тес-
ном контакте с углеродом, который используется в качестве эталона. В про¬
тивном случае возникнет дифференциальная зарядка и отсчет энергии связи
будет неверен.в. Устранение зарядового сдвига путем использования специальной
электронной пушки, нейтрализующей зарядку, представляет собой другой
подход, который часто использовался при попытках получения истинных
энергий связи для материалов типа нанесенных катализаторов (см. рис. 8.6).
Однако, как указал Виндэви [ 79] и обобщил Барр Г 80], такая методика,
даже если она прекрасно работает для подавляющего большинства сплош¬
ных систем, не будет действовать в случае несплошнух систем, подвержен¬
ных дифференциальной зарядке. Кроме того, как показали Льюис и Келли[ 76] и обобщил Барр Г 80], устранение зарядового сдвига сплошных систем
само по себе не способствует выравниванию уровней Ферми. Это означает,
что при любом способе определения работы выхода образца для получения
правильных значений энергий связи по-прежнему необходимо зафиксиро¬
вать уровень Ферми [ 83] или иметь точку отсчета энергий связи.г. Четвертый способ разрешения проблемы зарядки и установления по¬
лезных (и иногда истинных) значений энергий связи для материалов, ана¬
логичных металлическим нанесенным катализаторам, состоит в использо¬
вании параметров, характеризующих весь спектр, которые не зависят как
от зарядового сдвига, так и от смещения уровня Ферми. Существуют нес¬
колько таких параметров, каждый из которых, по-видимому, имеет свои
преимущества, которые, однако, не универсальны. Один такой метод сос¬
тоит в использовании оже-параметров (более детально описанных в других
главах этой книги). В этом методе для каждой системы регистрируются
характеристические значения (определяемые расщеплением между основным
фотоэлектронным пиком и оже-пиками), которые во многих отношениях ана¬
логичны химическим сдвигам, однако не зависят (в первом приближении для
сплошных материалов) от зарядки и смещения уровня Ферми. Эта методика
была введена в практику Вагнером (см., напримерД 37, 38]) и недавно полу¬
чила дальнейшее развитие в работе Кастла и Уэста (см., напримерД 84, 85]).
В работе [ 86], которая представляет особый интерес для тех, кто интересу¬
ется носителями катализаторов, Вагнер и др. распространили эту методику
на ряд кислородных соединений алюминия и кремния.
Т.П. Барр341Еще одна возможная методика состоит в регистрации ФЭС-спектров по¬
терь, которые были описаны в начале этой главы [29-31]. В литературе
описано успешное применение этого метода к SiOa [28], А1203 [ 32] и цеолитам [28].
В этих работах было показано, что сдвиги спектров потерь могут помочь в опреде¬
лении не только химического состава, но также и морфологии образцов. В первом
приближении сдвиги спектров потерь зависят только от электронных свойств
эмитирующих материалов, а также не зависят от положения уровней Ферми.При рассмотрении методов решений проблемы сдвигов, обусловленных
зарядкой, часто забывают, что, как и во многих других аналогичных ситуа¬
циях, могут возникать весьма полезные побочные эффекты. Например, бМло
показано [ 28], что из информации о наличии сдвигов, вызванных зарядкой
(и/или о неравенстве уровней Ферми), величине этих сдвигов и ее отклике
на изменение тока электронной пушки, компенсирующей зарядку, имеется
возможность микроскопически описать состояние поверхности (а иногда
и подповерхностных слоев). Это достигается путем сравнения эксперименталь¬
ных результатов с соответствующей моделью [ 80].8.3.2.3. Количественный анализ поверхностей катализаторовБыл опубликован ряд работ, посвященных использованию РФЭС исключи¬
тельно для определения количественных характеристик поверхностей ката¬
лизаторов. Однако полученные результаты указывают на необходимость
проведения дополнительных фундаментальных исследований.Основы количественного анализа методом РФЭС были изложены в гл. 5.
Поверхности катализаторов имеют ряд специфических особенностей, связан¬
ных с часто присутствующими у них неоднородностями (например, многофаз-
ность, изменение состава по поверхности, рост кристаллов, частичное клас-
терирование и т. д.). Брайнен [25] первым рассмотрел однородный случай
и некоторые из перечисленных неоднородностей. Кроме того, Шарпен [ 87]
подробно описал, насколько отсутствие дисперсности платины на Si02 мо¬
жет усложнить интерпретацию результатов количественных измерений. С тех
пор было разработано несколько моделей, рассматривающих проблему дис¬
персности, т. е. случаи, когда одна фаза (например, активное вещество)
плохо диспергирована в другой фазе (например, носителе), а также эффек¬
ты пористости носителя. Из-за недостатка места мы не будем описывать
эти модели, подробное их изложение заинтересованный читатель может най¬
ти в публикациях, посвященных исследованию дисперсности: Анжевен, Вар-
тули и Дельгас [ 88], Керкхоф и Мулин [ 89], Виндэви и Вагнер Г 78]. Фанг
[ 90] для объяснения характерных изменений формы недиспергированной фа¬
зы ввел дополнительные параметры в эти модели. Следует также ознако¬
миться с недавно опубликованными идеями Нефедова и др. [91—94], учи¬
тывающими влияние упругого рассеяния на результаты количественных
измерений.
3428. Применение электронной спектроскопии в гетерогенном катализе8.3.3. Катализ на платиновых металлах
8.3.3.1. Исследования Сомораи катализа "молекулярной структуры"Прежде чем приступить к изложению применения электронной спектро¬
скопии к прикладному катализу на платиновых металлах, поучительно про¬
анализировать предпринятые в этом направлении усилия Сомораи и его кол¬
лег. В основном эта работа выполнялась с использованием монокристаллов
платиновых металлов и была поставлена с целью получить представление
о некоторых определяющих моментах реакций типа риформинга. Среди всех
фундаментальных исследований, направленных на моделирование промыш¬
ленных катализаторов, эта работа, возможно, дает модель, ближе других
соответствующую реальности. В большинстве этих исследований различные
аспекты науки о поверхности используются в качестве определяющего
modus operand^, причем главную роль играет ДМЭ (и с недавних пор СХПЭЭ).
Использовалась также обычная электронная спектроскопия, особенно ЭОС
(в последнее время все чаще применяется РФЭС; см., например, [95]).В этих работах Сомораи и его коллеги пытались связать некоторые сторо¬
ны катализа на металлах с реакционным поведением большого числа ос¬
новных органических газов на полученных различными способами поверх¬
ностях платиновых монокристаллов (в первую очередь). Никаких носителей
в классическом смысле не использовалось; таким образом, грани кристал¬
лической платины действовали не только как катализирующий реагент,
цо также обеспечивали центры адсорбции. Практически во всех этих иссле¬
дованиях регистрировались следующие пять основных параметров: а) тип
органических реагентов и продуктов реакций; б) тип граней (с использова¬
нием специальных сколов для создания террас, ступенек и уступов); в) тем¬
пература (реакции проводились при температурах от 75 до 550 К); г) дав¬
ление Р газов, где 10~4 атм < Р< 100 атм (верхний предел был достигнут
благодаря специальной установке для работы при высоких давлениях); д) раз¬
личные параметры и расположение (на гранях монокристалла) удерживае¬
мых углеродсодержащих побочных продуктов реагентов, перечисленных в
"а". Регистрировалось также время жизни этих углеродистых слоев на
поверхности.Сомораи и его коллеги открыли, что (при используемых условиях) боль¬
шинство обычных реакций алканов и олефинов (дегидратация, циклизация,
изомеризация, гидрогенолиз и т. д.), по-видимому, более эффективно про¬
текает на гранях (монокристалла металлической Pt) с большими индекса¬
ми Миллера (низкая симметрия) [96]. Таким образом, если относительно
низкотемпературная хемосорбция и реакции не имеют места, например на
гранях (100), то они относительно легко пойдут на грани (111), особенно
при наличии ступенек и уступов. Сомораи и его коллеги открыли, что упо-Способ действия (лат.). - Прим. перев.
Т.Л. Барр343мянутый выше разрыв связей и процессы перестройки происходят избира¬
тельно на указанных гранях. Это означает, что грани монокристаллов плат
тины по-разному обеспечивают две основные характеристики хорошего ка¬
тализатора: а) энергию, необходимую для активации реакции, и б) селек¬
тивность, необходимую для выделения одной реакции из нескольких конкури¬
рующих процессов [ 8].Методом ДМЭ наблюдались дифракционные картины от указанных гра¬
ней. Подтверждение качественных и количественных характеристик адсор¬
бированных соединений осуществлялось методом ЭОС, а реагенты и продук¬
ты реакций определялись транспортными методами [ 98]. Как упоминалось
выше, исследования при высоких давлениях стали возможны благодаря ис¬
пользованию уникальной СВВ-системы со встроенной в нее камерой высоко¬
го давления [ 98].В частности, было обнаружено, что грань Pt (111) обладает значительно
большей реакционной способностью к дегидроциклизации, чем грань Pt (100)
[99]. Утверждалось также, что реакция гидрогенолиза предпочтительно
идет на уступах этой грани, в то время как дегидроциклизация, по-видимо¬
му, предпочитает ступенчатые участки [100, 101]. Сомораи и Заира сум¬
мировали все эти особенности в недавно опубликованном обзоре [ 102]. В этой
публикации были рассмотрены аналогичные реакции на гранях различного
типа (рис. 8.8) и для каждой из них были представлены данные об относи¬
тельной активности и селективности (рис. 8.9) [ 102]. Заметим, что количест-Рис. 8.8. Идеализированная атомная структура плоских поверхностей Pt (111) и
rt (100), ступенчатая поверхность Pt (755) и поверхность Pt (10, 8, 7)
с уступами [102].
3448. Применение электронной спектроскопии в гетерогенном катализеРис. 8.9. а - кривая зависимости накопления толуола при превращении n-гептана на
поверхностях Pt(111) и Pt (100) от времени реакции, б- зависимость де¬
гидроциклизации при давлении 15 мм рт. ст. для я-гептана, 480 мм рт. ст.
для водорода и при температуре 573 К от структуры поверхности [ 102].во атомов платины, расположенных на террасах (над ступеньками и уступа¬
ми), также играет некоторую роль.Было обнаружено, что указанные реакции катализируются при наличии
тонких упорядоченных слоев углеродистых побочных продуктов [ 103]. С дру¬
гой стороны, толстый аморфный слой этого "кокса”, по-видимому, отрав¬
лял (блокировал) активные центры катализатора, особенно если углерод
терял свою подвижность. Аналогичное поведение наблюдалось для похожих
реакций другими исследователями [104]. Проведя довольно тонкие экспери¬
менты с 14С [ 105], Сомораи и его коллеги смогли обобщить эти положения
и включить в рассмотрение предложенные ими состояния деградации (после¬
дующей дегидрогенизации) этого углеродистого налета [ 106]. Исходя из
этих соображений, они построили модели каталитически активных, подвиж¬
ных групп С2Н3, которые при дальнейшем нагреве или их использовании
становятся неподвижными, похожими в этом отношении на группы С2Н в
верхнем слое углеродистого налета. В конце концов адсорбированные гра-
Т.П. Барр345фитовые "хлопья” начинают расти и скорости всех реакций, по-видимому,
уменьшаются экспоненциально с ростом концентрации этих образований
[ 102]. Рис. 8.10 служит наглядным изображением влияния этого углеродис¬
того налета*Было показано также, что селективное введение некоторых металлов
(например, олова) может препятствовать гидрогенизации и тем самым уси¬
ливать дегидроциклизацию. Этот процесс может вызываться блокировкой
уступов оловом и отводом электронов от углов, что приводит к активации
ступеней [ 107]. Эти особенности могут иметь отношение к поведению, при¬
писываемому биметаллическим катализаторам, которые необходимы при
производстве высокооктанового бензина. Недавно Захтлер и др. [108] по¬
пытались глубже связать эти исследования монокристаллических катали¬
заторов с биметаллическими катализаторами. Были исследованы структуры
слоев золота на платиновых монокристаллах (и наоборот). Кроме некоторых
изменений структуры, было замечено, что золото на Pt (100) заметно
увеличивает скорость дегидрогенизации циклогексана в бензол. Кроме того,
золото на Pt (111), по-видимому, усиливает изомеризацию n-гексана (при
575 К), в то время как дегидроциклизация и другие реакции замедлялись
этим слоем золота [ 109] (рис. ft. 11).Недавно было описано влияние некоторых дополнительных реагентов,
включая кислород. Было показано (методом ЭОС), что предокисление из¬
меняет скорости упомянутых выше реакций, усиливая дегидрогенизацию,
очевидно, в связи с тем, что сильно связанный кислород изменяет элект¬
ронную структуру Pt (111) на уступах или около них [ 110]Сомораи и Заира [ 102] недавно распространили исследования такого
типа также на другой платиновый металл, родий. Они спланировали свои
эксперименты таким образом, чтобы выявить эффекты, связанные с изме¬
нениями состояния окисления этого металла. В этом случае вместе с мето¬
дом ЭОС для исследования восстановления N0 молекулой СО на Rh (331)(эта реакция представляет интерес для химии загрязнений воздуха) исполь¬
зовался также метод СХПЭЭ [111].Молекула N0 диссоциирует, образуя, по-видимому, в промежуточном
состоянии адсорбированный атом кислорода. Кроме того, Уотсон и Сомораи
[112] использовали РФЭС, а также ЭОС для изучения гидрогенизации СО на
родиевых катализаторах (эти реакции представляют интерес в технологии
син-газа). Метод РФЭС показал, что Rh° дает в основном алканы и олефи-
ны, тогда как для получения альдегидов и других окисленных продуктов необ¬
ходим Rh203. Однако обезвоженный Rh2Oa легко восстанавливается до
металлического состояния, замедляя реакции окисления. С другой стороны,
гидрированный Rh203 только частично восстанавливался в этом процессе,
приводя к образованию на поверхности мозаики из окисла и металла. При
расширении этих исследований недавно было открыто, что La RhOs стабили¬
зирует реагенты, приводя к образованию почти исключительно окисленных
3468. Применение электронной спектроскопии в гетерогенном катализе
T.J1. Барр347продуктов. Вероятным промежуточным продуктом, по-видимому, является
Rh+1 [ 113]. Недавно были предприняты первые попытки, используя метод
СХПЭЭ, распространить эти исследования на родий, находящийся на носите¬
ле А1203 °.Рис. 8.11. Изменения активности и избирательности реакций конверсии n-гексана в
Зависимости от степени покрытия золотом системы, представляющей со¬
бой сплав золото - Pt (111) [102]. Hj/HC = 10, Рполн= 220 мм рт. ст.Т = 573 К*Рис. 8.10. а - относительная доля чистой поверхности платины, измеренная методом
адсорбции - десорбции СО, в зависимости от степени покрытия углеродом
граней платины (100), (111) и (13, 1,1). Проведено сравнение между погло¬
щением СО, измеренным после исследования реакции п-гексана (7),и по¬
глощением СО, измеренным, когда СО адсорбировался совместно с "гра¬
фитовым" поверхностным углеродом при 673 К (2). б - состав и реакци¬
онная способность этилена-14С, хемосорбированного на Pt (111) при тем¬
пературе в диапазоне от 20 до 370°С; относительное количество адсорби¬
рованных молекул определялось методом меченых атомов, который пока¬
зал прекрасное согласие со средним содержанием водорода (Н/С) в сильно
связанных с поверхностью соединениях, в - модель образования рабочей
поверхности платинового восстанавливающего катализатора [102]. 1 - трех¬
мерный углеродный островок; 2 - углеродистый верхний слой; 3 - непокры¬
тая совокупность узлов Pt.l)Koel В.Е., частное сообщение-
3488. Применение электронной спектроскопии в гетерогенном катализеРис. 8.12. а - чувствительность структуры поверхности для случая синтеза аммиака
с использованием железного катализатора; б - идеализированная атомная
структура плоскостей железа с низкими значениями индекса Миллера: Fe
(100), Fe (111) и Fe(110) [102].Создание описанной выше комбинированной камеры высокого и низкого
давления позволило также изучать другие важные химические реакции на
кристаллах неплатиновых металлов. Например, ЭОС и упомянутая выше ка¬
мера использовались для анализа возможности использования железа в ка-
чесгве катализатора для получения NH3 из N2 и Н2 при 798 К и 20 атм [ 114].
Было обнаружено, что эта реакция предпочитает исключительно грань Fe
(111), а не грани Fe (100) и Fe (110) (рис. 8.12).Сомораи в своем последнем обзоре высказывал предположения о том,
как упомянутая информация может привести к промышленным вариантам
"высокоэффективных молекулярных катализаторов"» Такое предсказание
Т.П. Барр349может показаться немного преждевременным, однако практическое исполь¬
зование этих идей в будущем кажется вполне вероятным. В то же время
впечатляет обилие полученной информации, особенно относительно селектив¬
ного стимулирования реакций в узлах с низкой симметрией и когда за эта¬
пом стимулирования следует отравление катализатора вследствие образова¬
ния слоев "кокса".В.3.3.2. Введение в совоеменный катализ на платиновых металлахПлатина используется в качестве катализатора в течение многих лет,
однако лишь после открытия Хензелем [115] процессов риформинга стали
очевидными особые каталитические свойства платины. Эта каталитическая
система постепенно эволюционировала в систему, в которой платина в фор¬
ме комплекса помещается на носитель с большой площадью поверхности,
часто на уА1208 [116]. Эти системы впоследствии восстанавливаются, об¬
разуя, очевидно, микроскопические частицы Pt°, которые сильно дисперги¬
рованы по поверхности окиси алюминия. Частицы Pt° настолько малы, что
их невозможно было обнаружить обычным методом дифракции рентгеновских
лучей [ 117]. Эту каталитическую систему часто усиливали ионами галогенов,
что увеличивало кислотность окиси алюминия и приводило, таким образом,
к созданию бифункционального катализатора, пригодного для избиратель¬
ного стимулирования реакций дегидроциклизации, необходимых для увеличе¬
ния октанового числа высококачественного бензина [ 116, 117]. Эта система
в немного измененном виде также находит применение в развивающейся вы¬
сокими темпами нефтехимической промышленности, особенно в качестве де-
гидрогенизирующего катализатора при производстве олефинов [ 116L Таким
образом, к середине 60-х годов катализ на платиновых металлах, использу¬
емый для производства (методом риформинга) высокооктанового бензина,
нефтехимических продуктов и даже для некоторых видов окисления стал глав¬
ной независимой ветвью катализа. Однако эти каталитические системы ри¬
форминга не были лишены недостатков, поскольку они имели тенденцию те¬
рять активность и избирательность при их использовании для восстановле¬
ния низкокачественного сырья при низких давлениях и высоких температу¬
рах [ 117]. Поэтому даже до того, как была понята природа чисто платиново¬
го катализатора, ученые и инженеры, работающие в области катализа, для
улучшения этой каталитической системы экспериментировали с различными
добавками. Как следствие этих усилий в конце 60-х годов появились биме¬
таллические катализаторы.Многие из наиболее важных промышленных катализаторов представляют
собой полиметаллические системы, работоспособность которых ряд исследо¬
вателей связывал с их способностью образовывать сплавы [ 74]« Это полнос¬
тью справедливо для систем VIII - 1В, где первый член системы являет¬
ся катализатором, а второй работает в качестве модификатора. Утвержда-
3508. Применение электронной спектроскопии в гетерогенном катализелось, что избирательное поведение этих катализаторов зависит в равной
степени как от числа "смежных участков поверхности”, так и от их распо¬
ложения [ 118]» Таким образом, например, утверждается, что биметалличес¬
кие системы ведут себя одним способом, если их можно схематично предстаг
вить в виде -/4-Я-/4-#-Л-(где А -элемент группы VIII, а В - эле¬
мент группы 1£), и другим способом, если их можно представить в виде
-А-В-А-В-А-А-А-В-А [119]. Первая полностью дисперсная систе¬
ма, по-видимому, способствует разрыву или образованию -С-Н-свя-
зей, в то время как вторая система (с кластерами А) способствует разрыву
и/или перестройке связи С-С [74, 119]. С другой стороны, очевидно, что
соседние относительно большие кластеры атомов элементов VIII группы
(например, платины) будут усиливать реакции, которые, возможно, вызы¬
вают процессы "коксообразования”, быстро деактивирующие катализатор,
тогда как катализаторы, представляющие собой в основном микрокристаллы
платины (разделенные друг от друга сплавами (?) с элементами группы 1 В или с
элементами типа рения), по-видимому, обладают сильно выраженными дегидрогенизи-
рующими свойствами [74, 116]. Носитель этих биметаллических сплавов (как
правило, окись алюминия), возможно, также имеет кислотные участки, ко¬
торые могут независимо стимулировать избирательную циклизацию (арома¬
тизацию) [ 116]. Эту кислотность можно увеличить добавкой галогена и соот¬
ветствующей термической обработкой [116, 120]. Тагом образом, эти систе¬
мы можно считать биметаллическими бифункциональными катализаторами
[ 74]. Добавки элементов группы 1В и VII В (а иногда группы IV А) часто
называют модификаторами, так как они увеличивают долговечность и избира¬
тельность катализатора, в то же время подавляя (ослабляя) побочные реак¬
ции, которые дезактивируют каталитическую систему [119].Следует отметить, что ряд экспериментаторов опубликовали убедитель¬
ные результаты исследований, которые оспаривают понятие сплава в биме¬
таллических платиновых катализаторах [121]. Основное отстаиваемое ими
положение заключается в том, что модификаторы сохраняют состояние окис¬
ления после восстановления катализатора и что они взаимодействуют с пла¬
тиной таким способом, который предотвращает группировку атомов платины
в большие кристаллиты низкой активности [121].Информация по характеристикам этих катализаторов сильно рассеяна
в имеющейся литературе [118]. Относительное количество металлических
активных добавок в основном очень мало, и исследователям, работающим в
области катализа, долгое время не удавалось идентифицировать кислотные
участки на носителях [ 121]. Имеется широкий спектр вопросов о составе,
макро- и микроструктуре, химических и металлургических характеристиках
катализатора до, в процессе и после их использования [118]. От специалистов
по электронной спектроскопии, например, требовалось представить информа¬
цию о таких важных характеристиках, как а) степень сплавления (если вооб¬
ще этот процесс имел место) [118], б) тип и расположение кислотных цент-
Т.Л. Барр351ров [ 121], в) состояние(я) окисления металлов, входящих в катализатор
[ 118], г) идентификация роста кластеров [121] и д) химические и физические
характеристики углеродистых отложений [74, 102]. По-видимому, удовлет¬
ворительный ответ был получен лишь на очень немногие из этих вопросов.
Сложная природа промышленных катализаторов, а также разнообразие их
применения и широкий спектр входящих в них ингредиентов приводили к
различным имитациям, включая использование неактивных носителей (нап¬
ример, Si02 [ 122]) и нереально большого количества добавок. Хотя получен¬
ные до сих пор результаты оказались полезными, однако достаточно деталь¬
ное объяснение катализа на "реальных" катализаторах группы платиновых
металлов еще не получило необходимого освещения в литературе [ 123].Исследования катализа на платиновых металлах методами электронной
спектроскопии, которые будут описаны, делятся на пять категорий в зависи¬
мости от основного анализируемого параметра. Отметим, что понятие "пла¬
тиновый металл11 было расширено таким образом, что оно включает в себя не
только триады легких и тяжелых платиновых металлов, но также и тяжелые
"монетные" металлы (серебро и золото) и (для целей риформинга) рений.
Медь и никель не рассматриваются.8.3.3.3. РФЭС-спектры ключевых элементов и их окисловДо недавнего времени было проведено только несколько исследований
методом РФЭС платиновых металлов, который представляет интерес для
изучения катализа. Виноград и др. (см., например [124-127]) получили
несколько интересных результатов по платине, палладию, серебру и их окис¬
лам. Автор настоящей главы также опубликовал подробное исследование
чистых металлов палладия, платины, родия, их окислов и гидроокисей [ 128].
Кроме того, Чимино и др. [ 129] представили подробные результаты для ре¬
ния и его многочисленных окислов, что является первым этапом последую¬
щего исследования рениевых катализаторов на носителях. Вероятно, самые
лучшие результаты для золота содержатся в упомянутом выше обзоре под
редакцией Поуэлла, Эриксона и Мейди [21]. Многие из этих исследований
суммированы с результатами других работ по энергиям связи в недавней
публикации отделения физической электроники фирмы "Перкин-Эльмер"[130].Все эти исследования имеют важное значение для катализа, так как в
этой области важнейшей проблемой является обеспечение обратимости тех
изменений, которым подвергаются чистые металлы и окислы на стадиях
окисления и восстановления, обычно используемых во время приготовления
катализатора. Кроме того, необходимо детектировать эффекты, связанные
с различной продолжительностью взаимодействия с воздухом. Упомянутые
выше исследования в нашей лаборатории показали, что многие металлы,
3528. Применение электронной спектроскопии в гетерогенном катализевключая в значительной степени и большинство рассматриваемых здесь,
окисляются в атмосфере 02 или в воздухе, образуя окисные слои высокой
степени окисления; эти окисные слои в воздушной среде в конце концов
превращаются (пассивируются) в тонкие слои гидрированных окислов
или гидроокисей [128]. Был опубликован [128] примерный перечень
"естественных пассивирующих" слоев, которые в случае рассмат¬
риваемых металлов образуются в воздухе при комнатной температуре. За¬
метим, что платина вообще слабо пассивируется и что в умеренном вакуу¬
ме (например, Ю-8 ммрт. ст.) скорость (и возможно, степень) этой пасси¬
вации уменьшается, однако не до нуля. Поэтому указанные наблюдения
имеют важное значение для исследования катализаторов, поскольку большин¬
ство катализаторов контактирует с воздухом в процессе их работы (с пло¬
хой герметизацией) или же при транспортировке в рабочую камеру. Таким
образом, за исключением золота (и возможно, платины), для всех метал¬
лов, входящих в катализатор, следует ожидать некоторого окисления по¬
верхности просто вследствие контакта с воздухом. Необходимо, однако,
отметить, что результаты, полученные на металлических фольгах, подобные
результатам работы [ 128], не обязательно точно переносятся на металлы,
рассеянные на поверхности носителей большой площади. По этой причине
необходимо соблюдать осторожность для правильной интерпретации истин¬
ного состояния катализатора и/или манипулирования их состоянием, осо¬
бенно при попытке оценить изменение степени окисления используемых
металлов. Однако вопреки возражениям, высказываемым некоторыми кри¬
тиками, эти проблемы не приводят к невозможности проведения анализа по¬
верхности катализаторов, так как часто можно избежать, игнорировать,
устранить или вычесть величину этого окисления из-за контакта с возду¬
хом. Важно помнить, что загрузка, использование и удаление промышлен¬
ных катализаторов не производятся с соблюдением условий, при которых
будет предотвращено некоторое окисление из-за упомянутой пассивации на
воздухе. Поэтому процессы, протекающие в сверхвысоком вакууме, который
поддерживается в обычных камерах для анализа поверхностей, могут не
воспроизводить соответствующее состояние промышленных катализаторов.
Учет перепада давлений, как это обсуждалось (отрицалось) в работе Мейдик-
са [ 49] и было использовано Сомораи и Заирой [102], является поэтому су¬
щественным моментом при обсуждении указанных вопросов. Значения наи¬
более важных энергий связи, полученные в вышеупомянутых исследованиях,
суммированы в работе [ 130]. Спектры соединений рения, представленные
в работе Чимино и др. [ 129], возможно, требуют некоторой поправки в свя¬
зи с упомянутыми проблемами контакта с воздухом. Дополнительно к этому
по крайней мере два "типа" PtO были зарегистрированы в экспериментах
Винограда и сотр. [124-127j. Исследователи из этой лаборатории сообщали
только о Pt110, однако были обнаружены признаки существования еще
двух "состояний". Одно из них с меньшим химическим сдвигом, PtOa , воз-
Т.Л. Барр353можно, связано с неполным окислением Pt°. Этот момент может иметь су¬
щественное значение для эффекта "извлечения электрона" в катализе на
платиновых металлах, как это описывалось Кларком и Кринером [123], а
также Берчем [ 121].8.3.3.4. Исследование метопом РФЭС активных добавок
катализаторов риформингаНесколько экспериментов по моделированию промышленных платиноме¬
таллических катализаторов риформинга дали полезную информацию, несмот¬
ря на ограничения, связанные с очень небольшой "наблюдаемостью" неплот¬
но распределенных активных добавок и естественной пассивацией, произво¬
димой окислом носителя. Эскар и др. [ 131], например, использовали РФЭС,
термогравиметрический анализ и рентгеновскую дифракцию (РД) для иссле¬
дования состояния поверхности гексахлороплатиновой кислоты (H2PtCl6)
при термических условиях, используемых при ее нанесении на платиновые
катализаторы как на воздухе, так и в инертной атмосфере (H2PtCle являет¬
ся стандартным источником платины для промышленных катализаторов)[ 121]. При использовании методов сравнительного спектрального анализа
было обнаружено, что поверхностные группы H2PtCl6 подвержены интен¬
сивному термическому разложению. Анализ энергий связи показал, что это
разложение явно приводит к образованию смеси, состоящей в основном из
Pt11С12 и Pt° , "взаимодействующей" с адсорбированным кислородом» Ана¬
лиз методом РД показал, что разложение главным образом, но не полностью
ограничено поверхностным слоем. Поэтому кажется, что разложение может
происходить отчасти из-за взаимодействия с атмосферой и отчасти под дей¬
ствием анализирующего пучка; хотя этот эффект ограничен по глубине, он
будет влиять на окончательную структуру катализатора. Чаран, Финстер и
Шнабель [ 132] изучали этот же вопрос; они попытались, используя древес¬
ный уголь и алюмосиликаты в качестве носителей, более точно смоделиро¬
вать процесс приготовления реальных катализаторов. Они также наблюдали
упомянутое выше разложение (даже на двух носителях!) на компоненты
Pt° и Pt11. Было также обнаружено, что степень этого разложения зависит
от состояния и типа носителя. Очевидно, что степень дисперсности сильно
влияет на степень разложения. Эти авторы заметили также, что Pt(NH8)4Cl2,
другой традиционный источник платины для платиновых катализаторов ри¬
форминга, не разлагается после обмена на алюмосиликатном носителе.Бозон-Вердурас и др. [ 133] также применили РФЭС и спектроскопию
ультрафиолетового и видимого диапазонов для исследования продуктов, об¬
разующихся как после нанесения, так и после окисления и восстановления
PdCl на Si02, Ti02 и А1203. В этом случае авторы считали, что они
обязаны использовать нереально большие концентрации платинометалличе¬
ских активных добавок, чтобы облегчить анализ методом РФЭС- Основной
3548. Применение электронной спектроскопии в гетерогенном катализемомент заключался в детектировании взаимодействия между металлами и
носителем. В этом случае оказалось, что комплекс PdClсохранялся в
окисленном образце. После восстановления имелись признаки наличия ква-
зи-комплекса, образованного с участием содержащегося в носителе (?) С1 ”
Они постулировали это, так как наблюдалось заметное уширение линий
Pd Ы для восстановленного катализатора. Носитель, по-видимому, играет
явную, но еще не полностью понятную роль в образовании этого комплекса.
Было обнаружено различие концентраций этого комплекса на разных носи¬
телях, причем из этих трех носителей наибольшая концентрация была най¬
дена для А1203# Было обнаружено также, что самая "комплексная” система
(Pd — А1203) в восстановленной форме наиболее слабо поддерживает окис¬
ление этилена.Для аналогичных целей Гриффитс и Эванс [ 134] исцользовали РФЭС,
электронную микроскопию и электронную дифракцию для изучения изменений
в частицах серебра на графите при попытках произвести их окисление. Эти
результаты продемонстрировали увеличенную реакционную способность уг¬
лерода, что является следствием образования границы раздела фаз серебро-
кислород - углерод. Однако авторы отрицали важность сдвига спектра Ag
3d после восстановления как достаточного (по моему мнению) доказательст¬
ва окисления серебра. В этой работе, как и во многих других аналогичных
исследованиях, сдвиг вследствие зарядки мог играть не до конца учтенную
роль [ 28].Батиста-Лкаль и др. [ 135] также использовали метод РФЭС для изучения
изменений, происходящих в АиС13 и других золотосодержащих системах при
их термической обработке как в чистом виде, так и в случае их нанесения на
А120^. Нагрев в воздухе до 350 °С приводил к полному восстановлению
Аиш до Аи°, в то время как нагрев до 110 °С восстанавливал большую часть
Аиш до Au1. С другой стороны, АиС13, нанесенный на А1203, при комнат¬
ной температуре вовсе не восстанавливался. Авторы обсуждаемой работы
полагают, что эти эксперименты необходимо проводить более тщательно,
так как они обнаружили некоторое восстановление, индуцируемое рентгеновс¬
ким излучением.Шипиро и др. [136] применили РФЭС для исследования состояний различ¬
ных соединений Re™, нанесенных на носители Si02 и А1203<> Было обнару¬
жено, что степень восстановления определенных соединений рения на
у-А1203 выше, чем для тех же соединений на Si02. Как считают Шипиро и
др. [136], это является следствием значительно большего взаимодействия
металлов с носителем для Revn в первом (А1203) случае. Восстановле¬
ние этой системы в Н2 при 300 °С давало промежуточные состояния окисле¬
ния рения, возможно, ReIv. Нагрев в воздухе до 400 °С систем на носите¬
лях как из измельченного А1203, так и Si02, по-видимому, вновь окисля¬
ет рений до Re™ . Все эти особенности продемонстрировали применимость
Т.Л. Барр355РФЭС для описания изменений электронных свойств металлов, диспергиро¬
ванных на "активных" (А1203)и "неактивных" .(Si02) носителях. Очевид¬
но, эти особенности являются ключевым фактором, определяющим поведе¬
ние катализатора.Было опубликовано несколько работ, в которых электронная спектро¬
скопия использовалась для исследования многокомпонентных платинометал¬
лических катализаторов, моделирующих современные промышленные вос¬
становительные каталитические системы. По-видимому, наиболее интерес¬
ные и тщательные работы начаты в исследовательской лаборатории фирмы
"Шелл" в Амстердаме в группах Захтлера, Билона и других группах. Были
исследованы как система Pt -Re -S -Si02, так и система Pt -Ge -А120«,
однако в обоих случаях авторы из-за "проблемы наблюдаемости сигналов" бы¬
ли вынуждены использовать активные добавки в значительно больших коли¬
чествах по сравнению с их количеством, обычно используемым в соответ¬
ствующих промышленных системах. Исследования методом РФЭС восстанов¬
ленной системы Pt -Ge -А120^ [122, 137} показали наличие спектра
Pt 4f, который, как это было проинтерпретировано, возникает от Pt°, обед¬
ненной электронами. Состояние окисления германия в этом случае, по-ви-
димому, определяется как Ge2+ и Ge°, если система восстанавливалась вЩ при 625 °С, и как Ge4+ и Ge2+ для процессов восстановления при 550 °С
в присутствии платины. Эти работы были первыми детальными исследования¬
ми РФЭС, в которых представлены данные об образовании биметаллическо¬
го сплава (рис. 8,13, табл. 8.4). Были получены также данные, явно указыва-Рис. 8.13. Положение фотоэлектронных линий Ge 3d для a) Ge - А1203 и б) Pt,Ge -А1203 (точка отсчета:С Is = 284,6 эВ). 1 - прокаливание, 525°С; 2 -
прогрев в водороде, 450 °С; 3 - прогрев в водороде, 550 ^С; 4 — прогрев
в водороде, 650 °С [137].
356 8. Применение электронной спектроскопии в гетерогенном катализеТаблица 8.4. Положение линии Pt 4dm в некоторых стандартныхсистемах а [126]СистемаЭС, эВАЭС6ДЭС”Металл314,10РЮ2318,03,9Н2РЮб317,53,4Na2Pt04316,72,6PtGe0 72 сплав
W/y-A^O,315,51,40313,5-0,6Pt/ ai2o3После прокаливания
при 525 °С317,02,93,5После нагрева
в водороде при 650 °С
Ft, Ge/Al203314,60,51,1После прокаливанияпри 525 °С316,72,63,2После нагрева в водородепри 650 °С315,11,01,6а Энергия перехода С 1s равна 284,6 эВ
6 Эталон: металлическая платина.в Эталон ”Pt°”/7 л AL203, полученный путем измерения расстояния между линиями (Au —Р{)объем 4с,5/2 и измерения положения линии Au 4dg/2 для 0,1 монослоя золота на поверхности
у - А1?03.ющие на диффузию обоих металлов внутрь носителя S Ю2. Методы ИК и РФЭС,
а также методы хемосорбции показали, что сера способствует выде¬
лению атомов платины в катализаторе, представляющей собой сплав (?)
Pt -Re -S -Si02 [ 138]. Авторы иллюстрируют это положение следующим
образом:На такое превращение, по-видимому, указывали изменения в РФЭС-спектре
Re 4/, наблюдавшиеся после добавления сульфида. Авторы заметили также,
что создание и сохранение "дисперсных11 атомов платины, по-видимому, га¬
рантируют каталитическую избирательность и стабильность процесса вос¬
становления. Замена традиционного носителя из А1203 носителем из Si02
была произведена для того, чтобы избавиться от кислотности А1203, а так¬
же чтобы устранить влияние на линии Pt 4/ линий А1 2р. Такая замена силь¬
но изменяет каталитическое поведение этой системы по сравнению с типич¬
ным поведением стандартной промышленной (с носителем из А1203) систе-
Т.П. Барр357мы, так как на функционирование последней системы значительное влияние
оказывает кислотность окиси алюминия.В области невосстановительного катализа Анжевен и Дельгас [139] с ис¬
пользованием РФЭС изучили катализатор Ru -Мо на Al20j и Si02. Этот ка¬
тализатор использовался в реакциях метанолирования. При изучении пред¬
шественников этого катализатора, а именно систем Ru -Si02 и Ru -А1203,
было обнаружено, что первая система допускает полное восстановление
(нагревом в водороде до 450 °С) рутения до Ru°, в то время как в случае
второй системы (А1203) 45% рутения остается в окисленном состоянии.
Анжевен и Дельгас [ 139] связывали этот эффект с сильным взаимодейст¬
вием металл - носитель между рутением и А1203, которое отсутствует в
случае Si02. С другой стороны, в системе MoSi02 при тех же условиях
восстанавливался только до MoIV, тогда как в системе Ru -Мо - А1203 молиб¬
ден восстанавливался до Мо 0. По мнению авторов, это означает, что биметалличес¬
ки эффет(между рутением и молибденом)превышает взаимодействие металл -
носитель.8.3.3.5. Изучение образования кластеров и других морфологических
изменений платинометаллических катализаторовМетоды РФЭС, УФЭС и ЭОС совместно с РД, электронной микроскопи¬
ей, ДМЭ и методами термической спектроскопии использовались для полу¬
чения информации о микро- и макроморфологии как активных дооавок, так
и носителей промышленных и модельных платинометаллических катализато¬
ров. Эти исследования имеют важное значение, так как было показано, что
поведение таких катализаторов решающим образом зависит как от степени
спекания подложки, так и от степени кластерирования активных добавок.В частности, было обнаружено, что использование платинометаллических
катализаторов в условиях экстремальных физических (например, термичес¬
ких) напряжений может способствовать росту кристаллов платиновых метал¬
лов. По мере роста этих кристаллов коренным образом изменяются как сте¬
пень, так и тип последующих каталитических реакций. Для продолжительно¬
го поддержания реакций дегидроЦиклизаЦйи (основного производителя вы¬
сокооктанового бензина в методе риформинга) необходимо (как это упоми¬
налось выше) затормозить рост этих кристаллов. Поэтому любая комбина¬
ция упомянутых выше методов исследования, с помощью которой можно вы¬
делить задачи этого типа, имеет важное значение для промышленного ка¬
тализа. Вплоть до последнего времени электронная спектроскопия не игра¬
ла существенной роли в этой области, однако теперь, по-видимому, ситуа¬
ция изменилась.
3588. Применение электронной спектроскопии в гетерогенном катализеПроблема роста кристаллитов, а также некоторые попытки ее исследо¬
вания (хотя и не с помощью электронной спектроскопии) были прекрасно опи¬
саны в работе Даутценберга и Вальтерса [ 140]. Изучая влияние термичес¬
кой обработки на степень дисперсности платины на А1203, эти авторы ис¬
пользовали методы РД, ТЭМ и (БЭТ) хемосорбции Н2. Для увеличения
" наблюдаемости” использовалось большое количество платины (2 вес. %).С помощью метода РД авторам этой работы удалось обнаружить рост крис¬
таллитов металлической платины до кластеров с размером более 30 А. Ана¬
логичный диапазон размеров частиц был определен методом ТЭМ. Было так¬
же обнаружено, что пока отношение концентраций водорода и платины было
порядка единицы, платиновые кристаллиты имели малый размер. Рост этих
кристаллитов (до среднего диаметра 100-150 А) сопровождался уменьше¬
нием отношения H/Pt до ~ 0,5 по линейному закону. (Было проведено мно¬
жество независимых измерений отношения Н /Pt, и в большинстве случаев
значения порядка 1 соответствовали дисперсной каталитической активной
металлической платине, а значение 0,5 - кластерированной, каталитичес¬
ки неактивной металлической платине. Джордано и др. [ 141] усовершен¬
ствовали эти H/Pt -измерения и включили добавку хлоридов как в окисли¬
тельной, так и в восстановительной атмосфере. Они также представили
подробный критический обзор существующих моделей адсорбции водорода
на платине (рис. 8.14). Рост кристаллитов металлической платины, по-видимо¬
му, происходит вследствие поверхностной миграции Pt° при нагреве в возду¬
хе свыше 500 °С. ДаутЦенберг и Вальтере [140] обнаружили, что термичес¬
кая обработка в атмосфере Н2 до температуры 650 °С уменьшает отноше¬
ние H/Pt, однако без наблюдаемой агломерации металлической платины; та¬
ким образом, измерения методом хемосорбции водорода без привлечения
других методов не обязательно дают истинный размер кристаллитов метал¬
лической платины.Вследствие очевидных ограничений на диапазон детектируемых разме¬
ров кристаллитов методами РД, ТЭМ и хемосорбции ряд исследователей ста¬
ли использовать как средство изучения этой проблемы электронную спектро¬
скопию. Успехи в этом деле можно считать лишь относительными. В ука¬
занных исследованиях интерес представляют несколько связанных между
собой проблем, включая а) возможность получения достоверных количест¬
венных характеристик для поверхности и объема, б) определение размеров
пор носителя, в) обнаружение сегрегаций металлов, г) рассмотрение при¬
меров поверхностной диффузии.Как упоминалось ранее в этой главе, Брайнен и Шмит [142] в качестве
тестовой системы для исследования распределения металлических активных
добавок, в частности с точки зрения отношения концентраций на поверх¬
ности и в объеме, использовали родий на углеродном носителе. Эти авторы
подробно исследовали приводящие к неверным выводам изменения в коли¬
чественных данных, которые возникают вследствие плохой диффузии родия
Т.П. Барр359Рис. 8.14. Различные соединения Н - Pt и их стехиометрия, а- адсорбция молекул
Н2 приводит к слабой связи, б - одновременная адсорбция, как в случаях
"д" и "е" (см. ниже), в - водород сильно хемосорбирован из-за наличия со¬
седних гидроксильных групп; эта ситуация ярче выражена на менее дегид-
роксилированных поверхностях. В связывании участвует и Pt4+, согласно
схемег - некоторые авторы рассматривают связывание и диссоциацию молекул
водорода на Pt или на других металлических поверхностях, имеющих
d-электроны, как следствие сверхобмена или непрямого обмена - извест¬
ного явления для магнитного взаимодействия в диэлектрике. Связь вклю¬
чает систему, состоящую из двух соседних атомов Pt и молекулы 1^, т.е.
взаимодействие между неспаренными d-электронами Pt (спин "вверх"
или "вниз") и спаренными эпектронами молекулярного водорода (спин
"вверх" или "вниз"). Подразумевается, что в систему входят четыре элек¬
трона и три частицы. Такие хемосорбированные водородные соединения
легко удаляются в вакууме при комнатной температуре, хотя их можно де¬
сорбировать уже и при —20 °С методом температурно-программируемой
десорбции, д - водород распопагается на поверхности из атомов Pt бла¬
годаря слабой ковалентной связи с незначительной ионной составляющей
(обратимая хемосорбция), е - водород располагается в промежутке меж¬
ду поверхностными атомами ппатины, приводя к необратимой хемосорб¬
ции. Связи не локализованы только на двух конкретных атомах Pt
[141].и связанного с этим изменения результирующей глубины выхода фотоэлект¬
ронов.Стодарт, Мосс и Поуп [143] использовали ЭОС для изучения распреде¬
ления металлов на катализаторе, представляющем собой сплав Pd -Ni на
Si02, при этом в методе ЭОС они применили растрируемый пучок малого
размера. Поскольку наблюдалось прекрасное согласие для данных об объем¬
ной концентрации, полученных ЭОС и РФЭС, то никакого повреждения элек¬
тронным пучком не предполагалось. Результаты ЭОС указали на несколько
важных диффузионных эффектов, так, например, взаимодействие с возду¬
хом, по-видимому, приводит к переходу никеля из объемного сплава на по¬
верхность с образованием NiO.Аналогичные исследования методами РФЭС и сканирующей электронной
микроскопии (СЭМ) проведены для платиновых катализаторов окисления.
Например, Контур и др. [ 144] проанализировали распределение металлов
3608. Применение электронной спектроскопии в гетерогенном катализев системе Pt -Rh, используемой для окисления аммиака. Методом растро¬
вой электронной микроскопии было доказано, что во время горения происхо¬
дит раздельный рост кристаллов как платины, так и родия. Количественные
РФЭС-данные указали на то, что при окислении происходит сегрегация
Rh203 на поверхность, и, возможно, это было связано с Pt Of. Авторы от¬
мечали, что эти эффекты могут иметь отношение к окислению аммиака.Такасу и др. [145] использовали комбинацию методов РФЭС и УФЭС
совместно с электронной микроскопией для описания степени сегрегации пал¬
ладия в различных местах его расположения на Si02. Эти авторы достигли от¬
личного микроскопического разрешения, обеспечивающего регистрацию ме¬
таллических частиц размером вплоть до ^ 10 А. Они смогли заметить изме¬
нения в данных УФЭС и РФЭС, которые, по-видимому, были связаны с раз¬
мерами частиц. В качестве возможной причины результирующих (довольно
малых) изменений в РФЭС-энергиях связи и ширинах линий был предложен
матричный эффект Кима и Винограда [ 146]. Авторы указанной работы от¬
метили также, что эти эффекты согласуются с изменениями интенсивности
поляризации, к которым приводят расчеты Чини [ 147]. Однако не следует
исключить возможность того, что некоторые эффекты, обнаруженные Та¬
касу и др. [ 145], могли быть связаны с избирательной зарядкой, которая
может возникать вследствие неоднородности распределения частиц и нера¬
венства их размеров (см. ниже). Следует признать неудачным решение этих
авторов начать свою прекрасную работу с утверждения, что "методы иссле¬
дования поверхности, такие, как фотоэлектронная спектроскопия ..., не¬
пригодны для изучения металлических катализаторов на носителях". В де¬
сятках промышленных электронно-спектроскопических лабораториях было
показано, что это утверждение не корректно. Если бы авторы заменили вы¬
ражение "непригодны” выражением "трудны”, то их утверждение было бы
справедливо.Таусик и др. [ 148, 149] пытались охарактеризовать дисперсию метал¬
лов на поверхности термообработанной системы PdCl - А1203 при помощи
ЭОС и РФЭС. В этой работе авторы добавляли металлы в количестве, ти¬
пичном для промышленных катализаторов, чтобы избежать вредных дис¬
персионных проблем, связанных с чрезмерным количеством добавляемых
металлов. Как ЭОС, так и РФЭС показали, что количество палладия на по¬
верхности во время прокаливания и восстановления в грубом приближении
не изменяется, тогда как количество поверхностного С1 “значительно умень¬
шается. Данные РФЭС показали, что в атмосфере Н2 стадию восстановле¬
ния поверхностного палладия можно считать успешной весьма относительно,
тогда как при использовании гидразона происходило более интенсивное вос¬
становление палладия на поверхности.Мейсон и Бетцольд [ 150] опубликовали интересную статью, которая кос¬
венно связана с возможностью РФЭС оказывать содействие в обнаружении
Т.Л. Барр361роста кристаллических кластеров платины и других металлов. Изучая рост
металлических кристаллитов серебра на углеродном носителе, эти авторы
смогли описать некоторые изменения в РФЭС-энергиях связи серебра, ко¬
торые, по-видимому, прямо связаны с размером частиц кристаллов.Фанг [ 90] разработал количественные модели (см. выше), которые мо¬
гут увеличить применимость метода РФЭС в этой области путем сочетания
концепций Брайнена и Шмита [ 142] и Контура и др. [ 144] и связанных с раз¬
мером частиц эффектов, которые были предположены Мейсоном и Бетцоль-
дом [150], а также результатов измерений локализации частиц Такасу и др.
[145]. В этих моделях относительная интенсивность РФЭС-пика непосредст¬
венно связывается с характерными размерами, формой и распределением
источников этого пика. Фанг продемонстрировал, как можно использовать
эту модель при расчете степени агломеризации платины на Si02 после
восстановления. В настоящее время рассматриваются возможности распро¬
странения этих полезных моделей на многие другие прикладные области.8оЗаЗо6. Изучение различных отравляющих примесей
платиновых катализаторовОдна из основных задач, которая первоначально ставилась перед элек¬
тронной спектроскопией при изучении катализаторов, состояла в обнаруже¬
нии и классификации каталитических ядов. В случае катализа на платиновых
металлах эта задача очень сложна, поскольку некоторые соединения дейст¬
вуют как промоторы в одних условиях и как каталитические яды во многих
других условиях. Углерод, сера и некоторые элементы, используемые в ка¬
честве промоторов образования сплавов (например, Sn°), по-видимому, иног¬
да стимулируют каталитические реакции, однако они могут в некоторых
других условиях (обычно когда рассматриваемые элементы имеются
"в избытке11) мешать каталитическому действию платины [ 121]. Сера яв¬
ляется интересным примером; она используется в качестве промотора сов¬
местно с рением [138]; известно также, что сера селективно подавляет де¬
гидрогенизацию циклогексана в бензол на платиновом бифункциональном ка¬
тализаторе [151]. Поэтому можно ожидать, что будут разработаны методы
анализа поверхности, специально предназначенные для изучения серы на
платиновых катализаторах.Первоначально надеялись, что в этих исследованиях можно использо¬
вать ЭОС, поскольку этот метод обладает исключительно высокой чувстви¬
тельностью при исследовании поверхности, малым размером растрируемого
пучка и дает возможность легко проводить послойный анализ по глубине.
Таким образом, предполагалось, что оже-метод можно использовать для оп¬
ределения поверхностной локализации, распределения по глубине и типично¬
го окружения отравляющих примесей. В значительной степени в связи с
3628. Применение электронной спектроскопии в гетерогенном катализепроблемами разрушения электронным пучком эти надежды не полностью
оправдались, однако несколько интересных исследований уже проведено.
Бхасин [ 152], например, использовал ЭОС, пытаясь определить состояние
целого ряда отравляющих (?) примесей промышленного гидрогенизирующе-
го катализатора И ~А1203. Этот катализатор анализировался после его
использования и значительной потери активности. Был обнаружен ряд при¬
месей, включая серу и хлор. Однако количество измеренного поверхност¬
ного железа указывало на то, что оно могло служить каталитическим ядом
(если такой яд имелся) для этой системы. ЭОС также использовалась сов¬
местно с ДМЭ в работе Шимерской и Липски [ 153] для изучения сегрегации
серы на поверхности Pd (100). Это исследование показало, что восстановле¬
ние в Н2 приводит к миграции серы к поверхности палладия из объема, вы¬
зывая таким образом наблюдаемое уменьшение способности палладия раст¬
ворять водород. Кроме того, ЭОС, РФЭС, УФЭС и СХПЭЭ использовались
Мацумото и др. [ 154] для изучения взаимодействия серы с поверхностью пал¬
ладия. Применялись поликристаллические палладиевые фольги и было обна¬
ружено, что в этом случае сера легко мигрирует из объема к поверхности.В этом конкретном исследовании результаты РФЭС указывают на возможный
переход электрона от палладия к сере, в то время как для этого немонокрис-
таллического образца данные СХПЭЭ кажутся неубедительными.8.3.3.7. Изменения химических и физических характеристик
катионов платиновых металлов после их обмена
в цеолитахРФЭС также использовалась для изучения катионов платиновых метал¬
лов на цеолитах. Принципиальными моментами в этом случае, по-видимому,
являются а) степень обмена катионов платиновых металлов, б) изменения
состояния окисления этих катионов в процессе создания и использования
цеолитов, в) степень взаимодействия между этими катионами и цеолитом,
г) конкретные реакции, в которых участвуют цеолиты.Например, Ведрен и др. [ 155] применили РФЭС для наблюдения обмена
палладиевых и платиновых ионов в цеолите типа У. Было обнаружено, что
импрегнирование палладия приводит к положительным сдвигам линий в
спектре Pd Ы, свидетельствуя, по-видимому, об обмене иона Pd2+ на ионы
Na+. Последующее восстановление этой системы дает Pd°, однако резуль¬
тирующий спектр Pd 3d указывал также на наличие некоторого количест¬
ва палладия с недостатком электронов, возмото, связанного с образоваг
нием Pd+1. Как оказалось, импрегнированная платина успешно участвует
в обмене и также имелись некоторые доказательства "недостатка” элек¬
тронов после восстановления. Казалось, что металлы с "недостатком” элек¬
тронов жертвуют электронами из-за кислотной (?) природы цеолитов, тем
не менее никаких заметных изменений в РФЭС-спектре цеолитов не проис-
Т.Л. Барр363Рис. 8.15. Электронные спектры Rh 3d некоторых Rh-содержащих образцов (для каж¬
дого спектра первой указывается температура, при которой проводился
анализ).a)[Rh(NH3)sci]Gl2, 10°С; [— Л ). Rh(NH3)^:lX];О -80°С;
в) -80 °С, после 1 ч анализа; г) 400 °С, вакуум (400 С, 0,5 ч); д) -60 °С,
вакуум (400 °С, 0,5 ч); е) 25 °С, Q, или N2 (400 °С, 0,5 ч, 1 атм);
ж) 25 °С, СЮ (100°С, 17ч, 1 атм) или СО + Ciy (100°С, 15 ч, 225 + 225)
после 02 (450°С, 4^ 5 ч, 1 атм); з) 25°С, СО + 1^0 (стандартные
объем и давление при 25 °С) (100 °С, 20 ч, 1 атм) после 02 (450 °С, 2 ч,1 атм); и) 25 °С, (340 °С, 1,5 ч, 1 атм) после 02 (400 °С, 0,5 ч, 1 атм);
к) Rh - С (5%), 25 °С, вакуум (400 °С, 2 ч) [157].
3648. Применение электронной спектроскопии в гетерогенном катализеходило. Авторы выдвинули другое объяснение этих результатов, также выс¬
казав предположение об образовании комплекса переноса заряда между
платиновым металлом и цеолитовой кислотной группой* Они также отмети¬
ли, что этот недостаток электронов будет усиливать электронно-донорную
способность "частично восстановленных” катионов.Окамото и др. [ 156] также использовали РФЭС для изучения У-цеоли-
тов после обмена ионов Na + с Rh111. Восстановление этой системы в ва¬
кууме приводит к различным смесям Rh1 и Rh°. Эти результаты были по¬
лучены благодаря сравнительному анализу энергий связи. Предполагалось,
что Rh1 является активным центром для адсорбции этилена и последую¬
щей гидрогенизации, в то время как ацетилен присоединяется только к Rh°.
Проблема сдвига пиков вследствие зарядки, возможно, ставит под сомнение
некоторые из этих качественных и количественных выводов. На основе ме¬
тодов, аналогичных предложенным Брайненом [ 25], были проведены расчеты
отношений Cl /Rh на поверхности. Полученные результаты наводят на мысль,
что некоторая часть родия могла мигрировать в объем во время нагрева.
Возможно, привлечение более строгого подхода Керкхофа и Мулина [ 89]
может изменить это предположение. Одновременный анализ методом ЭПР
также показал, что добавка НС1 может конвертировать большую часть Rd°
в RhH и RhCl, очевидно, способствуя димеризации этилена.Наконец, интересное РФЭС-исследование, также включающее рассмот¬
рение цеолитов с добавкой родия, было недавно опубликовано Андерсоном
и Скаррелом [157]. Это исследование уникально в том смысле, чго в нем
предполагалось, что цеолит действует прежде всего как носитель. Перед
этими учеными стояла задача осуществления реакций карбонилирования спир¬
тов. Эта система была создана путем обмена хлораминового комплекса ро¬
дия на ^-цеолите и окисления результирующей "смеси” для удаления аммиач¬
ных лигандов. Нагрев этой "смеси" в N2 до 600 °С или в Н2 до 340 °С вос¬
станавливал родий, однако конечное состояние окисления родия было неопре¬
деленным. Измерение энергий связи показало, что, вероятно, имелось неко¬
торое количество Rh° (рис. 8.15). "Стандартный" количественный РФЭС-ана-
лиз указал на некоторую предпочтительную концентрацию Rh(Cl)3X на по¬
верхности, причем данные по энергиям связи наводят на мысль о возмож¬
ном наличии мостиков С1". Однако отсчет энергий связи, использованный для
получения этих результатов, может быть сомнительным. Авторы также пред¬
полагали, что 6 присутствии карбонилов может иметь место восстановление
Rh111 до Rh1. Замена этого цеолита носителем с меньшей кислотностью (нап¬
ример, Si02) сильно уменьшала сдвиги энергий связи и активность системы
к карбонилированию, указывая на значительное взаимодействие (обмен ?)
между родием и носителем в присутствии цеолита.
Т.П. Барр3658.3.4. Цеолиты: каталитический крекинг и другие процессы
S.3.4.i. Введение в теорию цеолитов и каталитический крекингИз всех веществ, описанных в этой главе, кристаллические алюмосили¬
каты, называемые цеолитами, или молекулярными ситами, вероятно, имеют
наиболее многообразную историю применений (и потенциальные возможности).
Э70Т факт обусловлен как их уникальными регулярными структурными ха¬
рактеристиками, так и их способностью сильно изменять свои адсорбцион¬
ные и реакционные свойства, хотя при этом необходим обмен катионов для
компенсации отрицательного заряда алюмосиликатной (структурной) ячейки.
Этот отрицательный заряд возникает, естественно, потому, что алюминий
вынужден захватывать дополнительный электрон для " дублирования” крем¬
ния в алюмосиликатной решетке. Вследствие регулярной кристаллографичес¬
кой структуры этих решеток алюминий занимает вполне определенные узлы,
и, для т ,»го чтобы обеспечить локальную неотрицательность, катионы должны
находиться в непосредственной близости от этих мест, как правило, лока¬
лизуясь в замкнутых полостях различного размера в "сверхрешетке" [158].
Простые катионы (например, Н+ Na+, Ва2+ и т. д.) значительно различают¬
ся по размеру и заряду. Таким образом, катионный обмен может представлять
собой чрезвычайно избирательное явление, поскольку конкретные ячейки час¬
то слишком ограничены по размеру для размещения в них больших и/или мно¬
горазрядных катионов [158]. Поэтому при определенных условиях можно конт¬
ролировать распределение катионов в некоторых цеолитах и таким образом
управлять некоторыми их свойствами. Кроме того, повторяющаяся, регуляр¬
ная структура базовых блоков цеолитов создает сверхрешеточные структуры,
имеющие каналы, окна, полости и поры различного размера (рис. 8.16). По
этим причинам конкретные цеолиты с конкретными катионами можно исполь¬
зовать для селективного адсорбирования и селективных реакций [ 158]. Таким
образом, цеолиты дают возможность управлять определенными аспектами
гетерогенного катализа [116] путем контроля не только за соединениями,
способными адсорбироваться (и десорбироваться) на различных участках, но
также и за временем их пребывания на этих участках. Поскольку различные
цеолиты сильно различаются между собой, возникла целая новая область -
"конфигурационная селективность” [ 159]1 >.Из всех областей каталитического использования цеолитов, несомненно,
наиболее важным до сих пор являлся каталитический крекинг [ 116]. Осущест¬
вление непрерывного контроля за каталитическим крекингом доставляло
много трудностей ученым и инженерам в течение многих лет из-за способ¬
ности этого процесса приводить к быстрому закоксовыванию используемого
катализатора и тем самым к разрушению его селективности и в конечном сче¬
те к потере активности [ 116]. Современная сырая нефть часто содержит мно-Haensel V., частно© сообщение, 1982.
3668. Применение электронной спектроскопии в гетерогенном катализего серы, и, по мере того как температура структурных блоков, участвующих
в каталитическом крекинге, повышается для усиления активности катализа¬
тора, отложения серы и углерода становятся существенной проблемой. Про¬
цесс крекинга включает в себя избирательный разрыв связи С — С с после¬
дующей гидрогенизацией для выделения полезных (прежде всего "бензиновых”)
молекул из более тяжелых фракций. Ранее (вплоть до конца 30-х годов) при¬
менялся термический крекинг, однако этот довольно неэффективный процесс
был почти полностью вытеснен каталитическими процессами [116]. Для под¬
держания упомянутых выше реакций необходимо наличие на катализаторе
кислотных участков. Вначале в качестве катализатора использовались раз¬
личные глины, однако постепенно они были вытеснены искусственными аморфными
кремнеземно-глиноземными системами [116]1*.В середине 60-х годов в качестве катализатора стали применяться ис¬
кусственные фожазиты (см. рис. 8.16) и сейчас около 90% всех процессов
каталитического крекинга проводится на цеолитах [1161. Наилучшие ката¬
лизаторы для крекинга, по-видимому, состоят из фожазитов типа У (см.
рис. 8.16), в которых исходный катион натрия заменен смесью про¬
тонов с редкоземельными [160] или щелочноземельными катионами. Эти
катионы, по-видимому, приводят к очень большому увеличению (селективной) кис¬
лотности У-систем, определяя значительную активность этих систем, которую мож¬
но поддерживать в течение длительного времени [116,160]. Цеолиты используют¬
ся также (как описано в других разделах этой главы) для других каталити¬
ческих процессов, к которым относятся дегидрогенизация, окисление и по¬
лимеризация (например, процесс метанолирования синтез-газа и димериза-
Ция этилена) [116]; они также широко используются при производстве ад¬
сорбентов. Введенное недавно фирмой "Мобил" понятие "конфигурационная
селективность" Г1591 и предложенные ею цеолиты типа ZSM-5 [161] до¬
пускают распространение использования цеолитов в таких областях, как
избирательный риформинг и алкилирование (например, отделение низкоок¬
тановых углеводородов и дегидрополимеризация метанола в бензин).Фожазиты образуются из содалитных блоков и имеют кристаллическую
структуру типа изображенной на рис. 8.16. Si /А1-отношение R для фожази¬
тов заключено в диапазоне —1 < R < 3 с соответствующим изменением чис¬
ла катионов. (Отметим, что существует возможность "декатионировать" фо-
жазитовую систему путем разрыва некоторых алюмосиликатных связей и
удаления этих катионов Г158]. В этом случае в решетку вносятся как поло¬
жительный, так и отрицательный заряды. В этой системе, очевидно, заклю¬
чены большие каталитические возможности [116]. Фожазит типа У, обычно
синтезированный как Nay, имеет отношение Si /А1 порядка 2,5.Среди вопросов, касающихся цеолитов, которые часто задают ученым,
работающим в области физики поверхности, имеются вопросы, относящиеся
к установлению и количественному описанию следующих фактов: а) мерыHaensel V., частное сообщение, 1982.
Т.Л. Барр 367Рис. 8.16. Схематическое изображение структурных блоков некоторых цеолитов, а -
содалитовый куб; б - структура типа А; в - структура фожазита. Каж¬
дая вершина представляет собой атом Si или А1, соединенный с внеосе¬
вым кислородом. Места распопожения катионов не указаны.успеха в упомянутом выше обмене катионов (в противоположность получению
новых катионов простым "диспергированием" на другой поверхности); б) из¬
менениям, происходящим в этих катионах после использования цеолита, и
в) целостности поверхности самого цеолита во время его использования. Пос¬
леднее особенно важно, поскольку у многих цеолитов проявляются обратные
поверхностные эффекты, такие, как деалюминирование в процессах термо¬
обработки и эксплуатации [158]. Кроме того, ненужные продукты, возникаю¬
щие вследствие неполной кристаллизации или внутреннего повреждения цео¬
лита, могут накапливаться на внешней поверхности или в порах цеолита, пре¬
пятствуя нормальному течению реагента и продукта. Этот факт можно обна¬
ружить, используя РФЭС, путем определения различий между поверхностью
цеолита и поверхностью глинозема или кремнезема и т. д. [28, 42].Упомянутые выше цеолиты ZSM-5 также имеют строго задаваемую струк¬
туру, которая заметно отличается от структуры фожазитов (см. рис. 8.16).Si/А1 -отношение R для системы ZSM-5, по-видимому, изменяется в преде¬
лах ~ 15 < R < 100 ■*-. Эти цеолиты часто изготавливают с включением ка¬
тионов органических аминов.
3688. Применение электронной спектроскопии в гетерогенном катализе8.3.4.2. Изучение апюмосипикатных структурных блоковХотя ряд интересных свойств цеолитов на первый взгляд кажется спе¬
циально созданным для ЭОС, исследование поверхности цеолитов этим ме¬
тодом было в значительной степени безрезультатным, что связано, вероят¬
но, с эффектами повреждений электронным пучком. Изучение цеолитов ме¬
тодом РФЭС берет начало с пионерской работы Дельгаса, Хьюза и Фэдли
[ 12]. Вскоре за ней последовала работа Миначева и др. [ 162], в которой
были получены результаты, указывающие на степень обмена катионов. Эти
ранние надежды были до некоторой степени опровергнуты упомянутыми вы¬
ше результатами работы Темпре, Делафоса и Контура [ 17], в которой при
помощи РФЭС были получены доказательства постепенного роста Si/А1 «от¬
ношения Rs на поверхности, которое значительно превышало увеличение
аналогичного отношения Rb, происходящее в объеме, хотя обе эти величины
были сравнимы для Л-систем (Si /А1 = 1) (табл. 8.5, а). Эта работа со всей
очевидностью указывает на наличие обширных обогащенных кремнием клас¬
теров на поверхности в случае систем с большим Si /А1 -отношением Rb, что
предполагает либо поверхностную деалюминизацию используемого вещества,
либо, возможно, избирательное разрушение поверхности, происходящее во
время анализа. Некоторые исследователи подвергали сомнению различные
аспекты этих количественных данных и степень замеченного постепенного
изменения (табло 8,5, б). В любом случае очевидно, что количественные дан¬
ные для поверхности и объема цеолита могут обнаруживать заметное рас¬
согласование. Результаты применения метода РФЭС в работе Кнехта и
Сторка [ 152], в которой были представлены количественные данные, указы¬
вающие на поверхностную деалюминизацию некоторых цеолитов типа А,
включая их разновидности с заменой на медь (см. табл. 8*5, в), еще больше
обострили эти проблемы*Следующий непонятный момент был выявлен исследователями из лабо¬
ратории автора этой главы [ 164], когда в 1978 г. было опубликовано, что
можно ожидать достижения значительной точности количественных измере¬
ний методом РФЭС для всех цеолитов, построенных из основной структурной
ячейки содалита. Было показано, что отношение Rs на поверхности растет
быстрее, чем Rb , однако отношение Rs /Rb для У-цеолитов, как было обна¬
ружено, составляет ^1,2 (см„ табл. 8е5, б) по сравнению со значением ~ 2,7,
измеренным Темпре, Делафосом и Контуром [ 17L Если степень деалюмини-
зации на поверхности представляет собой нестабильное, довольно тонкое
явление, зависящее от множества факторов, таких, как состояние гидратации,
то эти расхождения вполне допустимы.При исследовании методом РФЭС различных систем [28, 42, 164] автор
этой главы также обнаружил постепенный сдвиг энергий связи всех элемен¬
тов, входящих в цеолиты. Такие сдвиги сопровождались для этих цеолитов
Т.Л. Барр369Таблица 8.5.а. Соотношение кремния и алюминия в объеме и на поверхности раз¬
личных искусственных цеолитов [17]Молекулярное сито ЛиндеЦеолон НортонаNa/4 Na XNaYNaZ(Si/AI)b 1 1,252,55(Si/Al)s 2 2,66,511,5б. По данным
Барра [42]Отношение интенсивностей (Si/AI)1)
фотоэлектронных линий,
рассчитанное по объемной концентрацииОтношение (Si/AI)(2p)1)
из высот пиков,NaX 1,81,9NaY 3,74,0NaA 1,51,551^Для этих результатов трудно указать какое-либо значение их относительной погрешности
Исходя из применяемого метода и обсуждавшихся выше приближений, можно считать, что
эти погрешности не хуже 10%в. По данным Кнехта и Сторка [163]ОбразецОтношение в объемеОтношение на
поверхностиЦеолит А № 1
Цеолит А № 2Na/Al/Si = 0,96 : 0,99 : 1
Na/Al/Si = 0,90: 0,90: 1Na/Al/Si = 0,99 : 0,65 : 1
Na/Al/Si = 0,95 : 0,67 : 1довольно значительным уменьшением ширин соответствующих РФЭС-линий
кислорода, алюминия, кремния и других металлов по сравнению с аналогич¬
ными линиями "первичных” Si02- и А120,-системоВсе наши результаты были проанализированы и обобщены автором этой
главы в нескольких публикациях [ 28, 42]. Была обнаружена (во время РФЭС-
измерений) изменяющаяся степень деалюминизации поверхности, которая
действительно возрастает с ростом Rb , однако, как отмечалось выше, в
менее заметной степени, чем сообщалось в работе Темпре, Делафаса и Кон¬
тура [ 17]о Причина этой деалюминизации все еще неизвестна, но ее обнару¬
жение является специфичным и важным моментом для описания состояния
наружных пор этих цеолитовых систем* Было показано, что описанные выше
сдвиги энергий связи происходят для вполне определенных образцов, что
позволяет уверенно идентифицировать конкретные цеолиты, даже если их
содержание относительно невелико. Были обнаружены такие характерные
энергии связи и ширины линий для некоторых видов кремноземов и глино¬
земов, что, таким образом, позволяет проводить точное отождествление
Таблица 8.6.а. Типичные энергии связи в электронвольтах (±0,1 эВ). Сдвиги вследствие зарядки устранены (C1s = 284,4 эВ)Примесь Вследствие неполного обмена в цеолитах присутствует также небольшое количество других катионов6. Ширины линий для цеолитов, приведенных в табл. 8.4, в электронвольтах (±0,15 эВ)Точность уменьшена до ±0,3 эВ
Т.Л. Барр371компонент смеси после деалюминизации и других "перемешивающих” про¬
цессов* Некоторые из этих результатов представлены в табл» 8.6 и на
рис* 8 о17. Отметим, что при получении приводимых значений использовалась
как коррекция с помощью нейтрализационной пушки, так и калибровка по
линии С Is. Обсуждение корректности такой процедуры приведено в начале
этой главы. Были проведены также исследования характеристических ва¬
лентных зон и спектров потерь для этих систем Г 28, 42]0 Результаты иссле¬
дований были объединены с упомянутыми выше данными по энергиям связи
и ширинам линий с целью получения конкретных с точки зрения химии обос¬
нований для измеренных характеристик» Тот факт, что энергии связи дляРис. 8.17. Типичные сдвиги энергий связи (Si 2р) в цеолитах, а - в результате из¬
менения структуры, А -* Х-+ Y; б - в результате изменения катионов
Na - Са [28].
3728. Применение электронной спектроскопии в гетерогенном катализеэлементов, включая данные для катионов, кремния, алюминия и кислорода,
сдвигаются в одинаковом направлении, был вначале несколько непонятен»
Однако потом стало очевидным, что истинные "химические" сдвиги вызва¬
ны не изменением в энергиях связи этих элементов, а скорее всего изме¬
нением структуры кластеров (групп элементов). Таким образом были иден¬
тифицированы группы Si -О и катион -А1 -О, Эти идеи были подкрепле¬
ны появлением аналогичных групп при селективном (остаточном)
разрушении цеолитов в процессе травления ионами Аг+[28, 42]. На ос¬
нове этих идей вариации в результатах РФЭС-измерений, замеченные для
разных цеолитов, были соотнесены с различием следующих параметров:
а) отношения Si /А1, б) структурного блока цеолитов, например А -> Х-* Y,
а) катионов, например Na + -► К+ Са 2+, и г) поля заряда Демпси [ 165].8.3о4е 3. Исследования катионного обмена и
каталитических процессовБольшинство исследований цеолитовых систем методом РФЭС не было
сконцентрировано на самих молекулярных ситах в процессе каталитическо¬
го крекинга. Обычно упомянутые выше катионы служили основным объектом
исследований для РФЭС, результаты которых предназначались для реакций
восстановления водородом и полимеризации. Таким образом, РФЭС использо¬
валась в работе Вихтерловой и др. [ 166] в качестве одного из методов ис¬
следования замещения А1(и последующей "стабилизации" водяным паром)
в цеолите NH4 Y типа У. Было сообщено, что А18+ уменьшает способность
стабилизированных водяным паром У-цеолитов к дегидроксилированию. Ре¬
зультаты измерений методом РФЭС не использовались широко в этой рабо¬
те, за исключением значений Rs, которые указали на некоторую деалюми¬
низацию, по величине близкую к деалюминизации, считающейся типичной в
этой лаборатории.Браво, Дуайер и Замбаулис [ 167] также привлекли РФЭС для исследова¬
ния применения окиси углерода для восстановления NaCunX. Дополнительно
к ожидаемым структурным изменениям были получены доказательства вос¬
становления меди. Предполагалось, что структурные изменения приводят к
очень большому уширению, обнаруженному в спектре О Is после окислитель¬
но-восстановительной обработкио Эти изменения были отнесены на счет
"окклюзии" соединений активного кислорода в решетку цеолита.В предыдущих разделах обсуждалось несколько работ, в которых катио¬
ны платиновых металлов заменялись катионами натрия в молекулярных си¬
тах типа Хи У, В этих случаях электронная спектроскопия использовалась
прежде всего для описания изменений в состоянии платиновых металлов.
Другие иселедовате in также использовали РФЭС для изучения аналогичных
систем, однако они интересовались изменениями в цеолитах во время та¬
ких реакций, как получение метана или изменение соотношения двуокиси
Т.Л. Барр373углерода и водорода в водяном газе (С02 + Н2)0 Например, Педерсон и
Лансфорд [ 168] проанализировали систему Ru У методом РФЭС не только с
целью получения данных о замене Rus+ на Na+, но также для изучения дейст¬
вия всего цеолита» Результаты РФЭС показали, что обмен происходит толь¬
ко частично с компенсацией рутения, адсорбированного на внешней поверх¬
ности. (Эти предположения основывались на данных о Rs, которые были
аналогичны данным, полученным автором этой главы, что, возможно, свя¬
зано с применением обычной ЭСХА-установкИо) Таким образом, в присут¬
ствии 02 были обнаружены Ru02 на внешней поверхности и некоторое ко¬
личество Ru° в цеолитовых полостях в том случае, когда 02 был исключен
(рисо 8.18). Оказалось, что в этих условиях рутений окисляется значительно
легче, чем платина [169]. Все эти выводы были основаны на данных об из¬
менениях энергий связи, которые были одними из первых, использованных
для суждения об успехе или неудаче катионного обмена в цеолитахв В пос¬
ледующих исследованиях Густафсон и Лансфорд [ 170] применили РФЭС дляРис. 8.18. в- РФЭС-спектры уровней Ru 3d /2 и Ru 3dB/2 + С 1s 2%-ного RuNa :1 - дегазация в потоке Не при 400 С с последующим восстановлением в
потоке Н2 при 400 °С в течение 16 — 18 ч: 2 - обработка, как в случае 1,
с заменой Не на N2; 3 — обработка, как в случае 1, с последующим на¬
гревом в потоке смеси СО + при 280 °С в течение 24 ч, а затем в
при 300 °С в течение 18 — 20 ч; 4 - обработка, как в случае 1, с заме¬
ной Не на воздух и 5- импрегнированный 1%-ный RuNaY, обработан¬
ный также, как в случае 1.6 — РФЭС-спектры уровней Ru3 / + С 1s
2%-ного RuNaY : 1 - после дегазации в потоке воздуха до 200 °С со ско¬
ростью нарастания 100 °С/ч; 2- до 300° С или 400 °С и 3 - после де¬
газации в воздухе до 400 °С с последующим восстановлением в при
400 °С в течение 16 — 18 ч [168].
3748. Применение электронной спектроскопии в гетерогенном катализеанализа других систем RuY. В этой работе они опубликовали данные о ру¬
тении в решетке цеолита (не обязательно после обмена) как до, так и после
восстановления (Ru111 до металлического состояния). Было обнаружено,
что соответствующие участки каталитически активны. Сравнение с Ru -А1203
и Ru-SiOa не позволило проследить связь между кислотностью носителя и
каталитической активностью*,Файула, Энтони и Лансфорд [171] также привлекали РФЭС для анализа
цеолита PdY, используемого в качестве катализатора синтез-газа» Они об¬
наружили, что получение метанола, по-видимому, зависит от наличия
кристаллитов Pd° малого размера, обнаруживаемых с помощью РФЭС» С дру¬
гой стороны, получение метана, как оказалось, вызывается прежде всего
кислотностью цеолитов.Обмен платиновых металлов в цеолитах также использовался для уско¬
рения реакций, связанных с преобразованием углеродного скелета (полиме¬
ризации). Как было описано выше, Окамото и др. [156] изучали с помощью
РФЭС RhУ„ пытаясь описать для этой системы как изменения в состоянии
окисления, так и вариации в степени заполнения активных центров. В анало¬
гичных исследованиях Кузницки и Эйринг [ 172] открыли (используя РФЭС),
чтоР h111, помещенный в Y-решетку, может в значительной степени восста¬
навливаться до металлического состояния при менее жестких условиях,
чем те, которые необходимы для восстановления других платиновых метал¬
лов. Эти авторы полагают, что наличие этого цеолита увеличивает скорость
восстановления. Однако родий в RM не восстанавливался, и, более того,
этот цеолит не стимулировал полимеризацию. Поэтому предполагалось, что
восстановление ионов родия до металлического состояния является процесс
сом, активизирующим каталитическую полимеризацию. Аналогичное явление
наблюдалось также в случае палладия и рутения»В еще одной работе Ларкине и др. [ 173] изучали обмен платины на лан¬
тан и натрий в системах с У-цеолитом» Они опубликовали данные, получен¬
ные методом РФЭС, относительно недостатка электронов, обнаруженного в
платине и вызванного, очевидно, различием электростатических полей в
разных катионных узлах (т. е. полей Демпси [ 165])» На эти результаты мо¬
гут влиять размер частиц и эффекты неполного обмена. Нехватка электро¬
нов у платины может активно катализировать дегидрогенизацию и другие
каталитические процессы, приводящие к полимеризации (рис. 8о19).Бадринараджанан и др. [ 174] использовали РФЭС для определения состоя¬
ния никеля после его обмена на цеолите ZSM-5, который сохранил значитель¬
ную кислотность. Этот катализатор предназначался для ускорения как реак¬
ции полимеризации, так и реакции алкилирования» С помощью РФЭС было об¬
наружено, что Ni 11 не восстанавливается до Ni° (на что указывало от¬
сутствие сдвигов энергий связи) при условиях, аналогичных условиям, ис¬
пользовавшимся ранее для родия. Авторы этой работы отнесли отсутствие
восстановления на счет сильного взаимодействия Ni11 и цеолита через гид-
Т.П. Барр375Рис. 8.19. а - фотоэлектронный спектр образцов (Nа) Y-цеолита в области линий А12р
и Pt 4/. Спектр а: линия А1 2р (Na) Y-цеолита. Спектр В линия А1 2р и
Pt 4f 3%-ного (по весу) Pt(Na) Y-цеолита. Спектр С: разностный Pt 4/-
спектр, полученный вычитанием нормированного спектра А из спектра в.
б- профили фотоэлектронных линий Pt 4/?/2 g/2 3%-ного Pt(Na) Y-це¬
олита (спектр л) и 3%-ного (Na)Y-ueonnTa (спектр в). Показано разложе¬
ние на компоненты трех дублетов. Эти дублеты можно представить в сим-
метрикой форме Pt°(a,a'>,Pt+(b,b') и Pt2+(c,c') [173].роксильные группы. Поэтому, так как этот катализатор был активен в реак¬
циях полимеризации, авторы рассматриваемой работы не принимали в рас¬
чет необходимость восстановления обменных катионов (в данном случае ни¬
келя), чтобы создать активный (полимеризующий) каталитический промотор.8.3.5. Кобальт-молибденовые катализаторы: обессеривание
8.3.5.1. введение; применения и особенности катализатораКак описано ранее в этой главе, до 1975 г, был опубликован ряд работ
по каталитической системе Со -Мо -А12Оа (с серой и без серы)» Эта сис¬
тема имеет широкую область применения, прежде всего для гидрогенизации
и удаления серы из сырья, идущего на переработку, нефтяных остатков и ка¬
менного угля (путем гидрообессеривания) [ 116L Хотя присутствие серы иног¬
да полезно, как правило, она является вредной примесью, которая, к сожале¬
нию, загрязняет в неприемлемой степени большинство из известных в нас¬
тоящее время мировых источников углеводородного топлива (например, сырой
саудовской нефти, западных месторождений каменного угля США)о Поэтому
глубокое понимание и правильное использование катализа реакции гидрообес*
серивания (ГОС) важно с экономической точки зрения*
3768. Применение электронной спектроскопии в гетерогенном катализеЗадолго до появления современной науки о поверхности было показано,
что Мо08 на глиноземе катализирует процесс ГОС, и после нескольких лет
проб и ошибок было открыто, что окислы некоторых других переходных ме¬
таллов действуют аналогичным образом. Было также показано, что кобальт
и никель в присутствии сульфида "стимулируют" этот процесс [ 116]» По мно¬
гим причинам для этого процесса обычно предпочитают использовать систе¬
му Со-Мо, хотя другие системы, в частности система Ni -W, также
часто используются для обессеривания [ 116]о Было открыто, что правильная
подготовка системы Со -Мо требует строгого контроля не только количест¬
ва активных добавок, но также и последовательности, в которой они вносят¬
ся [ 175].С точки зрения РФЭС эта система кажется очень привлекательной, так
как кобальт, молибден и сера имеют большое число обычных состояний окис¬
ления, что приводит к значительным сдвигам энергий связи* Кроме того,
система Со -Мо обычно действует по типу катализатора, "сильно обогащен¬
ного активной добавкой" (в отличие от риформинг-катализатора), в котором,
как правило, используется 3-5 вес. % СоО и 12-15 вес. % Мо03 [175].
Обычно для активации этой системы добавляется также значительное коли¬
чество серы [ 116]. Вскоре была открыта и отрицательная сторона такого
подхода, состоящая в том, что в процессе приготовления и использования
этого катализатора его существенные составляющие подвержены ряду слож¬
ных взаимодействий и других изменений, что иногда приводит к сильным
сдвигам результирующих энергий связи, тогда как в других случаях энергии
связи изменяются лишь незначительно [ 175]„ Эти затруднения включали воз¬
можное сильное взаимодействие между легирующими добавками и носите¬
лем (с образованием таких соединений, как Со А1^04) и сложное предпочти¬
тельное наслаивание одной легирующей добавки на другой [175]« (Последнее
обстоятельство может вызвать недоверие ко многим из ранних количествен¬
ных измерений на этой системе с использованием РФЭС«)Со временем большинство этих проблем было уточнено (и они сами ста¬
ли объектом многих экспериментов с использованием РФЭС!). В РФЭС-литера¬
туре, посвященной прикладным каталитическим системам, наибольшее коли¬
чество работ относится к системе Со -Мо - В последние годы число этих
публикаций растет со все возрастающей скоростью. Существует несколько
очевидных причин для этого "предпочтения": а) упомянутое выше изобилие
активных добавок; б) естественная тенденция "проводить перепроверку слож¬
ных материалов современными методами”, в) кажущаяся на первый взгляд
легкость создания этой системы,. Например, ее можно изготовить методами
пропитки, а не, по-видимому, более сложным методом ионного обмена (в цео¬
литах) и техникой совместного осаждения (риформинг-катализаторы на осно¬
ве платиновых металлов), которые необходимы для многих других катализа¬
торов*, Эта "простота изготовления" представляет собой значительное прей-
Т.П. Барр377мущество для ученых, изучающих поверхность, которые часто не имеют опы¬
та в искусстве приготовления катализаторов*. И наконец, г) приоритетные
ограничения, которые часто препятствуют публикации результатов исследо¬
ваний, касающихся промышленных катализаторов; это (по непонятным причи¬
нам) не распространяется на систему Со -Мо. Последний момент является
важным для академических ученых, изучающих поверхность, которые часто
не знакомы с системой патентных ограничений, практикуемой в промышлен¬
ном катализе. Тем не менее не следует полагать, что поскольку исследова¬
ниям поверхности системы Со -Мо посвящено больше всего публикаций, эти
исследования неизбежно являются самыми исчерпывающими.» Возможно, в
промышленных лабораториях тщательнее изучаются другие, более прибыльные
системы. Плоды этих неопубликованных исследований вначале появятся (ес¬
ли они были успешны) как составная часть программы по изучению конкрет¬
ного катализатора и лишь затем станут доступны научной общественности.Все возрастающее укрепление позиций науки о поверхности в промышленных
исследованиях является, конечно, косвенным средством контроля успешного
завершения таких начинаний.Как упоминалось выше, система Со - Мо имеет многочисленные примене¬
ния, однако в этой главе будет описано только гидрообессеривание (ГОС)0 Ис¬
пользование этой системы в качестве катализатора реакций гидрогенизации
обсуждается в следующем разделе, где также будет рассмотрена система
Ni -W -А1208 , являющаяся основным заменителем системы Со -Мо.8.3о5.2. Справочные РФЭС~данные, необходимые для
изучения системы Со -МоПрежде чем начать серьезный анализ РФЭС-работ по системе Со -Мо,
следует ознакомиться с РФЭС-результатами для следующих веществ: Со0,Мо°, СоО, Cof04, Со208, Мо02, Мо205, Мо03, гидроокисей, соответст¬
вующих сульфидов, алюминатов и смешанных Со Мо О-систем. Энергии связи
и ряд спектров чистых металлов, некоторых окислов и небольшого числа
гидроокисей были получены автором этой главы в процессе общего исследо¬
вания естественной пассивации [ 128]0 Отметим, что эти исследования содер¬
жат неопределенность относительно того, какой именно окисел, Со203 или
Со304, образуется на поверхности СоО в результате взаимодействия с воз-
духомо Данные, полученные другими авторами, по-видимому, указывают на то,
что этим окислом является Со304 [ 176LБрандл [178, 179] провел детальный анализ молибдена в чистом состоя¬
нии и изменений его состояния после адсорбции контролируемых доз 02, СО и
НзО. Этот анализ использует как УФЭС-, так и РФЭС-данные и представляет
собой более тщательное исследование начала окисления, чем предыдущие ра¬
боты этого же автора. Чимино и де Ангелис [180] также изучали различные
состояния окислов молибдена и добавили к этому важное обсуждение вопроса
3788. Применение электронной спектроскопии в гетерогенном катализео том, как изменяются энергии связи и форма пиков при помещении этих
окислов на носители из у-А1208 и кремнезема. Кроме того, эти авторы ис¬
следовали важные с точки зрения катализа соединения Na2Mo04 и СоМо04.Одно из первых исследований важных соединений кобальта было про¬
ведено Фростом, Мак-Доуэллом и By леи [ 1813» В этой работе были пред¬
ставлены данные по структуре пиков и энергиям связи Со110 и ConS.
Чистый кобальт и СоО являлись также двумя основными объектами иссле¬
дования в работе Фирманса, Хугевийса и Венника [ 176], в которой было
проведено систематическое изучение методом РФЭС окислов переходных
металлов. Ким [ 182] опубликовал детальное РФЭС-исследование электрон¬
ной структуры СоО, представив подробный и очень полезный с точки зре¬
ния катализа анализ ключевых сателлитных спектров. Эти же сателлиты
являлись характерной особенностью РФЭС-спектров СоО, Со ООН и Со(ОН)2?
опубликованных Макинтайром и Куком [183], которые отметили и подроб¬
но описали важный вопрос о малых различиях расщепления сателлитов.Хуанг, Брандл и Ван дет [ 184] также зафиксировали малые сдвиги этих же
сателлитов; кроме того, они изучили важные изменения, возникающие вслед¬
ствие последующей пассивации, обусловленной взаимодействием с возду¬
хом, а также химические изменения, индуцируемые бомбардировкой ионами
Аг+. Некоторые важные результаты этих исследований, в частности, с точ¬
ки зрения их последующего использования для анализа катализаторов мож¬
но найти в обсужденных выше работах.8.3.5.3. РФЭС-результаты для кобальт-молибденовых
каталитических системМногие осложнения интерпретации систем Со -Мо -S -А1208 возни¬
кают в связи с тем, что очевидный подход к решению этих задач не срабаты¬
вает» Например, чтобы избежать возможного образования Со А1204 (вред¬
ного продукта сильного взаимодействия металл - носитель), в качестве
носителя можно использовать Si 02. Это, однако, как оказалось, стимули¬
рует образование СоМо04 - другого вредного соединения. Аналогичные
трудности, по-видимому, возникают повсеместно при исследовании методом
РФЭС каталитической системы Со -Мо.Было очевидно, что, для того чтобы правильно оценить состояния и из¬
менения, которым подвержена эта сложная система, экспериментаторы долж¬
ны анализировать отдельно каждую ее компоненту при самых разных усло¬
виях ее использования. Хабер и др. [185] опубликовали результаты исследо¬
вания такого типа, в котором РФЭС использовалась для анализа изменений,
происходящих в Мо08, Мо02 и СоМо04, когда эти соединения применялись
для окисления альдегидов через соответствующие кислоты до С02# К удив-1} Prims R частное сообщение, 1980.
Т.Jl. Барр379лению, оказалось, что Мо02 является самым активным окислительным реа¬
гентом, что, очевидно, обусловлено возросшей способностью "восстанов¬
ленной поверхности” адсорбировать и удерживать не только альдегиды, но
также и кислотные промежуточные продукты; было обнаружено, что соеди¬
нение СоМо04 совершенно бесполезно для этих реакций.Деклерк-Гримс и др. [ 186] методом РФЭС изучили поведение двух от¬
дельных окисных катализаторов СоО -А1203 и Мо03 -А1203, прежде
чем исследовать, используя тот же метод, комбинированный катализатор
в присутствии сульфида [187]. Методом РФЭС они доказал^ что восстановле¬
ние в Н2 без S 2“ приводит к превращению Со 2+ -> Со 0 в системе Со -> А120^
тогда как в присутствии молибдена и сульфида, восстановление в Н2, по-ви¬
димому, превращает Со 2+ в Co9S3. При слишком большом количестве кобаль¬
та в первоначальной системе Со -Мо -S -А12о8 из его избытка, веро¬
ятно, образуется Со Мо04. Утверждалось, что это соединение является
отравляющей примесью [187].В 1977 г. Окамото и др. [188] начали серию детальных исследований
методом РФЭС системы Со -Мо - А1203, используемой для реакции ГОС
в смеси тиофена и Н2. Они представили с некоторыми подробностями до¬
вольно странную смесь объяснений, существовавших в то время для акти¬
вированных смесей реагентов, включая: а) синергическое взаимодействие
между кобальтом и молибденом, б) межплоскостное внедрение Со 2+ в
MoS2 , в) неясный в некотором смысле "комплекс" кобальта и молибдена
и г) так называемую модель поверхностного обогащения, (Как мы увидим
позднее, некоторые модификации почти всех этих интерпретаций сохраня¬
тся и войдут в более четкие модели, принятые в настоящее время») Окамо¬
то и др. [188] считают доказанным, что каталитическое поведение смешан¬
ных окисных катализаторов связано с молибденом, в то время как Со2*
действует как "промотор". РФЭС-спектр окисного катализатора, по-видимо¬
му, указывал на то, что молибден в свежем (готовом к использованию) катали¬
заторе представляет собой смесь Мо5+ иМо4+, тогда как большая часть Со2*вос¬
станавливается до металлического состояния Со0, а основное количество
кобальта, по-видимому, превращается в неактивное (стабилизированное по¬
верхностью) соединение СоМо04. Некоторые данные, полученные методом
РФЭС, по-видимому, подтверждают предположения Гримбло и Бонеля
[ 189], что ионы молибдена расположены в октаэдрических, а ионы кобаль¬
та - в тетраэдрических узлах шпинелевидных образований в структуре
окиси алюминия. Массос [ 190] опубликовал критическую статью по поводу
упомянутой выше работы Окамото и др. [ 188], в которой он подверг сомне¬
нию универсальность интерпретации Окамото и др., поскольку в условиях
их эксперимента содержание серы было чрезвычайно мало» (Никаких суль¬
фидов не добавлялось!). В частности, Массос связывает наличие Со0 с
этим отсутствием потока сульфида. Отвечая на эту критику, Окамото,
3808. Применение электронной спектроскопии в гетерогенном катализеИманака и Тераниси [ 191] согласились, что увеличение содержания серы
будет стимулировать образование сульфидов кобальта, а также MoS2. Од¬
нако они защищали свои выводы и схему своего эксперимента, связывая
низкое содержание серы с влиянием носителя. Для проверки этих выводов
Окамото и др. [ 192] использовали метод РФЭС для изучения влияния суль¬
фида на Со2+ и Мо6+ в восстанавливающей среде при различных потоках
сульфида. Это исследование показало, что при восстановлении в атмосфе¬
ре Н2 в присутствии (только) тиофена Мо6 + восстанавливался до Мо4+, при¬
чем тиофен, по-видимому, приводил к образованию MoS2, никаких следов
CoS не было обнаружено. Однако при добавлении к восстанавливающему
потоку H2S образуется CoS: Исследуя эту задачу далее, Окамото и др.[ 193] сообщили, что процесс приготовления катализаторов, по-видимому,
в значительной степени изменяет отношение Со/Mo на поверхности и в
объеме и что это отношение очень сильно изменяется при восстановлении,
однако это явление существенно зависит от того, использовался при этом
H2S или нетоОкамото и др. [ 194] проконтролировали некоторые другие промежуточ¬
ные явления, используя РФЭС для детального изучения восстановления сис¬
темы Мо - А1208 при различных потоках сульфида. Используя довольно
точный количественный метод, аналогичный методу Брайнена [ 25], они об¬
наружили, что А1208, по-видимому, "не задействован" в процессе сульфи¬
рования Мо03, тогда как Мо03, очевидно, перед восстановлением превращается
из окисла в сульфид. Активность молибдена в реакции ГОС тоже, по-видимому,
линейно возрастала с образованием MoSоднако наблюдался явный максимум актив¬
ности, после которого следовал быстрый (почти линейный) спад ГОС-актвнос-
ти. Таким образом, эта система характеризуется классической вулканооб¬
разной зависимостью [ 50] (рис, 8.20).Детально анализ Окамото и др* [ 194] методом РФЭС молибденовых сое¬
динений, образованных при различных концентрациях молибдена, также ука¬
зывал, что монослой сульфидов молибдена, возможно, занимал тетраэдри¬
ческие узлы глинозема при содержании молибдена ~ 12%, что принималось
без доказательств Джордано и др0 [ 195], которые использовали методы,
отличные от РФЭС Если процентное содержание молибдена превышает
это значение, то Окамото и др0 [ 194] предсказывают, что избыток молибде¬
на приводит к образованию "отравляющих" слоев Мо03 и Al2(Mo04)s* Они
также утверждают, что предварительное сульфирование во время восста¬
новления приведет к восстановлению большого количества молибдена с об¬
разованием MoS, часть которых будет располагаться в октаэдрических
узлахо Активными будут только те молекулы MoS, которые попадут в тет¬
раэдрические узлы. Основываясь на подобных количественных измерениях
и наблюдениях искажений некоторых РФЭС-пиков, Окамото и др. [ 194] так¬
же утверждают, что эти тетраэдрические узлы ненасыщены серой и содер-
Т.П. Барр381Рис. 8.20. Корреляция между собственной активностью молибдена в реакции гидрообес-
серивания тиофена при 400 °С и степенью сульфирования молибдена в ката-
лизаторе Мо03/А1203 : 1 - Мо03 - А1203 после отжига при 550 °С и не-
полного восстановления; 2 - без предварительной обработки; 3 - после
предварительного сульфирования; 4 - отожженный при 700 °С Мо03/А120
после неполного восстановления [194].жат анионные вакансии, т0 е. образуются вакансии следующего вида:где □ - анионная вакансия. Эдмондс и Митчел [ 196], проводя аналогичные
РФЭС-исследования системы Мо - А1203, довели до конца свой более ран-
ний анализ [10] ГОС-системы и также установили, что Мо08 имеет тенден¬
цию образовывать многослойное покрытие окиси алюминия, причем насыще¬
ние степени покрытия достигается при ~ 9 вес. % молибдена. Они также
заметили, что образование многослойных молибденсодержащих соединений,
вероятно, не происходило до тех пор, пот не завершалось формирование
этого монослоя.Еще одна серия подробных исследований этих катализаторов была про¬
ведена в исследовательской лаборатории Давида Эркюля0 Первоначально
[197] эти исследования касались эффектов восстановления на системе
Со —Мо -А1203 как при наличии предварительного сульфирования, так и
без него- Восстановление без H2S приводило к ожидаемому уменьшению
Мо 71 и соответствующему возрастанию как Мо v, так и Мо IV о График
этих изменений представлен на рис» 8.21. Предварительное сульфирование
с восстановлением, очевидно, приводило к образованию Мо S2 и, по-видимо-
3828. Применение электронной спектроскопии в гетерогенном катализеРис. 8.21. Зависимость концентрации молибдена в различных состояниях окисления от
времени восстановления. 1 - Мо VI. 2 - Мо v; 3 - MoIV . Все образцы
катализаторов восстанавливались в течение указанного времени в атмос¬
фере водорода высокой чистоты при 500.°С. Мольные доли определялись
путем разложения на составляющие перекрывающихся компонент линий
Мо 3р и Мо 3d с использованием графического решающего устройства
Дюпона модель 310, дополнительного сумматором. Разложение 20 спектров дает
следующие значения энергий связи уровня Mo3d5/2 : 233,0 ± 0,1, 231 ± 0,1,229,9 + 0,1 эВ. Связь этих значений с Мо VI MoV и MoIV была уста¬
новлена путем сравнения со спектрами Мо03 и Мо02 » Для которых бы¬
ло обнаружено, что расщепление линий составляет 3,1 ± 0,1 эВ [207].му, влияло на кобальт, однако авторы этой работы не были уверены в типе
конечного соединения кобальта, они только отметили, что изменения кобаль¬
та в этом катализаторе, вероятно, происходят быстрее, чем в случае
Со А1204 [ 198]. Было подвергнуто сомнению наличие дискретных фаз суль-
фидовоЦинг и др. [ 199] немного позже использовали РФЭС для изучения ка¬
талитической системы Мо08 -А1203) добавив к этим результатам данные,
полученные методами спектроскопии рассеяния медленных ионов (СРМИ) и
лазерного комбинационного рассеяния. Эти исследователи при изучении
отожженного (предварительно восстановленного) катализатора получили
доказательство существования четырех возможных состояний Мо VI, вклю¬
чая поверхностный монослой, по-видимому, "активного" Мо^ в тетраэд¬
рических узлах, таким образом, подтверждая предположения как Эдмонд¬
са [ 175], так и Окамото и др. [ 194]„ Однако Цинг и др. [ 199] обнаружили
также некоторое количество активного Мо щ в октаэдрических ячейках. Эр-
Т.П. Барр383кюль и его группа также обнаружили некоторое количество неактивного сое¬
динения А12(Мо04)8, которое, очевидно, образуется в результате взаимодей¬
ствия между Мо^ и носителем в тетраэдрических узлах. Восстановление
этой системы, как оказалось, приводит к образованию упомянутых выше
М отсоединений (я тетраэдрических узлах)к Мо^ -соединений (в октаэдри¬
ческих узлах)* Если концентрация молибдена поддерживается на уровне
ниже 15 вес. %, то большая часть Mov в тетраэдричезких узлах, по-видимо-
му, активна к ГОС тиофена и, возможно, находится в виде "поверхностного
молибдата":После увеличения весовой концентрации молибдена свыше 17% эти "молибда-
ты", по-видимому, становятся неактивными. При ^20 вес» % молибдена
полагалось, что вышеупомянутый монослой насыщается. По-видимому, в
этом случае образуется значительное количество октаэдрического MoIV с
концентрацией в диапазоне 7 < вес. % молибдена <20, однако лишь неболь¬
шое количество этого молибдена активно» При концентрации выше 20 вес.%
оказывается, что молибдаты опять образуются в тетраэдрических узлах,
однако теперь они, вероятно, представляют собой в основном неактивную
объемную разновидность. Можно предположить, что добавка сульфида может
преобразовать в тетраэдрических ячейках поверхностный молибдат Цинга и
др. [ 199] в активные сульфиды, описанные Окамото и др. [ 194]» Оба этих
возможных соединения, по-видимому, "оптимизируются" в одинаковом диа¬
пазоне концентраций. Тем не менее следует отметить, что Цинг и др. [ 199]
предсказывают, что необходимо иметь соединения молибдена как в октаэд¬
рических, так и в тетраэдрических узлах для того, чтобы окисленный ката¬
лизатор впоследствии имел достаточную активность. Эркюль и соавторы в
дальнейшем сделали эти доводы более четкими, однако пе^д анализом этих
(обзорных) публикаций было бы полезно рассмотреть некоторые исследова¬
ния других авторов, повлиявшие на современные воззрения этой группы.Например, Дельво, Гранж и Дельмон [200] использовали РФЭС для изу¬
чения ГОС-системы, но они начали без окиси алюминия, что предпочтитель¬
нее, чем опускать одну из металлических добавок. Они обнаружили, что при
"правильном" Со/Мо-отношении (?) и достаточном количестве сульфида об¬
разуются Co9S8, MoS2 и соединения с вакансиями Мо3+. Последнее вещест¬
во можно отождествить с соединениями, имеющими сульфидные вакансии,
описанные ранее в работе Окамото и др. [ 194]. Ключом к созданию этих
объектов, согласно Дельво, Гранжу и Дельмону [200], по-видимому, будет
сочетание термической обработки (аналогичной описанной в работе Цинга
и др. [ 199]) и правильных отношений H2S/H2 и Со/Мо. Тем не менее еле-
3848. Применение электронной спектроскопии в гетерогенном катализедует помнить, что эти факторы могут изменяться при наличии А1203. Исхо¬
дя из этого, можно объяснить, почему Брайнен и Армстронг [ 201] полагали,
что при определенных условиях может образовываться Со а не упомянутый
выше сульфид. (Отметим также уже упоминавшуюся полемику между группа¬
ми Окамото [188, 191] и Массоса [ 190].)Для дальнейшей проверки этих положений Делане и др, [202] использо¬
вали РФЭС и электронную микроскопию при исследовании различных смесей
полной каталитической системы Со -Мо -S -А1203. В этой работе они от¬
метили, как и ранее, важность баланса концентраций (на поверхности), причем
любой избыток кобальта явно приводил к покрытию наружной поверхности сис¬
темы неактивным соединением Со 304. Микроскопическое исследование, как
оказалось, подтвердило, что при оптимальных термических условиях "подхо¬
дящая” смесь кобальта, серы (и молибдена) приводит к образованию CoeS8o
Делане и др. [ 202] также пре/щолагали, что основным соединением молибде¬
на, которое образуется лосле сульфирования, является MoS2, однако они не
пытались обнаружить каких-либо вакансий, аналогичных описанным ранее в
работе Окамото и др. [ 194], и строили свои предположения на основе старых
работ группы Дельмона и Делане. Они утверждали, что некоторое количест¬
во избыточного кобальта может образовать неактивное соединениеСо А1204,
имеющее структуру шпинели. Если относительная концентрация поддержива¬
ется на таком уровне, что этого не происходит, то, согласно Делане и др.
[202], соединения Co9S8 могут располагаться вблизи MoS2 с возможным
образованием сложного смешанного сульфида. Это наводит на мысль, что
синергическая модель может описывать стимулирующие (промоторные) свой¬
ства кобальта. Термическая обработка, по-видимому, восстанавливала в
этом эксперименте поверхностную концентрацию молибдена.Ликоргиотис и др. [ 203] рационализировали такой подход, указав, что
многие из этих идей частично зависят от природы кислотных участков А1208.
Для проверки этого положения они добавили Na + к описанной выше (пред¬
варительно обработанной серой) системе. В первой публикации авторы сооб-
щили данные о влиянии Na + на кобальт, отметив, в частности, что щелочной
катион, вероятно, препятствует диспергированию кобальта. В результате
явно образуется избыточное количество (вредных) Со304 и Со А1204. Во вто¬
рой статье Ликоргиотис и др. отметили, что Na+ также, по-видимому, пре¬
пятствует образованию Mov после восстановления Мо03 - А1208»Гаярдо, Гранж и Дельмон [ 204] также использовали РФЭС и спектро¬
скопию диффузного отражения для анализа различных систем Со - Мо и
проверки представления о стабильном молибденовом монослое. Они получи¬
ли доказательства существования этого монослоя в смешанных системах
и высказали предположение, что кобальт, диспергированный на у-А12оз
(сверху молибдена), действует как промотор, помогая распределению молиб¬
дена (рис. 8.22). Однако добавка слишком большого количества кобальта,
Т.П. Барр385Рис. 8.22. Предположительная структура катализатора СоМо -у-А12оз [204]. 1 -
Мо(VI) тетр. в монослое (при низком и среднем количестве Мо); 2 -
Мо(VI ) окт. в многослойном покрытии; 3 - Со (И ) тетр. В СоА1204;4 - Co (III ) окт. [ и Co (II ) окт.? ], взаимодействующий с Mo (VI ) тетр,
монослой (двойной слой); 5- Со(II) тетр. и Со(Ш ) окт. в Со304.как оказалось, приводила к образованию Со А1203 и многослойных структур,
что препятствовало диспергированию монослойного молибдена.Многие из упомянутых выше результатов и гипотез были подкреплены
выводами других экспериментаторов. Аптекарь и др. [205], например, в сво¬
их первых РФЭС-исследованиях восстановленных катализаторов MoOt - А12С^
зафиксировали образование молибдена в трех состояниях окисления (Мо71,
Mov и MoIV). Некоторые из этих идей обсуждались в уже упоминавшемся об¬
зоре Эдмондса [175]. Особый интерес в этой работе представлял анализ из¬
менений энергии связи при термической обработке и изменений процентно¬
го содержания молибдена в катализаторе Мо03_ Al р^Многие из этих результатов и идей были недавно проанализированы в
обзоре Эркюля и Клейн [206]о Затем последовало быстрое развитие этих
результатов группой Эркюля [207] и важное подкрепление их гипотезы, ос¬
нованной на этих результатах» В своей обзорной статье Эркюль и Клейн
[ 206] указали, что с помощью тщательного анализа методом РФЭС теперь
можно выявить, следя за возможным образованием комплексной вакансии
на поверхности, различие между полезными взаимодействиями металлов с
носителем, например, как это наблюдали Окамото и др. [ 194]:и отрицательными или вредными взаимодействиями металлов с носителем,
т. е. за образованием соединений объемного типа Со А1204 или А12(Мо04)3.
3868. Применение электронной спектроскопии в гетерогенном катализеЭркюль и Клейн [206] отметили также, что РФЭС-исследования позволяют по¬
лучить необычно глубокое представление об (Со - Мо)-катализаторе, в част¬
ности благодаря осуществлению селективных, иногда едва заметных изме¬
нений химических состояний и концентраций соединений на поверхности.Так, например, надежно установленное наличие активного монослоя молибде¬
на, открытое сразу несколькими группами, работающими методом РФЭС,
превосходно согласуется с количественными воздействиями при избиратель¬
ном увеличении активных добавок. Кроме того, указывалось, что добавка
кобальта сверх установленного в настоящее время предела явно приводит к
образованию отравляющих поверхностных слоев Со304 и, возможно, СоА1204„
Тот факт, что Со2+, как оказалось, сначала занимает тетраэдрические, а
затем октаэдрические узлы и что каталитическое значение этих узлов из¬
меняется, недавно подтвержден с помощью РФЭС,хотя результаты и детали
этих измерений все еще содержат некоторые неопределенности.В обзоре Эркюля и Клейна [ 206] также содержатся исследования мето¬
дами РФЭС системы Мо -А1203, показавшие, что именно РФЭС позволила
впервые обнаружить несколько до сих пор неясных Мо™ -соединений на
окисленном катализаторе. Привлечение данных, полученных методом лазер¬
ного комбинационного рассеяния, позволило построить гипотезы об актив¬
ных поверхностных Мо™-соединениях и неактивном соединении А12(Мо04)3,
которые образуются в зависимости от условий внесения добавок и темпера¬
туры отжига. Восстановление соединений Мо71, по-видимому, приводит к
двум различным состояниям окисления молибдена, зависящим от местополо¬
жения Мо^ на окиси алюминия, т. е.:Мо™ -у Мо v тетраэдрические узлы,Мо™ -» МIV октаэдрические узлы.Обычно считают, что первое состояние является главным источником ката¬
литической активности [ 206]. Этот момент, однако, как мы увидим, все
еще дискутируется.Чин и Эркюль [207] наглядно обобщили эти аргументы, проведя деталь¬
ные исследования каталитической системы Со - Мо -А1203 при фиксиро¬
ванной концентрации молибдена (15 вес.%) переменной концентрации кобаль¬
та (1-9 вес. %) и осуществив восстановление без H2S и с H2S. Методами
РФЭС, СРМИ и фотоакустической спектроскопии проводились тщательные
измерения для демонстрации, кроме прочего, также того факта, что избы¬
точное содержание Со2+ (сверх 7 вес. % при 15 вес. % молибдена), по-ви¬
димому, приводит к образованиюСо304. Они заметили, что это последнее
соединение нельзя обнаружить, поскольку оно, очевидно, превращается в
СоМо04, если молибден также берется в избытке (свыше 20 вес.%)(рис. 8. 23)„ Соединение СоМо04, как было показано, является отравляю¬
щей примесью для реакции ГОС и оно естественно образуется при осаждении
Т.П. Барр387Рис. 8.23. а - ЭСХА-спектр дублета Мо 3d и участка спектра S 2s сульфированных
катализаторов. Все образцы исследовались после взаимодействия в тече¬
ние 1 ч с 15%-ной смесью H,S + Н, при 250 °С: 1 - ВА15; 2 - АЦЗ 315;
3-ALp5l5;4-AU3 915; 5-ALP 915. б - участок ЭСХА-спектра Со 2рз/2 сульфи¬
рованных АЦЗ-катализаторов. Образцы исследовались после взаимодейст¬
вия в течение 1 ч с 15%-ной смесью I^S + Н2 при 250 °С: 1 - АЦЗ 315; 2-
АЦЗ 515; 3 - АЦЗ 715; 4 - АЦЗ 915. Концентрация СоО и Мо О (в вес.%) составляла
соответственно: ЗА (0; 15,6), АЗЦ 315 (2,8; 16,5), АЦЗ 515 (4,8;14,2), АЦЗ
715 (6,7; 14,2) и АЦ3915 (8,9; 14,5) [207J.добавок молибдена и кобальта. Чин и Эркюль [ 207] предположили, что "поло¬
жительное” влияние кобальта не наблюдается до тех пор, пока его не до¬
бавляют в количестве более 1 вес.%, так как это начальное количество ко¬
бальта идет на образование неактивного соединения Со А1204 в тетраэдри¬
ческих узлах окиси алюминия. Количество этого неактивного соединения
может также слегка увеличиваться при концентрации кобальта от 1 до
3 вес.%, однако при концентрации выше 3% концентрация "активных" соедине¬
ний кобальта становится доминирующей. Восстановление, согласно Эркюлю
и сотр., не приводит к образованию значительного количества Со0 до тех
пор, пока не будет "избытка кобальта” (свыше 7 вес.% при 15 вес.% молиб¬
дена). Этот факт используется для доказательства присутствия в последнем
случае СоМо04, а не СоА1204, который трудно восстановить.Проводя совместные наблюдения, Чин и Эркюль [207] сделали важный
вывод, что СРМИ-результаты, по-видимому, указывают на то, что молибден
3888. Применение электронной спектроскопии в гетерогенном катализе(монослой ?) формируется в активном катализаторе на кобальте (7 вес.%
кобальта и 15 вес.% молибдена), в то время как неактивная аналогичная
система не имеет такой слоистой структуры.Предварительное сульфирование, согласно Чину и Эркюлю [207], определенно
способствует образованию MoS2; возможность вакансии MoS2-комплекса присоединять¬
ся по механизму Окамото и др. [ 194] к окиси алюминия в тетраэдрических узлах не
рассматривалась.» Предполагалось, что сульфирование кобальта приводит
к превращению некоторого количества кобальта в Со 9S8. Это предположение
основывалось на сдвигах энергий связи, а более доказательно - на коли¬
чественных данных по нарушению пропорциональности в MoS2o (Этот воп¬
рос будет прокомментирован позже») Сульфирование, как было показано,
не влияет на СоА1204; поэтому они утверждали наличие фазы кобальта
"сверху” t-CoAl204. Наличие кобальта, по-видимому, не влияет на суль•
фирование молибдена• Авторы сомневались относительно возможности об¬
разования значительного количества Со0 при этих условиях, хотя отмети¬
ли, что эта особенность может сильно зависеть от условий приготовления
катализатора (см. выше)оЧтобы объяснить эти результаты, Чин и Эркюль [207] предположили,
что кобальт (монослой) должен формироваться сверху *-Со А1204 и этот
слой находится в двумерном (близком) контакте с активным монослоем
молибдена, который лежит на немо (Таким образом, оба слоя образуют
квазибислойо) Стехиометрия этого Со - Мо-бислоя (КМС), как предполага¬
лось, равна 1 : 1о Было высказано предположение, что этот бислой являет¬
ся активной слоистой структурой для реакции ГОС в катализаторах, содер¬
жащих соединения Co9S8 и MoS2, образовавшиеся в этой структуре во вре¬
мя предварительного сульфирования. Таким образом, они считали, что мо¬
либденовый монослой в окисленном катализаторе занимает как тетраэдри¬
ческие (40%), так и октаэдрические (60%) узлы, в то время как активный
подслой Со2+ занимает тетраэдрические узлы между подслойным А1203 и
"верхним" монослоем молибдена. Этот подповерхностный слой (Со - Мо)
должен синергически влиять на каталитические свойства молибдена, но,
вероятно, он не влияет на его способность окисляться или реагировать с
серойоРассматриваемой проблеме изучения функционирования системы Со -Мо
было посвящено несколько других достаточно важных рабоТо Например,Хоуалла и Дзльмон [208] исследовали влияние на катализатор СоО - А1203
добавок бора. Бор, как предполагалось, увеличивает кислотность окиси алю¬
миния, что явно приводит к дальнейшей дисперсии кобальта и образ ованию
некоторого количества Со А1204 на поверхности. Аналогичным образом
Ведрен и др. [209] использовали метод ЭПР совместно с УФЭС и РФЭС для
изучения катализатора Мо03 -Ti04. В этой работе Мо™ вначале наносил¬
ся на анатазо Результирующее отношение концентраций поверхность/объем
Т.П. Барр389указывало на отсутствие сегрегации, т. е. Rs/Rb « 1. После прогрева на
воздухе анатаз превращался в рутил и значение Rs /Rb сильно возрастало.
Кроме того, объемный молибден, по-видимому, восстанавливался до Мо IV.
Это, очевидно, служило еще одним доказательством наличия сильного взаг
имодействия подложка - металл у Мо03, на которое влияют термические
условия. Для прямого сравнения с окисью алюминия необходимы дополни¬
тельные исследования.8.3.5.4. Сводка резупьтатовПоскольку рассмотренные выше результаты сложны и весьма сущест¬
венны для понимания функционирования каталитической системы Со — Мо,
в данном разделе мы приведем сводку этих результатов.а. Молибден в окисленном катализаторе Мо 03 -А1203 существует в
нескольких соединениях, содержащих Мо™, что зависит от концентрации,
температуры прокаливания и места его расположения. Указывалось [212]
на существование пяти таких соединений, причем некоторые из них, зани¬
мающие тетраэдрические узлы, как предполагается, имеют максимальную
каталитическую активность. Эти последние соединения, как утверждается,
существуют в виде комплекса поверхностного молибдата [199], имеющего
видОднако существование этого соединения на поверхности не было точно ус¬
тановлено. Надежно доказано, что каким бы ни было это активное соеди¬
нение, содержащее Мо™, оно с морфологической точки зрения диспергиро¬
вано в виде монослойного покрытия [ 195], которое насыщается при ~ 15 вес.%
молибдена. Дальнейшее увеличение Мо™ явно приводит к образованию вред¬
ного слоя Мо03 [207]. Утверждается, что в объеме также присутствует
отравляющее соединение А12(Мо04)3.б. Восстановление окисного катализатора Мо - А1203 (без S2+) приво¬
дит к образованию соединений, содержащих как Мо v, так и MoIV [ 205]. Пер¬
вые из этих соединений, как утверждается, занимают тетраэдрические уз¬
лы окиси алюминия, а вторые, как предполагается, возникают в октаэд¬
рических узлах [199].в. Комбинация восстановления и предварительного сульфирования ка¬
тализатора Мо03 - А1203, как было показано, приводит к образованию
MoS2 [206]. Были представлены некоторые доказательства [ 194] сущест-
3908. Применение электронной спектроскопии в гетерогенном катализевования комплекса с ненасыщенной серойоднако существование этого соединения не было подтверждено. Наличие мо¬
нослоя "активного" MoS2, по-видимому, было подтверждено в работе [206],
насыщение этим соединением тетраэдрических узлов окиси алюминия, веро¬
ятно, наступает при ~ 15 вес.% молибдена. "Избыточный” молибден оста¬
ется неактивным [ 206]. Присутствие Мо° после восстановления не было об¬
наружено, хотя автор этой главы ставит под сомнение [27] возможность
различить MoS2 и Мо°, исходя только из сдвигов РФЭС-энергий связи, так
как химический сдвиг (Зигбана) отсутствует. Это явление типично для
многих халькогенидов (см. также случай кобальта, описанный ниже). Что¬
бы можно было обнаружить любое из этих соединений, мы советуем де¬
тально исследовать индуцируемые РФЭС сдвиги потерь энергии или оже-
параметрьи Некоторые исследователи допускали возможность образования
соединений Мо111, однако имеется мало доказательств существования это¬
го необычного иона.г. Ряд исследователей также изучали информативную систему Со -А120^
[186]о В окисленной форме кобальт, как было показано, взаимодействуетс носителем из окиси алюминия, образуя, по-видимому, некоторое количест¬
во Со А1204 в тетраэдрических узлах. Тип образующихся соединений, как
было убедительно показано, зависит от количества активных добавок и тем¬
пературы прокаливания. Чин и Эркюль [207] утверждают, что начальное ко¬
личество, равное + 1 вес.% кобальта, попадает в тетраэдрические узлы
окиси алюминия в виде неактивного соединения f-Со А1204* Затем дальней¬
шее поступление кобальта приводит к образованию кобальта, по структуре
напоминающего монослой, и этот монослой насыщается при концентрациях,
которые, очевидно, могут зависеть от количества имеющегося молибдена
(в то время как обратное, очевидно, неверно)» Избыток кобальта может
поступать в октаэдрические узлы, существуя там, по-видимому, в форме
Со04 [207]оПродукты восстановления и предварительного сульфирования системы
СоА1204 не столь ясны, как аналогичные продукты катализатора
Мо -А1204. По-видимому, имеются данные об образовании Co9S8 [186, 190,
199J.д. На создание смешанного окисного катализатора СоО - Мо03 -А1203
вероятно, больше влияют возможное распределение и формы восстановления
соединений кобальта, а не молибдена. Восстановление этой "окисной" сис¬
темы, как было показано, приводит к образованию смеси соединений, в ко¬
торые входят Со11, Со Мо 71, Мо v и Мо1 v.
Т.П. Барр391е. Восстановление и предварительное сульфирование катализатора
Со -Мо -S -А1203 дают ряд своеобразных результатов, однако и в
этом случае присутствие кобальта опять, по-видимому, мало влияет на
соединения молибдена. Кажется доказанной значительная степень восстав
новления молибдена до MoS2. Это соединение, как утверждается, образует
монослойную структуру, которая насыщается при ^ 15 вес.% молибдена
[ 207L Некоторые данные, в частности, полученные методом СВМИ, строго
доказывают, что этот монослой покрывает любое активное соединение ко¬
бальта. Избыток молибдена, по-видимому, скапливается на наружной по¬
верхности в виде (ядовитого) Мо03. Имеются данные об образовании вред¬
ного объемного соединения СоМо04, однако выдвигаются также предполо¬
жения, основанные на РФЭС-данных и фигурах восстановления для этого
соединения, что оно образуется только в результате реакции избытка
Со304 сМо03. "Активный” кобальт в катализаторе Со -Мо -S — А1203
после (Н2 -H2S)-обработки, по-видимому, находится прежде всего в виде
Co9S8. Было получено доказательство существования значительного коли¬
чества Со0 [201], однако другие авторы полагают, что такой кобальт появ¬
ляется только во время восстановления любого избыточного количества
СоМо04, который образовался описанным выше способом. Отметим, что
разумному объяснению различий между Со9S8 и Со0 в методе РФЭС пре¬
пятствуют некоторые проблемы, упоминавшиеся ранее для MoS2 и Мо® [27].В любом случае количество кобальта, молибдена и серы, добавляемое во
время этих процессов, может существенно влиять на результаты. Со А1204>
очевидно, присутствует в восстановленном катализаторе Со -Мо -S -А12СЬ?
что обусловлено сопротивляемостью этого соединения процессу восстанов¬
ления» Чин и Эркюль [207] утверждают, что Со А1204 заполняет тетраэдри¬
ческие узлы глинозема при поступлении первого избыточного 1 вес% ко¬
бальта. Поступающий в дальнейшем кобальт, как предполагается, образует
монослойную структуру ниже монослоя MoS2 и, возможно, квазимостиковые
структуры между Мо S2 и тетраэдрическими узлами носителя из окиси алю¬
миния [207]. Предполагается, что этот кобальт находится в соединениях,
которые восстанавливаются до Co9S8 в потоке H2S. Хотя кобальт сам и не
влияет на поведение молибдена во время (Н2 - H2S )-обработки, именно этот
бислой MoS2, расположенный сверху "сандвича" Со 9S8, как утверждается,
обеспечивает уникальные ГОС каталитические свойства системы
Со —Мо -S -А1203 [207].Жо Взаимодействия носитель — металлы в этом катализаторе, как было
показано, имеют сильное положительное и отрицательное влияние на ката¬
лизатор. Эти взаимодействия прежде всего зависят от кислотности носите¬
ля, а также от его физической структуры [209]. Первый эффект исследо¬
вался путем добавки натрия [203] для подавления и бора для увеличения
кислотности окиси алюминия. Эти изменения, как было показано, заметно
3928. Применение электронной спектроскопии в гетерогенном катализевлияют на образование Со А1204 и Со03, а также на дисперсность и зна¬
чения Rs /Rb добавок. Необходимы дальнейшие исследования, чтобы внести
ясность в эти результаты.Наконец , следует отметить, что полный набор РФЭС-методов, исполь¬
зовавшихся для изучения этого катализатора, содержал не только традици¬
онные методы анализа спектров, но также и некот )рые из новых методов,
описанных ранее в этой главе. Например, Цинг и др. [199] учли зарядовый
сдвиг для объяснения небольшого уширения результирующего пика, что мог¬
ло неверно интерпретироваться другими исследователями.» Другие недавно
разработанные приложения метода РФЭС, возможно,способны оказать по¬
мощь в этих исследованиях. Например, проведение более точных структур¬
ных измерений по методикам Анжевена, Варту ли и Дельгаса [ 88], Керкхо-
фа и Мулина [89] и даже Фанга [90] приведет к улучшению анализа слоев0
Кроме того, использование оже-Параметров [27] или анализа сдвига пиков
потерь [ 32] может оказаться полезным при детектировании сульфидов*,В заключение отметим, что не следует забывать о тех серьезных последст¬
виях, к которым могут приводить даже малые изменения физических усло¬
вий. Это положение подтверждают результаты Уолтона [210].8.4. ЗаключениеВ настоящее время исследованиями гетерогенного катализа занимает¬
ся большое число специалистов в области электронной спектроскопии.
Объекты исследования в гетерогенном катализе чрезвычайно интересны,
так как они обладают сложными физическими и химическими свойствами*Как указывалось в предыдущих разделах этой главы, перед исследователя¬
ми вставали такие трудные проблемы, как зарядка образцов, обработка их
in situ, получениз относительных и количественных данных о неоднород¬
ных материалах, перемежающиеся химические сдвиги и т* д. Некоторые
из этих проблем обсуждались в упомянутых выше прогнозах Иейтса [ 2],
Иейтса и Мейди [ 31 и Фишера [ 4]. Тем не менее многие из них являются
новыми; особенно это относится к прикладному катализуиАвтор надеется, что в этой главе был показан вклад электронной
спектроскопии (особенно РФЭС) в исследование катализа. Несколько сущес¬
твенных достижений заслуживают особого упоминания*а* Возможно, наиболее ожидаемым достижением явилось использова¬
ние РФЭС для прямого детектирования химических сдвигов (особенно в
состоянии окисления) упомянутых выше добавок (например, металлов, окис¬
лов металлов, сульфидов металлов и То д«) в процессе приготовления и ис¬
пользования катализаторов* Были описаны многочисленные примеры восста¬
новления добавок, например: Rh111 -► Rh1 - Уотсон и Сомораи [ 112] - Re™1-
Шипиро и др. [ 136] — Pt и * Pt° (недостаток электронов в присутствии гер¬
мания ) — Боумэн и Билон [137] — Pd11 -> Pd° — Ведрен и др. [ 155] — Мо ^ 03 -♦
Т.П. Барр393-*Mov + WoIV - Цинг и др. [199], а также окисления добавок, на¬
пример, Ru° -♦ Ruiv02 на У-цеолитах - Педерсон и Лансфорд [ 168].б. Одним из самых главных недавних достижений в области приложения
электронной спектроскопии к изучению катализа явилось получение данных,
которые непосредственно связаны с взаимодействиями носитель — металл
(Н ~М) и металл - металл (М -М)о Были опубликованы многочисленные при¬
меры взаимодействия первого типа, особенно для систем с носителем из
А1203 (например, Ru -А1203) - Анжевен и Дельгас [139], Мо03/А1203А1 2(Мо04) 3- Окамото и др. [ 193, 194]0 В целом было установлено, что ког¬
да имеет место сильное М -Н- или М - М-взаимо действие, то способность
окисленных форм добавок восстанавливаться сильно уменьшается (напри¬
мер, Анжевен и Дельгас [ 139], Цинг и др0 [ 199]).в. Была установлена возможность наслаивания в катализе, и некоторые
ее аспекты были проверены методом электронной спектроскопии. Иногда
негомогенность, как было показано, имеет положительный каталитический
эффект, например при образовании селективных монослоев в системеСо -Мо -S (Гаярдо, Гранж и Дельмон [ 204], Чин и Эркюль [ 2071), а в
других случаях негомогенность отрицательно влияет на катализ, особенно
когда образуются кристаллы платиновых металлов (например, Сомораи и
Заира [102], Мейсон и Бетцольд [150]). Были также зарегистрированы се¬
лективная деградация цеолитов и осаждение на их поверхности значительно¬
го количества побочных продуктов (например, Барр и др0 [28, 42, 164]).г. РФЭС и ОЭС также успешно использовались для исследования свойств
и изменений каталитических промоторов. Одними из них были добавки
окислов металлов или сульфидов, например Re -S, в случае платиновых ка¬
тализаторов риформинга (Билон и др0 [ 138]); предполагалось, что другие
промоторы приводили к модификации носителя, например F~ на Al 203
(Скохарт и др. [211]) и Се+ на А1203 (Дефоссе и др. [212, 213]). В равной
степени впечатляющими были многочисленные ситуации, когда электрон¬
ная спектроскопия использовалась для обнаружения и измерения парамет¬
ров каталитических ядов, например углерода на платине (Сомораи и Заира[ 102]) и Na+на Со -Мо — А1203 (Ликоргиотис и др. [203]).Нет сомнения в том, что имеется возможность прямых контактов меж¬
ду "фундаментальным” и "прикладным" катализом. Сомораи и Заира [102],
а также Эртль [70] дали примеры трех типов таких экспериментов* Однако
до тех пор, пока подобные усилия не подкрепляются контролируемым влия-
нием добавок металлов, диспергированных на носителе с хорошо установ¬
ленными свойствами (возможно, с использованием СХПЭЭ), прямой пере¬
нос полученной информации будет в лучшем случае дискуссионным. Автору
настоящей главы представляется, что в области прикладного катализа так¬
же необходимы большие усилия со стороны искусных экспериментаторов, что¬
бы, например, получить информацию о необычных фазах носителей, обо-
3948. Применение электронной спектроскопии в гетерогенном катализегащенных специально подобранными металлами, которые, согласно ре¬
зультатам измерений по методу СТЭМ, диспергированы на микроскопа
ческом уровне, и подтвердить правильность описанных результатов. На
данном этапе можно с помощью электронной спектроскопии детектировать
изменения спектров для обнаружения любой химической перестройки, од~
новр\. ченно следя при помощи электронного микроскопа за физическим
состоянием системы• Усилия по обе стороны существующего в настоящее
время фундаментально-прикладного барьера позволяют надеяться на то,
что в текущем десятилетии будет достигнуто "проникновение" через этот
барьер*В заключение перечислим, к чему приведет использование электронной
спектроскопии в области гетерогенного катализа в предстоящие восемь
лет.а) к полезному использованию СХПЭЭ для изучения нанесенных катали¬
заторов;б) к получению прямой информации о связи металл - адсорбат в прик¬
ладном катализе;в) к квантовомеханическим расчетам, которые будут коррелировать с
химическими данными для модельных и даже некоторых промышленных
катализаторов;г) к достижению прямой корреляции между структурными изменениями
(например, дефекты, дисперсность, образование кластеров) и данными
электронной спектроскопии;д) к количественным методикам, точно фиксирующим эти химические
и структурные изменения;е) к разработке методов, которые дают информацию (без явного пов¬
реждения материалов) о химических свойствах слоев катализатора в попе¬
речном направлении и по глубине;ж) к получению полностью согласующихся между собой результатов
анализов с использованием многих методик;з) к проведению динамических исследований в области прикладного,а
также фундаментального катализа и, наконец,и) к осуществлению важных исследований в области прикладного ката¬
лиза, которые не только будут давать информацию об изучаемой системе,
но позволят также предсказывать будущие более практичные каталитичес¬
кие системы*Литература1. Siegban К., Perspectives and problems in electron spectroscopy, J. Elect¬
ron Spectrosc., 5, 3 (1974).2. Yates /«Т., Jn, Chem. Engr. News, August 1974# 19 (1974).3. Yates J»T*, Ju, Madey T.E., in Science, Technology and the Modem Navy,
Office of Naval Research, Arlington, Va., 1976, pp. 415 — 432.
Т.П. Барр3954. Fischer Т.Е., J. Vac. Sci. Technol., 11, 252 (1974).5. McCarroll J.T., Surf. Sci., 53, 297 (1975).6. Tompkins F.C., Proc. Int. Congr. Catal., 6, 32 (1977).7. McCarroll Edmonds T., Pitkethly R.C., Nature, 233, 1260 (1969).8. Carberry ].}., Kuczynski G.C., Martinez E., J. Catal., 26, 247 (1972).9. Van Santen R.A., Sachtler W.M.H* J. Catal., 33. 202 (1974).10. Stevens G.C%, Edmonds T., J. Catal., 37, 544 (1975).11. Bhasin MM., J. Catal., 34, 356 (1974).12. Delgass W.N., Hughes T.R., Fadley C.S., Catal. Rev., 4, 179 (1970).13. Brinen /.S., Melera A., J. Phys. Chem., 76, 2525 (1972).14. Brinen /.S., J. Electron Spectrosc. and Related Phenomena, 5, 377(1974).15. Matienzo L.J., Yin L., Grim S.O*> Swartz W.E., Jr., Inorg. Chem., 12,2762 (1973).16. Lindsay J.B., Rose H*J/г., et aU, Appl. Spectrosc., 27, 1 (1973).17. Tempere J»Fr., Delafosse D.M Contour J*P*, Chem. Phys. Lett., 33 „ 95(1975).18. Armour A.W., Mitchell P.C.H. et al.9 J.Less. Common Metals, 36, 361 (1974).19. Carmesm P., Grange P., C.R. Acad. Sci., Paris, 281, 757 (1975).20. Madey T.E., Wagner С. D., Joshi A., J. Electr. Spectrosc. and Related
Phenomena, 10, 359 (1977).21. Powell C.J., Erickson N.E., Madey T.E., J. Electron Spectrosc. and
Related Phenomena, 17, 361 (1979).22. Fadley C.S., Electron Spectroscopy: Theory, Techniques and Applications
(eds. C.R. Brundle^.D. Baker), Vol. 2, Academic Press, New York, 1978,
pp. 1 —156.23. Gadzuk J*W., Sunjic MPhys. Rev., В12, 524 (1975).24. NgK.T., Hercules DMJ. Phys. Chem., 80, 2094 (1976).2 5, Brinen J.S., ins Applied Surface Analysis (eds. T.L. Barr, L.E. Davis).
ASTM, Philadelphia, 1978, pp. 24 - 41.26. Hercules DM», Klein J.CApplied Electron Spectroscopy for Analysis
(eds. H. Windawi,F. Ho), John Wiley, New York, 1982, pp. 147 — 1980.27. Barr T.L., The Eleventh Annual Symposium Applied Vacuum Science and
Technology, AVS, Tampa, February 1982 (не опубликовано).28. Barr T.L., Appl. Surf. Sci., 15, 1 (1983).29. Barr T.L.V Greene J*E., Eltoukhy A.H., J. Vac. Sci. Technol., 16tf 517
(1979).30. Eltoukhy A.H>, Natarajan B.R., Greene /•£•, Barr T.L., Thin Solid Films,
69, 217 (1980).31. Barr T.L., Greene J.E., Eltoukhy A.H., Natarajan B.R., Thin Solid Films
(в печати).32. Barr T.L. (в печати).33. Larsen P.E., J. Electron Spectrosc, and Related Phenomena, 4, 214 (1974).
3968. Применение электронной спектроскопии в гетерогенном катализе34. Barr T.L., Hackenberg J.J., Appl. Surf. Sci., 10, 523 (1982).35. Barr T.L., Hackenberg J.J., J. Amer. Chem. Soc., 104, 5390 (1982).36. Barr T.L., Surf. Interface Analysis, 4, 185 (1982).37. Wagner C.D., Anal. Chem., 47, 1201 (1975).38. Wagner C.D., Gale C.D., Raymond R.H., Anal. Chem., 51, 466 (1979).39. Gland J»L. Sexton B.A., Fisher G., Surf. Sci., 95, 587 (1980).40. Kishi K., Roberts M.W., Proc. Roy. Soc., London, A352, 587 (1976).41. Thomas G.E., Weinberg W.H., Phys. Rev. Istt., 41 * 1181 (1978).42. Barr T.L.t \CS Symposium, Chicago, January 1982 (в печаш)о43. Miller J*N>, Ling D. T. et ah, Surf. Sci., 94* 16 (1980).44. McCabe R.W., Schmidt L.D., Surf. Sci., 60, 85 (1976).45. Adsorption at Solid Surfaces (eds. C.R. Brundle, C.J. Todd), North-Holland,
Amsterdam, 1975.46. Interactions on Metal Surfaces, (ed. R. Gomer), Springer-Verlag, Berlin,1975.47. Roberts McKee C.S., Chemistry of the Metal — Gas Interface, Clarendon
Press, Oxford, 1978.48o Somorjai G.A., Chemietry in Two Dimensions: Surfaces, Cornell U. Press,
Ithaca, 1981.49. Madix R.J., Surf. Sci., 89. 540 (1979).50. Madix R.J., Adv. in Catal., 29ff 1 (1980).51. Benziger J.B., Madix R.J., J. of Catal., 65, 36 (1980).52. Barteau M.A., Ко Madix R.J,, Surf. Sci., 102, 99 (1981).53. Matloob M.H., Roberts M.W., J. Chem. Soc., Faraday Trans., 73, 1393 (1977).54. Jonson D.W., Matloob M.H., Roberts M.W., J. Chem. Soc. Chem. Comm.,1978, 41 (1978).55. Roberts M.W., Adv. in Catal., 29, 55 (1980).56. Chen B.~H., Close J.S., White JM., J. Catal., 46, 253 (1977).57. Campbell C.T., White J.M., J, Catal., 54, 289 (1978).58. Golchet A., White JM., J. Catal., 53, 266 (1978).59. Lee НЛ., White J.M., J. Catal., 63, 261 (1980).60. Close J.S., White J.M., J. Catal., 36, 185 (1975).61. Matsushima Т., White JM., J. Catal., 40, 334 (1975).62. Matsushima Т., Mussett C.J., White JM., J. Catal., 41, 397 (1976).63. Shi S.K., White J.M., Hance R.L., J. Phys. Chem., 84 , 2441 (1980).64. Tseng F*H., White J.M., J. of Chem., Kinetics, 12, 417 (1980).65. Wu O.K.T., PhD. Dissertation, U. of Illinois, Chicago, 1979*66. Wu O.K.T., Bums R.P., J. Phys. Chem., 84, 1445 (1980).67. Wu O.K. Т., Bums R.P., Surf. Interface Anal., 3, 29 (1981).68. Ho P., PhD Dissertation, U. of Illinois, Chicago, 1981.69. Ho P., Bums R.P., Appl. Surf. Sci. {в печати).70* Ertl GChemistry and Physics of Solid Surfaces (eds. R. Vanselow,W. England), V. Ill, CRC Press, Boca Raton, Florida, 1982, p. 11.
Т.Л. Барр39771. Boudart Мь, Interactions on Metal Surfaces (ed. R. Gomer), Springer-Verlag,
Berlin, 1975, pp. 275 - 298.72. Briggs D., Appl. Surf. Sci., 6, 188 (1980).73. Kelley М., J. Catal., 57, 113 (1979).74. Clarke J.K.A., Chem. Rev., 75, 291 (1975).75. Kelley MThe Catalyst Club, Chicago, 1981 (не опубликовано).76. Lewis R.T., Kelly M.A., J. Electron Spectrosc., and Related Phenomena,
20, 105 (1980).77. Wagner C.D., J. Electron Spectrosc. and Related Phenomena, 18, 345(1980).78. Windawi H., Wagner C.D., in: Applied Electron Spectroscopy for Chemical
Analysis (eds* H. Windaw^F.F.L. Ho), John Wiley and Sons, New York,
1982, pp. 191 - 208.79. Windawi HJ. Electron Spectrosc. and Related Phenomena, 22, 373 (1981).80. Barr T.L., U. of Wisconsin, Milwaukee (в печати).81. Barr T.L., Chem. Phys. Lett., 430 89 (1976).82. Swift P., Surf. Interface Anal., 40 47 (1981).83. Stephensen D.A., Binkowski N*J*, J. Noncrystalline Solids, 22, 399 (1976).84. Castle J*E., West R.H., J. Electron Spectrosc. and Related Phenomena,18, 195 (1979).85. West R.H., Castle J.E., Surf. Interface Analysis, 4, 68 (1982).86. Wagner C.D., Six H.A., Jansen W.T., Taylor J.A., Appl. Surf. Sci., 9, 263(1981).87. Scharpen L.H., J. Electron Spectrosc. and Related Phenomena, 5, 369
(1974).88. Angevine P.J., Vartuli J.C., Delgass W.N., Proc. Int. Congress Catal.,6 , 611 (1977).89. Kerkhof F.P.J., Moulijn ].A.: J. Phys. Chem., 83, 1612 (1979).90. Fung S.C., J. Catal., 56, 454 (1979).91. Baschenko 0*A., Nefedov V.L*, J. Electron Spectrosc. and Related Phe¬
nomena, 13, 405 (1979).92. Nefedov VJ., Serguskin N.P., Band I.M., Trzhaskovskya J., Electron
Spectrosc. and Related Phenomena, 2, 383 (1973).93. Nefedov V.L et al., J. Electron Spectrosc., and Related Phenomena, 7,175 (1975).94. Nefedov V.L, Yarzhensky V.G., J. Electron Spectrosc. and Related Pheno¬
mena, 11, 1 (1977).95. Somorjai G.A., Adv. in Catal., 26, 1 (1977).96. Stair P.C., Kaminska T.J., Kismodel L.L., Somorjai G.A., Phys. Rev.,В11. 623 (1975).97. Lang B., Joyner R.W., Somorjai G.A., Surf. Sci., 30, 454 (1972).98. Blakely D.W., Sexton B.A., Kozak E., Somorjai G.A., J. Vac. Sci. Technol.
13, 1091 (1976).
3988. Применение электронной спектроскопии в гетерогенном катализе99. Gillespie W.D., Herz R*K*, Peterson E*ESomorjai /, Catal., 70, 147(1981).100. Gland J.L; Baron K*, Somorjai G.A., J. Catal., 36, 305 (1975).101. Blakely D*W; Somorjai G*A>, J. Catal., 42, 181 (1976).102. Somorjai G*A., Zaera FJ. Phys. Chem., 86, 3070 (1982).103. Lang ВJouner R*W; Somorjai G*A*, Proc. Roy. Soc. London, A331 (1975).104. Weinberg W*H*, Deams Я.Л., Merrill Я.Р., Surf. Sci., 41, 312 (1974).105. Davis S*M>, Gordon B,EPress M., Somorjai G.A., J. Vac. Sci. Technol.,
19, 231 (1981).106. Salmeron М», Somorjai G.AJ. Phys. Chem., 86, 341 (1982).107. Kismodel L«L., Falicov Solid State Comm., 16, 1201 (1975).108. Sachtler J»W.A., Van Hove М.Л., Biberian /.P., Somorjai G.A., Surf. Sci.,
110, 19, 43 (1981).109. Sachtler J*W,ASomorjai G.4. (в печати)0110. Smith C.Z., Biberian J.P., Domorjai G.A., J. Catal., 57, 426 (1979).111. Dubois L.H; Hansma P*K; Somorjai G.4., J. Catal., 65, 318 (1980).112. Watson F$R*, Somorjai G.4., J. Catal., 72, 347 (1981).113. Watson P»R., Somorjai G.4., J. Catal., 74, 282 (1982).114. Spencer N*D; Schoonmaker R.C., Somorjai G.A; J. Catal., 74, 129(1982).115. Haensel VUS Patent 2, 479, 109 (1949).116. Heinemann H*, in: Catalysis: Science and Technology (eds. J.R. Ander¬
son, M. Boudert), Springer-Verlag, Berlin, 1981, pp. 1— 41.117. Good M*L*, Hayes /*C., The Kilpatrick Lectures at IIT, October 1982.118. Sachtler W*M*H», Van Santen R.AAppl. Surf. Sci., 3, 121 (1977).119. Sachtler W*M»H», Van Santen R.A., Adv. in Catal., 26, 69 (1977).120. Boumouville J*P•, Martino Gin: Catalyst Deactivation, (eds. B. Delmon,
G.F. Froment), Elsevier, Amsterdam, 1982, p. 159.121. Burch R*, J. Catal., 71, 348, 360 (1981).122. Bowman R*, Biloen PProc. Seventh Intern. Vac. Cong, and Third Intern.
Conf. Solid Surfaces, Vienna, 1977, p. 1129.123. Clarke J*K*A*, Creaner A»C»M*, Ind. Eng. Chem. Prod, and Dev., 20, 575
(1981).124. Kim K*S; Winograd NDavis J%, Amer. Chem. Soc., 93, 6296 (1971).125. Hammond J»S*, Winograd NJ. Electroanal. Chem. Interfacial Electrochem.,
78, 55 (1977).126. Kim K.SGrossman H»FWinograd М., Anal. Chem., 46* 197 (1974).127. Gaarenstroom S.W., Winograd N., J. Chem. Phys ., 67, 3500 (1977).128. Ban T.L., J. Phys. Chem., 82, 1801 (1978).129. Cimino A., De Angelis B*A., Gazzoli D., Valigi MZ. Anorg. Allg. Chem..
460, 86 (1980).130. Wagner C.D., Riggs W.N• et ah, Handbook of X -ray Photoelectron Spec¬
troscopy, Perkin-Elmer Co, Eden Prarie, Minnesota, 1970.
Т.Л. Барр399131. Escard J., Pontvianne В., Chenebaux М.Т., Cosyns J., Bull. Soc. Chem.
Frances, 11, 2399 (1975).132. Czaran E., Finster ]., Schnabel K.H., Z. Anorg. Allg. Chem., 443, 175
(1978).133. Bozon-V erduraz F., Omar A., Escard J., Pontvianne B., J. Catal., 53,126 (1976).134. Griffiths R.J.M., Evans E.L., J. Catal., 36, 413 (1975).135. Batista-Lcal М., Lester J.E., Lucchesi C.A., J. Electron Spectrosc. and
Related Phenomena, 11, 333 (1977).136. Shipiro E.S., Avaev V.I., Antoshin G.V. et al., J. Catal., 55, 402 (1978).137. Bouwman R., Biloen P., J. Catal., 48, 209 (1977).138. Biloen P., Helle V.N. et al., J. Catal., 63, 112 (1980).139. 4ngevine P.T., Delgass V.N», Sixty-ninth Annual AIChE meeting, Chica¬
go, 1976, p. 55a.140. Dautzenberg F.M., Walters H.B.M., J. Catal., 51, 26 (1978).141. Giordano N., Bart J.C.J. et al., Anali di Chemico, 1980, 585 (1980).142. Brinen J.S., Schmitt J.L., J. Catal., 45, 274 (1976).143. Stoddart C.T.H., Moss R.L., Pope D., Surf. Sci., 53, 241 (1975).144. Contour J.P., Mouvier G., Hoogewys М., Ledere C., J. Catal., 48, 217
(1978).145. Takasu Y., Unwin R., Tesche B., Bradshaw A.W., Surf. Sci., 77, 219
(1978).146. Kim K.S., Winograd N., Chem. Phys. Lett., 30,91(1975).147. Cini М., Surf. Sci., 62, 148 (1979).148. Garbasi F., Tauszik G.R., J. Microsc. Electron, 2, 245 (1977).149. Mattagino G., Polsonetti G., Tauszik G.R., J. Electron Spectrosc. and
Related Phenomena, 14, 237 (1978).150. Mason M.G., Baetzold R.C., J. Chem. Phys., 64, 271 (1976).151. Haensel V., Sterba M.J., Ind. Eng. Chem. Prod. Res. and Dev., 15, (1976).152. Bhasin M.M., J. Catal., 38, 218 (1975).153. Szymerska I., Lipski М., J. Catal., 41, 197 (1976).154. Matsumoto Y., Soma М., Onishi Т., Tamaru K., J. Chem. Soc., Faraday
Trans. I, 76, 1122 (1980).155. Vedrine J.C., Dufaux М., Naccache СImelik B., J. Chem. Soc., Faraday
Trans. I, 74, 440 (1978),156. Okamoto Y., Ishida N., Imanaka Т», Teranishi S., J. Catal., 58, 82 (1979).157. Anderson S.T..T., Scurrell M.S., J. Catal., 71, 233 (1981).158. Breck D.W., Zeolite Molecular Sieves, Wiley-Interscience, New York, 1974.159. Weiz P.B., Frilette N.J., Maatman R.W., Mower E.B., J. Catal., 1, 307
(1962).160. Plank C.J., Rosinsky E.J., Hawthorne W.P., I and E.C. Prod. Res. Devel.,
3, 165 (1964).161. Angauer R.J., Landolt G.R., LLS. Patent 3702886 (1972).
4008. Применение электронной спектроскопии в гетерогенном катализе162. Миначев X. М., Антошин Шапиро ЕНаврузов Т*А>, Изв. АН СССР
сер. хим., 9, 2131 (1973)0163. Kneckt ]., Stork G., Z. AnaL Chem., 283, 105 (1977).164. Barr T»L», Amer. Chem. Soc. Div. Petroleum Chem., 23. 82 (1978).165. Dempsey Е», Molecular Sieves, Soc. Chem. Ind. London 1968, 293 (1968).166. Wichterlova S., Novakova Kubelkova LJiru P., Proc. Fifth. Int. Gonf.
Zeolites (eds. Rees, Lovat), 1980, pp. 373 — 381.167. Bravo F.O., Dwyer J., Zambaulis D., Proc. Fifth. Int. Conf. Zeolites, (eds.
Rees, Lovat), 1980, pp. 749 — 759.168. Pederson L.A», Lunsford J*H*, J. Catal., 61, 39 (1980).169. Gallezot P., Alacon-Diaz A., et ah, J. Catal., 39, 334 (1976).170. Gustafson B»L», Lunsford ]»H., J. Catal., 74, 393 П082).171. Fajula P., Anthony R.G., Lunsford J.H., J. Catal., 73, 237 (1982).172. Kuznicki S.M., Eyring E.M., J. Caul., 65, 227 (1980).173. Larkins P.P., Hughes M.E., Anderson J.R., Foger K>, J. Electron Spectrosc.
and Related Phenomena, 15,. 33 (1979).174. Badrinarajanan SHodge RJ. et ah, J. Catal, 71, 439 (1981).175. Edmonds Т», in: Characterization of Catalysis (eds. J.M. Thomas, R.M. Lam¬
bert), Wiley-lnterscience, John Wiley and Sons, New York, 1980.176. Fiermans L., Hoogewijs R>, Vermik Surf. Sci., 47, 1 (1975).177. Atkinson S»J%, Brundle C.R., Roberts M.W., Discussion of Faraday Soc.,58, 62 (1974).178. Brundle C.R., Surf. Sci., 48 99 (1975).179. Brundle C.R., Aspects of kinetics and dynamics of surface reactions, Lec¬
ture delivered at La Jolla Institute Workshop, San-Diego, 1979 ( неопубли-
ковано).180. Cimino ADeAngelis S.4., J. Catal., 36, 11 (1975).181. Frost D.C., McDowell С.Л., Woolsey LSMolec. Phys., 27, 1473 (1974).182. Kim K.S., Phys. Rev., В11, 2177 (1975).183. McIntyre N.S., Cook M.C., Anal. Chem., 47, 2208 (1975).184. Chuang TJ., Brundle C.R>, Wandelt Kt, Thin Solid Films, 53, 10 (1978).185. Haber Marczewski WStock J», Ungier Lb, Proc. Int. Cong. Catal.,6, 827 (1977).186. Declerck-Gnmce RJ., Canesson P., Friedman R.M», FripletJJJ. Phys.
Chem., 82, 885 (1978).187. Declcrck°Grimce R.L, Canessen P., Friedman R.M., Fnplet J. Phys.
Chem., 82, 889 (1978).188. Okamoto YNakano H. et ah, J. Catal., 50, 447 (1977).189. Grimblot ]», Bonnelle J.P., J. Electron Spectrosc. and Related Phenomena,9, 449 (1976).190. Massoth P.P., J. Catal., 54, 450 (1978).191. Okamoto Y., Imanaka TTeramshi S., J. Catal., 54, 452 (1978).192. Okamoto YShimokawa Т., Imanaka Т., Teramshi S., J. Chem. Soc.,Chem. Soc,, 47 (1978).
Т.П. Барр401193. Okamoto Y», Shimokawa T*, Imanaka Т», Teranishi S., J. Catal,, 57, 153
(1979).194. Okamoto Y•, Tomioka H. a/*, J. Phys. Chem., 84, 1833 (1980).195. Giordano NCastellan et ah, J. Catal., 37# 204 (1975).196. Edmonds Т», Р.С.Я., J. Catal., 64, 491 (1980).197. Patterson T.A., Carver J»C», Leyden D.C., Hercules DM», J. Phys. Chem.,
30, 1700 (1976).198. Patterson Т.Л., Carver J.C., Leyden D.E., Hercules DM*, Spectrosc.
Lett., 9, 65 (1976).199. Zmgg D.S., Makovsky L.E» et aU, J. Phys. Chem., 84* 2898 (1980).200. Delvaux GGrange P., Delmon В», J. Catal., 56, 991 (1979).201. Brinen J.S», ArmstrongW»D. J. Catal., 54, 57 (1978).202. Delannay FGajardo P., Orange P., Delmon ВJ. Chem. Soc., Faraday
Trans. 1,76, 988(1980).203. Lycourghiotis A., Defosse et ah, J. Chem. Soc., Faraday Trans. I, 76,
1677, 2052 (1980).204. Gajardo P., Grange P., Delmon ВJ. Catal., 63, 201 (1980).205. Aptekar E.L., Chudinov Alekseev AM; Krylov O.F., Reaction
Kinetics and Catal. Lett., 1, 493 (1974).206. Hercules DM», Klein ]%Cin: Applied Electron Spectroscopy for Chemi¬
cal Analysis (eds, H. Windawi, F.F.L. Ho), John Wiley and Sons, New York,
1982.207. Chin R.L», Hercules DM», J. Phys. Chem., 86* 3079 (1982).208. Houalla M., Delmon ВAppl. Catal., 1, 285 (1981).209. Vedrine /.С., Prilaud HMeriandeau PChe MSurf. Sci., 80,101
(1979).210. Walton R.A., J, Catal., 44, 335 (1976).211. Scohart P.O., Selim 5.Л., Demon J,PRouxet P»G• J. Coll. and Inter¬
face Sci., 70, 209 (1979).212. Defosse С», Canesson P», Rouxhet P.G., Delmon ВJ. Catal., 51, 269(1978).213. Defosse С», Canesson P., Rouxhet P., Delmon В», J. Catal., 57, 525(1979).
Г™/iciBci 9
ПРИМЕНЕНИЕ РФЭС
В ТЕХНОЛОГИИ ПОЛИМЕРОВД. Бриггс119.1. ВведениеПолимеры находят все более широкое применение в материаловедении:
в формовании пластмасс, листов, волокон и пленок, в композитах с неорга¬
ническими наполнителями, в защитных покрытиях, герметизирующих соста¬
вах, клеях и т. д. Метод РФЭС занимает значительное место в этой области
для определения характеристик материалов по двум причинам: во-первых,
благодаря возможности анализировать сравнительно трудные в обращении
материалы без необходимости специальной подготовки образца и, во-вто-
рых, благодаря его чувствительности к свойствам поверхности.В данной главе сначала обсуждаются различные аспекты аппаратуры,
работы с образцами и спектральной информации, имеющие особенное значе¬
ние при изучении полимерных материалов. Затем кратко рассматриваются
преимущества и недостатки РФЭС по сравнению с другими используемыми
методами. Оставшаяся часть главы посвящена примерам успешного приме¬
нения РФЭС в этой области. Основное внимание сконцентрировано на ис¬
следованиях модификации поверхности полимеров для улучшения адгезии,
поскольку благодаря этим исследованиям удалось многое прояснить в эм¬
пирически разработанных процессах, имеющих важное технологическое
значение.9.2. Работа с образцамиВ общем случае для работы с полимерами не требуется никаких специальных при¬
способлений. Вопреки ожиданиям многих исследователей, работающих с метал¬
лическими системами, при анализе полимеров не возникает серьезных проблем,
связанных с гажением образцов. Закрепленные или приклеенные двусторонней
клеящей лентой (которая сама по себе является полимерной) образцы легко^Do Briggs, Imperial Chemical Industries PLC Petrochemicals and Plastics Division
Wilton, Middlesbrough, Cleveland, UK.
Д. Бриггс403могут быть введены в сверхвысоковакуумную камеру. Часто выражаются опа¬
сения о возможности загрязнения кремнием от соединений, использующихся
в клеящих лентах. В работе Кларка [1] сообщалось об экспериментах, в кото¬
рых методом РФЭС изучалась миграция соединений кремния вдоль полимер¬
ных поверхностей и через полимерные пленки (не в условиях вакуума). Наб¬
людалась миграция обоих типов, причем скорость ее вдоль некоторых поверх¬
ностей оказалась довольно большой (например, она составляет несколько
сантиметров за десять дней вдоль поверхности полиэтилена высокого давления
(ПЭВД) при комнатной температуре). Следовательно, существует опасность
загрязнения при довольно длительном контакте образцов с клеящей лентой,
например при обеспечении сохранности образцов во время перевозки и при
нанесении на образцы клеящихся меток.Загрязнение поверхностей полимеров не представляет особой проблемы
при внимательном обращении с образцом. В общем случае коэффициент при¬
липания молекул остаточного газа довольно мал, тем не менее за время
эксперимента часто наблюдается рост сигнала углеводородных соединений
(этот сигнал часто использовался в качестве точки отсчета по энергетичес¬
кой шкале при изучении полимерных материалов, которые не имеют в спект¬
ре хорошо разрешенной "углеводородной" линии С Is; см. приложение 2),Эти загрязнения вносят искажения при количественных измерениях в первую
очередь при регистрации пиков в области малых кинетических энергий (эти
пики образованы электронами с малой глубиной выхода). Возможно, еще
более существенным эффектом воздействия загрязнений является искажение
профиля линии С Is. Источниками загрязнений практически всегда являются
окно или корпус источника рентгеновского излучения, располагающиеся в
непосредственной близости к образцу- При работе с полимерами рабочее
давление в спектрометре обычно существенно выше остаточного давления,
до которого откачивается система, вследствие медленного гажения и выде¬
ления летучих низкомолекулярных соединений Это приводит к усложнению
проблем, связанных с загрязнением образца- Лучшая предупредительная
мера в этом случае - это регулярная откачка системы с прогревом.По опыту автора, поверхности полимеров, даже содержащих добавки, яв¬
ляются достаточно стабильными (в том смысле, что спектры в значитель¬
ной мере остаются неизменными в течение времени анализа) о Длительное
облучение образца рентгеновским излучением с характерной плотностью по¬
тока часто приводит к его видимому разрушению (обесцвечиванию)» Сооб¬
щалось, что у политиокарбонилфторида происходит очень быстрая деполимери¬
зация, а в поливинилиденфториде постепешо диссоциируют связи HF и сшив¬
ки [ 2]. В работе [ 3] детально исследовались изменения, происходящие на
поверхности политетрафторэтилена (ПТФЭ) при длительном рентгеновс¬
ком облучении, и состав соединений, выделяющихся с образца- Среди широ¬
ко распространенных полимеров, по-видимому, наиболее чувствительными
к рентгеновскому облучению с точки зрения разрушения образца являют-
4049. Применение РФЭС в технологии полимеровся полимеры, содержащие хлор, и в первую очередь поливинилхлорид. Раз¬
рушение образцов происходит также вследствие теплового воздействия. Так
как источники рентгеновского излучения в некоторых приборах могут силь¬
но нагреваться, при анализе поверхностей хороших теплоизоляторов будет
происходить их разогрев до значительных температур. Это приведет к раз¬
рушению структуры поверхности, миграции и/или испарению добавок и т. д.
Уменьшению этих эффектов может способствовать охлаждение держателя
образцао9.3. АппаратураОсновной проблемой при анализе поверхности полимеров является на¬
личие эффектов зарядки, связанной с диэлектрическими свойствами этих
материалово Однако при использовании обычных источников рентгеновс¬
кого излучения величина эффекта обычно не превышает 5 В. Можно счи¬
тать, что эта величина особенно сильно зависит от состава поверхности
[ 4], все остальные факторы (в первую очередь поток рентгеновского из¬
лучения и положение образца) примерно равноценные Значительно хуже
обстоит дело с использованием монохроматического рентгеновского излу-
ленияо Это связано в основном с тем, что окно, отделяющее источник рент¬
геновского излучения от образца и обеспечивающее стабилизирующий поток
электронов на диэлектрические поверхности, в этом случае расположено
значительно дальше от образца и не может обеспечить достаточный поток
электроново В таком случае обязательно необходимо применять какой-ли¬
бо из методов нейтрализации заряда. Обычно используется компенсацион¬
ная пушка, облучающая поверхность образца потоком низкоэнергетичных
электронов (см. приложение 2)0В общем случае для возбуждения преимущественно используется линия
MgKa, поскольку при этом пики имеют наименьшую ширину. В тех случаях,
когда наблюдается перекрывание представляющего интерес пика фотоэлект¬
ронов с внутренней оболочки с пиком оже-электронов, имеет смысл исполь¬
зовать источник с двойным анодом (обычно MgKa и AlKa), Например, на
линии MgKa наблюдается сильное перекрывание между С Is и Na KLL и
между Na Is и С 1 LMM, Вопрос применимости монохроматизированного
рентгеновского излучения (монохроматизировать необходимо в первую оче¬
редь излучение А\Ка) при изучении полимеров является спорным. Дости¬
гаемое при этом улучшение разрешения незначительно по сравнению с макси¬
мальным разрешением на линии MgKa и осуществимо только при очень тща¬
тельной компенсации заряда. Времена сбора данных могут быть велики,
так как поток рентгеновского излучения уменьшается, однако устраняются
рентгеновские сателлиты, которые могут быть помехой для анализа спект¬
ров (см. гл. 3). С другой стороны, это легко осуществить при машинной
обработке спектров, полученных при использовании немонохроматизированно-
го излучения (например, MgKa;cM. приложение 3).
Д. Бриггс405Рис. 9.1. Схематичное изображение профилей концентрации кислорода по глубине,
полученных на основании измерений при разных углах сбора фотоэлектро¬
нов. Поверхности полимеров были окислены различными способами: а - по¬
лиэтилен, обработанный коронным разрядом; б - полиэтилен после горяче¬
го прессования; в - полистирол, окисленный плазмой [5].Особую ценность при анализе поверхности полимеров представляет
возможность задавать угол между поверхностью образца и направлением на
входную щель анализатора (см. гл. 2). Во многих интересных случаях на
анализируемой с помощью РФЭС толщине образца концентрация может за¬
метно изменяться. Метод травления ионами аргона неприемлем для получе¬
ния профилей состава в полимерах вследствие быстрых изменений струк¬
туры и состава. Поэтому неразрушающий метод профилирования путем ва¬
риации угла становится в этом случае особенно важным. Простой, но
выразительный пример приведен на рис. 9.1 <>9.4. Спектральная информация9.4.1. Эффективная толщина анализа образца
(информационная глубина)Толщина образца, с которой поступает информация, определяется сред¬
ней длиной неупругого рассеяния (ДНР) электронов в твердом теле (Л).Как обсуждалось в гл. 4, 95% наблюдаемого фотоэлектронного сигнала пос¬
тупает от слоя толщиной 3Xsinoc (для плоской поверхности), где а - угол
сбора электронов, отсчитываемый от поверхности1 *. По вопросу о значени¬
ях А для полимеров существовали спорные мнения. Это связано в основном
с тем, что два различных метода определения Л давали разные результаты.
Кроме того, считалось, что можно использовать данные, полученные на
пленках Ленгмюра - Блоджетта (ЛБ) органических материалов. НедавняяВ гл. 5 а = 90 — е. где 0 — угол относительно нормали к поверхности.
4069. Применение РФЭС в технологии полимеровработа по исследованию ДНР электронов в пленках ЛБ строго доказала не¬
справедливость этого предположения [ 6]. Прекрасный обзор литературы,
касающейся ДНР электронов в полимерах, дан в работе [7]. Там сделан
вывод, что ДНР электронов в полимерах примерно такая же, как в типичных
металлах и неорганических материалах.Как обсуждается в гл. 4, возможно неразрушающее определение профиля
состава по глубине. Для этого регистрируются изменения относительной
интенсивности пиков при изменении кинетической энергии фотоэлектронов
(см., например, работу Кларка [ 1], в которой для возбуждения электронов
с внутренних уровней полимеров используются линии Mg/ftx и Ti Ка ) или из¬
менения относительной интенсивности пиков фотоэлектронов с двух внутрен¬
них уровней, значительно различающихся по энергии, например О Is/О 2s
или F ls/F 2s. Последний способ особенно полезен при определении толщи¬
ны модифицированных слоев, содержащих кислород или фтор на подложках,
в которых эти элементы отсутствуют. Отношение пиков Is/2s минимально,
если модифицированные слои имеют толщину примерно Зл^^п а (пример¬
но равную значению для однородных материалов), и возрастает при уменьшении
толщины. Пример приводится в разд. 9.6.2.2.9.4.2. Энергии внутренних уровнейПо этому вопросу за последние 10 лет большое число работ было опуб¬
ликовано Кларком и сотр. [ 8]. Они изучали чистые полимеры и малые мо¬
дельные молекулы, проводя параллельно теоретические расчеты» Большинст¬
во этих работ упоминается в обзоре Дилкса [ 9]. На основании этих данных
можно сделать некоторые простые выводы.9.4.2.1. Энергии связи С Isа. Для углерода, связанного только с углеродом и/или с водородом, не¬
зависимо от типа гибридизации энергия связи С Is равна 285,0 эВ. Это
значение часто используется для калибровки шкалы энергий связи.б. Замещение атомами галогенов приводит к сдвигу линии С Is в сторо-
рону больших энергий связи. Можно выделить первичный эффект замещения
(на атом углерода, непосредственно связанный с галогеном) и вторичный
эффект замещения (на соседний или соседние атомы углерода). Соответ¬
ствующие химические сдвиги в расчете на один атом галогена приведеныв табл. 9.1.в. Замещение атомом кислорода приводит к сдвигу в сторону более вы¬
соких энергий связи примерно на 1,5 эВ в расчете на одну связь С - О
(так, группы О-С-О и > С=*0 имеют близкие значения энергий связи
С 1 s и т. д.) Вторичный сдвиг, обусловленный X в случае С — О - X, мал
(± 0,4 эВ), кроме случая Х= N02 (нитратные сложные эфиры), когда соз¬
дается дополнительный сдвиг на 0,9 эВ.
Д. Бриггс407Таблица 9.1.ГалогенПервичный химический
сдвиг, эВВторичный химический
сдвиг, эВF2,90,7С11,50,3Вг1,0<0,2г. Первичный эффект замещения азотсодержащими функциональными
группами заметно зависит от свойств конкретной группы. Так, химичес¬
кие сдвиги линии С Is для групп -N(CH3)2, -NH2, -NCO и -N02 равны
0,2; 0,6; 1,8 и 1,8 эВ соответственно. Сдвиги для обоих атомов углерода
в -CH2-CssN составляют примерно 1,4 эВ. Сдвиг линии С Is, вызван¬
ный группой — 0N02, составляет около 2 эВ.9.4.2.2. Энергии связи OlsЭнергия связи О Is в большинстве функциональных групп изменяется
в узком диапазоне порядка 2 эВ около энергии 533 эВ. Предельные значе¬
ния наблюдаются в карбоксильной и карбонатной группах. В этих группах
наибольшее значение энергии связи Ols имеет кислород, связанный с од¬
ним соседним атомом.9.4.2.3. Энергии связи N IsДля многих обычных азотсодержащих функциональных групп энергия
связи N Is находится в узком диапазоне 399 - 401 эВ. К ним относятся
-CN, -NH2, -OCONH и -CONH2. При образовании четвертичных соеди¬
нений азота (например, -NHg) происходит увеличение энергии связи N Is
примерно на 1,5 эВ по сравнению с энергией связи для свободного амина
в основном благодаря воздействию контриона.Окисленные функциональные группы азота имеют значительно большие
энергии связи N Is: - 0N02 ( « 400 эВ), -N02 (*407эВ)и -0140(»405эв).9.4.2.4. Внутренние уровни других атомовСреди атомов, часто присутствующих в полимерах, наряду с упомянуты¬
ми выше только сера и кремний могут иметь различную степень окисления.
Первичное влияние атома серы на энергию связи С Is очень мало ( * 0,4 эВ
в полисульфоне). Энергии связи для уровня S 2р, однако, заметно варьиру¬
ют: R-^S-Д (ш 164 эВ), R-SO2-R ( « 167,5 эВ), R -S08H ( * 169 эВ).Было опубликовано несколько сообщений об энергиях связи Si 2р. Для
типичных кремнийорганических полимеров, имеющих структуру полисилокса-
на -OSiR20-, энергия связи Si 2р порядка 102 эВ.
4089. Применение РФЭС в технологии полимеровСледует также заметить, что ионы галогенов можно отличить от ковалент¬
но связанных галогенов. В случае фтора энергия связи F 1 s для отрицательно¬
го иона F “ примерно на 4 эВ меньше, чем для С-F (типичное значение 689 эВ),
а энергия связи С1 2р для С1 - примерно на 2 эВ меньше, чем для С-С1 (типич¬
ное значение 201 эВ).9.4.3. Сателлиты электронной "встряски”(shake-цэ)Как отмечалось в гл. 3, наличие сателлитов "встряски” ожидается
у всех полимерных систем, имеющих ненасыщенные связи. Однако они бы¬
ли детально исследованы только у углеводородных полимеров с аромати¬
ческими группами, в первую очередь у замещенных полистиролов. Эти ис¬
следования показывают, что сателлиты "встряски" обычно располага¬
ются на 6-7 эВ ниже по шкале кинетической энергии по отношению к ос¬
новному пику С Is, имеют асимметричную форму, иногда проявляются
в виде отчетливого дублета и составляют до 8% от интенсивности основ¬
ного пика. Такая структура однозначно соответствует двум переходам с
возбуждением электрона, находящегося на одной из двух заполненных
орбиталей Ъ t ^ и а 2 на незаполненную орбиталь Ь J* (т.е. тт -* тг*) [ 9].
Следовательно, наличие этих сателлитов является указанием на присут¬
ствие ароматических групп в исследуемом материале. В работе [ 10] по
пикам сателлитов наблюдали за разрушением поверхности полистирола
при анализе ее методом ВИМС (рис. 9.2).Рис. 9.2. Сателлит ’’встряски", связанный с электронным тт -*тт*-переходом, в
спектре C1s полистирола (а); полное исчезновение сателлита "встря-
ски” после бомбардировки ионами аргона (энергия 4 кэВ, доза 1,6'1014ион/см2)
(б). Обратите внимание на уширение основного С1 s-пика [Ю].
Д. Бриггс409Были предприняты попытки использовать тт -»тт*-сателлиты "встря¬
ски" для определения структуры поверхности в тех случаях, когда регист¬
рация пиков фотоэлектронов только с внутренних оболочек не подходила
для этой цели. Примером является работа Кларка и Дилкса [11] по изуче¬
нию сополимеров алканов и стирола. В таком подходе требуется, чтобы
отношение интенсивности сателлитов и первичного пика не зависело от
изменений матрицы (а именно, изменений состава сополимера, степени
кристаллизации и т» д.). На то, что это требование может не выполняться,
указывалось в работе [ 12], посвященной изучению блок-сополимеров поли¬
стирола и окиси этилена.Наличие сателлитов "встряски", связанных с ненасыщенной связью
винильной группы и сдвинутых примерно на 7 эВ от первичного пика С Is,
использовалось в работе [ 13] для определения структуры полигексафтор-
бутина-2 и в работе [ 14] для выяснения причин расхождения стехиометри-
ческого соотношения F/С в полимере, образующемся при полимеризации
в плазме фторэтиленов [ 14]. Некоторую ценность с аналитической точки
зрения может представлять тот факт, что атомы, непосредственно связан¬
ные с ненасыщенными центрами, также будут иметь в спектре фотоэлект¬
ронов сателлиты электронной "встряски".9.4.4. Фотоэлектронные спектры валентных оболочекВ гл. 3 уже говорилось о возможности различать близкие по составу
полимеры по фотоэлектронным спектрам валентных оболочек. Пример при¬
веден на рис. 9„3. Видно четкое различие спектров различных углеводород¬
ных полимерово В целом данный вопрос внимательно исследовался как с
экспериментальной, так и с теоретической точки зрения Пиро и сотр. [ 15].
Эти исследователи опубликовали спектры высокого разрешения* получен¬
ные с помощью рентгеновского монохроматора, для полимеров, содержащих
кислород, фтор и хлор, и углеводородных полимеров. Они также продемон¬
стрировали чувствительность спектров валентных оболочек к различным
типам изомеризации (структурной, конформационной, стерео), а также к
наличию регулярности в молекулярной структуре и геометрической конфор¬
мации.9.4.5. Образование производных соединений для пометки
функциональных группКак отмечалось в п. 9.4.2.1, энергии связи для внутренних уровней не¬
достаточно специфичны, многие функциональные группы характеризуются
близкими величинами химических сдвигов. Эта проблема становится осо¬
бенно острой в тех случаях, когда на поверхности имеется несколько таких
групп. Отдельные пики при этом перекрываются и происходит уширение
спектра вследствие вторичного эффекта замещения. Некоторые исследовате-
4109. Применение РФЭС в технологии полимеровРис. 9.3. Спектры валентных оболочек углеводородных полимеров: а - полиэтилена
высокого давления (низкой плотности); б - полипропилена и в - полибуте-
на-1. Эти полимеры имеют идентичные C1s-спектры.ли пытались обойти эти трудности, используя селективные реагенты для
образования производных функциональных групп. Реагируя только с одной
функциональной группой, конкретный селективный реагент помечает ее
атомом элемента, легко идентифицируемым в РФЭ-спектрах. Этот метод
может иметь дополнительное преимущество: в том случае, если у нового
элемента сечение поглощения рентгеновского излучения больше, чем,нап¬
ример, у углерода, азота или кислорода, то его введение вызовет увеличе¬
ние чувствительности детектирования. В табл. 9*2 перечислены исследован¬
ные к настоящему времени реакции.Для знакомства с условиями реакций и оценками возможности их ус¬
пешного использования читателю рекомендуется обратиться к оригиналь¬
ной литературе. Кроме селективности, реакции образования производных
должны отвечать еще двум требованиям: во-первых, они должны быстро
протекать в мягких условиях, и, во-вторых, любой необходимый раствори¬
тель должен быть безопасен для полимера. Трудно найти реакцию, удовлет¬
воряющую этим двум последним условиям. Реакции, быстро протекающие
при комнатной температуре в растворе, часто стерически запрещены в по¬
верхностных слоях полимеров. Растворители, проникающие в полимер, по
всей вероятности, ускоряют реакцию, но могут при этом привести к пере¬
стройке поверхности, например к миграции функциональных групп в объем.
Эти аспекты образования производных изучались в некоторой степени
Эверхартом и Рейли [27, 28].
Д. БриггсТаблица 9.2. Реакции образования производных соединений411ФункциональнаягруппаРеагентПродуктЛитература9.5. Сравнение РФЭС с другими методами [29]До появления РФЭС практически единственным методом, пригодным
для анализа поверхности полимеров, был метод отражательной инфракрас¬
ной спектроскопии [ либо с нарушенным полным внутренним отражением
(НПВО), либо спектроскопии многократного внутреннего отражения]. При
его использовании необходимы достаточно большие образцы с плоской
либо легко деформируемой поверхностью. Получаемая информация харак¬
теризует слой материала толщиной около 1 мкм. За последние годы удалось
улучшить поверхностную чувствительность и уменьшить требуемые разме¬
ры образца благодаря использованию методов инфракрасной фурье-спектро-
скопии, но даже в наиболее благоприятных случаях инфракрасная фурье-
спектроскопия не может конкурировать по чувствительности с РФЭС. Одна¬
ко при анализе систем, имеющих переменный состав на глубине 0-1 мкм,
эти два метода дополняют друг друга и могут эффективно использовать¬
ся совместно (см., например, работу [30]).
4129. Применение РФЭС в технологии полимеровВозможность применения ЭОС в этой области анализа материалов обыч¬
но не принималась во внимание на основании того, что при электронном
облучении скорости разрушения образца неприемлемо велики. Однако фак¬
тически этот вопрос нельзя было изучать из-за сильных эффектов заряд¬
ки образца, препятствующих получению спектров. Недавно было показано
[31], что можно проводить анализ малых областей образца, если сначала
покрыть его пленкой золота определенной толщины и затем удалить ее с
помощью ионного травления. В этом методе можно получить СЭМ-изображе-
ние образца in situ перед распылением и исследовать элементный состав
поверхности различных структур на этом изображении. По-видимому, в
результате такой процедуры происходит графитизация поверхности, поэто¬
му данный метод имеет очевидные ограничения. Его применение, однако,
возможно в важной области анализа полимеров - анализе неорганических
частиц в полимерной матрице. И конечно, данный метод в принципе имеет
важное преимущество перед РФЭС - с его помощью можно получить изо¬
бражение с высоким пространственным разрешением. Аналогичная зада¬
ча ставилась в работе Бриггса по изучению применимости ВИМС для ана¬
лиза поверхности полимеров.Результаты систематических исследований [10] практических проблем
позволяют надеяться, что будет возможно получить пространственное раз¬
решение «100 мкм» Недавно было продемонстрировано [32] получение кар¬
тины распределения молекул на поверхности с таким разрешением. Масс-
спектроскопия с бомбардировкой быстрыми атомами [МСББА], в которой
вместо пучка ионов (в методе ВИМС) используется нейтральный пучок атомов,
дает эквивалентные результаты без необходимости компенсации заряда
[ 33]о Она, однако, не может использоваться для работы с большим простран¬
ственным разрешением. Дальнейшие исследования [ 34], проведенные к нас¬
тоящему времени, показывают, что ВИМС и МСББА позволяют различать мо¬
лекулярный состав поверхности и потенциально являются более чувствитель¬
ными, чем РФЭС. С точки зрения предельной поверхностной чувствительнос¬
ти перспективным является метод СНРИ (спектроскопия низкоэнергетич-
ных рассеянных ионов)» Так же как и ВИМС, этот метод только начинает
применяться для изучения поверхностей полимеров.Другой вид колебательной спектроскопии, имеющей большие возмож¬
ности в этой области анализа, - это спектроскопия комбинационного рас¬
сеяния (КР). Были получены спектры КР тонких полимерных пленок. С по¬
мощью КР-микроанализатора возможно анализировать полимерные материа¬
лы с высоким пространственным разрешением (* 1 мкм) при зондировании
первых нескольких микронов образца. Значительное преимущество мето¬
дов колебательной спектроскопии, описанных в этом разделе, - это возмож¬
ность давать информацию о строении полимеров (например, о конформации
цепи, ориентации и кристаллическом состоянии). Это очень важный аспект
структуры полимеров. Другие методы этой информации не дают.Несмотря на недостаточно высокое пространственное разрешение (мо¬
жет быть, оно не всегда будет таким), РФЭС является чрезвычайно важным
Д. Бриггс413методом анализа поверхности полимеров. Это несомненно, поскольку дан¬
ный метод без особых проблем позволяет получать количественную инфор¬
мацию об элементном составе и значительную долю структурной информа¬
ции, и при его использовании не возникает неразрешимых проблем, связан¬
ных с зарядкой образца или его радиационным повреждением. Кроме того,
на форму образца не накладывается никаких ограничений, поэтому можно
работать непосредственно с полученными материалами без дополнительной
подготовки (естественно, имеются определенные ограничения на габарит¬
ные размеры образца).9.6. Применение РФЭС в задачах анализаповерхности полимеров9.0.1. Некоторые общие положенияСпособность детектировать малые количества материала на поверх¬
ностях полимеров просто благодаря появлению в спектре фотоэлектронов,
соответствующих характеристическим внутренним уровням, может пока¬
заться тривиальной» Распространенные полимеры состоят из небольшого
числа элементов; более того, они имеют простые спектры рентгеновских
фотоэлектронов (обычно линия С Is и одна или две линии из ряда О Is,N Is, FIs и Cl 2s, 2p). Однако обычные добавки или загрязнения содер¬
жат дополнительные элементы, такие, KaKS, Р, Si , Al, Na, К, Вг, Sn, Cr,
Ni, Ti , Zn, Ca, Sb, Ge, и поэтому можно очень легко обнаружить эти
соединения. Способность детектировать эти элементы, особенно на непо¬
средственно полученных образцах малого размера или нерегулярной формы,
представляет особую ценность для контроля качества и изучения причин
неисправностей, связанных со свойствами поверхности. Сюда относятся
оптические свойства (например, наличие помутнений, глянца, пятен), адге¬
зионные свойства (такие, как смачиваемость, пригодность для печатания,
возможность склеивания, термосвариваемость, возможность выпрессовки
из форм), электрические свойства (например, возможность статической за¬
рядки), общие технологические свойства (такие, как коэффициент трения и
слипание пленок в рулонах)» Некоторые типы загрязнений могут также при¬
водить к образованию складок на поверхности и растрескиванию многих
пластиков»Добавки в пластиках и полимерных материалах служат для предотвра¬
щения окисления (теплового и фотохимического), нейтрализации кислотных
свойств, обеспечения огнеупорности, улучшения обрабатываемости.Обычно предполагается достаточно однородное распределение добавок
в объеме материала, но при определенных обстоятельствах может происхо¬
дить сегрегация у поверхности. В некоторых случаях предполагается миг¬
рация добавок к поверхности. Это требуется при введении средств для по¬
нижения трения, для повышения поверхностной проводимости и для предот¬
вращения слипания поверхностей. В одних случаях миграция может
не происходить, в других возможна миграция через границу контакта
4149. Применение РФЭС в технологии полимеровРис. 9.4. Обзорные спектры поверхностей образцов изделий, формованных из поли¬
этилена высокого давления: а - образец обнаруживает недопустимое рас¬
трескивание поверхности, б-нормальный образец. Поверхность растрес¬
кавшегося образца была загрязнена соединениями кремния, по-видимому,
от смазок, облегчающих выемку изделий из форм. Силиконовый компаунд
вызывает коррозию полиэтилена высокого давления под напряжением.с другой поверхностью. Частицы наполнителей и пигментов часто имеют
покрытие, материал которого может переходить в полимерную матрицу и
оттуда к поверхности.Часто наблюдается поверхностная сегрегация молекул эмульгаторов и
стабилизаторов. Обычные загрязняющие примеси - это остатки катализато¬
ра полимеризации, смазочные масла и консистентные смазки, присадки, об¬
легчающие выемку изделий из форм, пыль. Типичные примеры возможностей
простого детектирования элементов показаны на рис. 9.4 - 9.6.Более сложным является анализ тонких слоев материалов, не содержа¬
щих отличительных элементов. Например, на рис. 9.7 показаны спектры уров¬
ней С Is и О Is, полученные с двух сторон пленки полиэтилентерефталата,
химически модифицированной с одной стороны для улучшения адгезионных
свойств. Толщина модифицированного слоя слишком мала для анализа мето¬
дом отражательной ИК-спектроскопии. Так как после модификации в спектре
внутренних оболочек не происходит появления новых пиков, для наблюдения
Д. Бриггс415Рис. 9.5. Обзорные спектры поверхностей попипропиленовых пленок, применяемых
в качестве упаковочного материала. Пленки покрыты с обеих сторон сопо¬
лимером вин ил иденди хлорида (который уменьшает проницаемость для кис¬
лорода и воды) и на одной стороне отпечатан текст. Поверхность обратной
стороны используется для сварки с аналогичной поверхностью в операциях
упаковки пленочной тары. Оба приведенных спектра получены для этой по¬
верхности пленок.в - образец, поверхность которого практически потеряла способность
свариваться в стандартных условиях, б - нормальный образец. Потеря спо¬
собности свариваться звязана с присутствием на поверхности комплекса
титана, который входит в состав чернил для улучшения их адгезии с по¬
верхностью. Данный комплекс мигрирует из чернил на "чистую" поверх¬
ность во время контакта поверхностей при хранении пленок в рулонах. За¬
метное уменьшение интенсивности пика С2р и исчезновение пика C1LMM
(которому соответствуют фотоэлектроны существенно меньшей энергии)
свидетельствуют о значительной степени загрязнения "чистой" поверхнос¬
ти.различий в спектрах двух сторон необходимо регистрировать спектры высоко¬
го разрешения. Зная соответствующие факторы относительной чувствительнос¬
ти для детектируемых элементов, можно определить стехиометрические соот¬
ношения. Немодифицированная сторона пленки полиэтилентерефталата лег-
4169. Применение РФЭС в технологии полимеровРис. 9.6. Обзорные спектры поверхностей пленок из полиэтилентерефталата, исполь¬
зующихся в качестве упаковочного материала. На одну сторону пленки был
нанесен в вакууме тонкий слой алюминия для уменьшения газовой проница¬
емости и с декоративной целью. Оба спектра получены для неметаллизиро-
ванной стороны.а - образец с пониженной способностью образования сварных соедине¬
ний и малопригодный для печатания; б - нормальный образец. Отклонения
от нормы в данном случае являются результатом улучшения адгезии между
двумя поверхностями пленки (оксидом металла и полиэтилентерефталатом)
при хранении ее в рулонах (механизм не ясен). При последующем разматы¬
вании часть алюминийсодержащего материала остается на неметаллизиро-
ванной стороне, загрязняя поверхность полиэтилентерефталата.ко идентифицируется с помощью сателлитов электронной "встряски”
(см. разд. 9.4.3).Примеры подобного анализа методом РФЭС полимеров, имеющих важные
значения в биомедицинских приложениях, приводятся, среди прочих авторов,
Ратнером и сотр. Так, обнаружено, что состав поверхности материалов на
основе полиуретана (последние часто используются для прессования трубок)
заметно изменяется. Это обусловлено миграцией олигомерных фракций, име¬
ющих повышенное содержание одной из компонент сополимера, и концентра-
Д. Бриггс417Рис. 9.7. Спектры внутренних уровней двух сторон пленки полиэтилентерефталата. Сто¬
рона 1 модифицирована для улучшения адгезии. Немодифицированная сторо¬
на 2 имеет в спектре отчетливый пик тт тт*-"встряски".цией загрязняющих примесей у поверхности [35]. Было показано также, что
состав модельных поверхностей, образующихся при блок-сополимеризаЦии
из раствора очищенных полимеров, зависит от того, была ли поверхность
в контакте с подложкой (например, с чистой поверхностью стекла) или с
воздухом. Обнаружена корреляция состава поверхности, полученного с по¬
мощью количественного РФЭС-анализа, и ее свойств, например, в работе
[ 35] при изучении метаболизма тромбоцитов крови. Другим примером при¬
менения РФЭС в этой области анализа материалов является работа [ 36].В ней было показано, что состав поверхности гидрогелей, полученных ме¬
тодом радиационной графт-полимеризации, коренным образом изменяется
при изменении степени гидратации (конечно, эти изменения могут быть свя¬
заны с тем, что в установке поддерживается высокий вакуум; имеется опи¬
сание специальных методов охлаждения образца для разрешения этих проб¬
лем). Рис. 9.8 иллюстрирует этот эффект.В данной главе не ставится задача дать полный перечень применений
РФЭС в анализе поверхности полимеров. Это ранее было сделано автором в
обзоре [ 37], охватывающем литературу вплоть до 1977 г. (включая важную
область анализа волокон и тканей). Примеры, приведенные в этом обзоре,
пока достаточно полно представляют данную область. Список работ, выпол¬
ненных к настоящему времени, приводится в библиографии в конце главы.
Вместо обзора всех выполненных работ для наилучшей демонстрации воз¬
можностей и ограничений РФЭС в данной главе будет детально обсуждаться
одна определенная (и значительная) область анализа поверхности полиме¬
ров - анализ модификаций поверхности полимеров.
4189. Применение РФЭС в технологии полимеровРис. 9.8. С ls-спектр акриламида (гидрогель), привитого с помощью радиационной
полимеризации на силиконовой резине: а) 160 К (гидратированный); б)303 К (обезвоженный). Обезвоживание приводит к исчезновению сигнала
N1s (от акриламида) и существенному росту сигнала Si 2р (от силиконовой
подложки). Эго данные указывают на практически полную миграцию гидро¬
геля в подложку при обезвоживании [Зб].9.6.2. Модификация поверхности полимеров
9.6.2.1. Фторированные системыНа ранних стадиях применения РФЭС для структурного анализа полиме¬
ров исследовались преимущественно фторированные системы по той простой
причине, что фтор как наиболее электроотрицательный элемент вызывает
наибольший химический сдвиг в энергии связи С Is, Следовательно, наи¬
более просто идентифицировать различные структурные единицы по спект¬
рам С Is в сильно фторированных материалах. По этой же причине иссле¬
дования модификации поверхности полимеров первоначально касались воп¬
росов обработки политетрафторэтилена.
Рис. 9.9. Линии углерода, кислорода и фтора и краевые углы тефлона FEP(сополиме¬
ра перфторпропилена и тетрафторэтилена) до и после обработки поверхнос¬
ти разными методами [39].
4209. Применение РФЭС в технологии полимеровСклеивание политетрафторэтилена возможно после обработки несколь¬
кими различными способами. Наиболее часто для обработки пользуются
раствором натрия в жидком аммиаке и системой натрий - нафталин - тетра-
гидрофуран. Независимые исследования обоих способов, проведенные Брех¬
том, Мейером и Биндером [ 38], и первого способа, проведенные Дуайтом и
Риггсом [ 39], позволили заключить, что при обоих способах обработки про¬
исходит дефторирование поверхности и образование сильно ненасыщенно¬
го углеродистого слоя, который затем при взаимодействии с воздухом реа¬
гирует с кислородом и водой. Уже на этой ранней стадии исследований
стало понятно, что метод РФЭС не позволяет сделать каких-то выводов о
строении групп, содержащих только углерод (или углерод, связанный с во¬
дородом)* Поэтому обе группы исследователей использовали реакцию с Вг.
для получения количественной информации об уровнях ненасыщенности.
(Сейчас известно, что поверхностные группы, содержащие кислород, услож¬
няют ее интерпретацию.) Дуайт и Риггс [ 39] исследовали также дальней¬
шее поведение модифицированной поверхности - ее снашивание, разруше¬
ние в атмосферных условиях и т. д. (рис. 9.9). По-видимому, это было
также первым исследованием соотношения углов смачивания и состава
поверхности, в котором анализ поверхности проводился с помощью РФЭС.
Коллинс, Лоу и Николас [ 40] в это же время опубликовали результаты
исследований, посвященных обработке поверхности политетрафторэтиле¬
на тлеющим разрядом в аммиаке и менее детально в воздухе. Сигнал С Is,
полученный с поверхности при равновесии (разряд в аммиаке), состоял из
пяти компонент. Хотя его не удалось полностью интерпретировать, оче¬
видно было, что происходит дефторирование поверхности и присоединение
кислород-и азотсодержащих групп. Улучшение смачиваемости и адгези¬
онных свойств этой поверхности можно было вновь устранить обработкой
в смеси азотной и хлорной кислот. РФЭС показала, что это происходит бла¬
годаря удалению модифицированного слоя с немодифицированного поли¬
тетрафторэтилена.Изучение простых фторполимеров привело к лучшему пониманию и поз¬
волило перейти к более сложным системам. Так, через два года после упо¬
мянутых выше исследований Кларк и др. [41] занялись идентификацией сое¬
динений, образующихся при фторировании поверхности полиэтилена высокой
плотности (низкого давления). Рис. 9.10 иллюстрирует сложность формы ли¬
нии С Is в той степени, в которой верно разложение кривой при столь де¬
тальной идентификации. В этой работе была впервые сделана серьезная
попытка получить количественную оценку распределения модифицированных
групп по структуре и по глубине. С этой целью регистрировалось отношение
интенсивностей пиков F Is и F 2$, в частности, в зависимости от времени
реакции.Преимущества работы с фторированными системами также использова¬
лись Кларком и Дилксом [ 42] в модельном исследовании процесса сшива-
Д. Бриггс421Рис. 9.10. Линии С1 s и F1 s РФЭ-спектра поверхности полиэтилена низкого давления
(высокой плотности) после фторирования в F2/N2 [41].ния полимеров (образования связей между цепями) при воздействии на обра¬
зец возбужденными атомами инертных газов. В качестве модельного поли¬
мера использовался нерегулярный сополимер этилена и тетрафторэтилена
(52%). Данный сополимер имеет простой С Is-спектр, состоящий только из
двух пиков, которые соответствуют группам — СН2— и — CF2-k Для сшива¬
ния приповерхностных слоев полимера использовалась аргоновая плазма.За ходом процесса следили по удалению фтора с поверхности и образованию
групп CF. Регистрация отношения интенсивностей пиков F Is и F 2s, ис¬
пользование метода варьирования угла и количественной оценки интенсив¬
ности пиков позволили описать кинетику процесса на основе механизмов
прямой и радиационной передачи энергии соответственно на поверхность
полимера и в приповерхностный слой.Позднее Дилкс [9, 14] использовал РФЭС для изучения синтеза полиме¬
ров в тлеющем разряде (полимеризация в плазме). И вновь использовались
сильно фторированные полимеры. Задачей этой работы было изучение соот¬
ношения между структурой мономера, добавляемого в разряд, составом об¬
разующегося полимера (который осаждался в виде тонкой пленки) и пара¬
метрами, характеризующими разряд и геометрию системы. Можно было
также прояснить кинетику полимеризации. На рис» 9.11 приводятся типич¬
ные С Is-спектры и их разложение на отдельные пики, необходимое для ко¬
личественной интерпретации. В работе Милларда [43] обсуждается иссле¬
дование с помощью РФЭС полимеризации в плазме на поверхности шерстя-
4229. Применение РФЭС в технологии полимеровРис. 9.11. Линии F1 s и С1 s поверхностей полимеров, полученных полимеризацией
в плазме фтористого винила (а), 1,1-дифторэтилена (6) и трифторэтилена
(в) [14].ных волокон и обработки плазмой тканей с целью модификации свойств
поверхностей.9.6.2.2. ПолиопефиныХотя фторполимеры очень привлекательны для модельных иссле¬
дований методом РФЭС, они составляют лишь очень малую долю всего
спектра полимеров как по объему годового производства, так и по диапа¬
зону применения. Основным классом полимеров являются полиолефины (поли¬
этилен низкого и высокого давления, полипропилен и широкий диапазон
сополимеров, включающих этилен, пропилен и более высокие а-олефины).
Модификация поверхности этих материалов важна для их широкого и ус¬
пешного применения. Отчасти это связано с тем, что неполярные поверх¬
ности этих материалов имеют низкую поверхностную энергию и изначаль¬
но плохую смачиваемость и адгезионные характеристики. Причины этого
были предметом многочисленных споров в течение многих лет [ 44]»
Исследования с помощью РФЭС многое прояснили в этом вопросе.С 1976 г. Бриггс и его коллеги проводили систематическое изучение ком-
Таблица 9.3. Состав поверхности и адгезионные свойства полимеров после травления [44]ПолимерУсловия травленияНе проводилось
Конц. H2S04
1 ч при 70 °С
Смесь 8®5 с при 20 °С
Смесь А*1 мин при 20 °С
Смесь А30 мин при 70 °С
Смесь А6 ч при 70 °С
Не проводилосьКонц. H2S04
1 ч при 70 °С
Смесь В
5 с при 20 °С
Не проводилось
Конц. H2S04
1 ч при 70 °С
Смесь В5 с при 20 °С
Смесь А1 мин при 20 °С
Смесь А6 ч при 70 °С36,819826980,047,164,3145262382283261ОтношениеатомныхконцентрацийC/SОтношениеатомныхконцентрацийO/S3.29Д12,710.4
9,54.29.29,714.5
16,213.5Атомы Сс группамиSO-H, %
з ’ <Отношение
количества О
(не в группах
503Н) к полному
количеству С,
%0/СНапряжениесдвига,МН/м2Тип разру¬
шения6О Is/O 2sB11,613.0
9,2
9,918,225.135,922.713.70,553,34.87.57.69.5
0,383.57.00,281.12.84.7
11,20,250,63,13.69.3
13,90,521,84.30,252.63.3
4,6
4,02,70,50,41,32,11,60,70,40,30,40,4ПЭВД — полиэтилен высокого давления (низкой плотности), ПЭНД — полиэтилен низкого давления (высокой плотности); ПП — полипропилен
а Смесь А состоит из бихромата калия, воды и серной кислоты (весовые соотношения К2Cr2О^: Н 2 О: Н 2SO^ = 7:12:150) Смесь В состоит из тех же
компонент, концентрация К2Сг207 в 100 раз меньше.I — разрушение по границе раздела; М — разрушение материала образца.
в Для однородных образцов 01s/02s - 10 [30].
4249. Применение РФЭС в технологии полимеровмерческих способов обработки и других процессов, приводящих к улучшению
адгезионных характеристик, таких, как травление хромовой кислотой [30,45],
плавление на алюминии [ 17], тепловое окисление [46], обработка озоном
[46], пламенем [47] и электрическим (коронным) разрядом [21, 22, 48, 49].
Два из этих исследований будут ниже обсуждаться детальнее, так как они
демонстрируют некоторые важные аспекты применения РФЭС.Травление хромовой кислотой (KgCrgOT/HgO/HaSOj поверхностей
полиолефинов приводит к появлению значительной шероховатости. Краевой
угол (угол смачивания) при контакте поверхности с водой в случае полиэти¬
лена низкого и высокого давления по мере протекания процесса травления
быстро падает. В случае полипропилена краевой угол за короткое время
достигает минимума, затем начинает возрастать и выходит на плато при
значении, близком к величине краевого угла для контакта с водой необ¬
работанной поверхности. Отражательная ИК-спектроскопия этих поверхнос¬
тей указывает на химическую модификацию (окисление и сульфирование)
только поверхности полиэтилена низкой плотности. На основании этого мож¬
но предположить, что увеличение адгезии связано с ростом шероховатостей
или просто с удалением "слабого” (граничного) поверхностного слоя.В табл. 9.3 приводятся некоторые данные рентгеновских фотоэмиссионных
спектров, полученные с поверхностей этих полимеров после обработки раст¬
ворами хромовой кислоты и для сравнения после обработки концентрирован¬
ной серной кислотой [30, 44]. Величина энергии связи для пика S 2р, ко¬
торый появляется в спектре после обработки, говорит о присутствии групп
-S03H. Количественные оценки, приведенные в таблице, сделаны в пред¬
положении, что вся сера на поверхности находится в такой форме. Данные
по адгезии (напряжение сдвига соединений, склеенных эпоксидным клеем)
четко коррелируют со степенью окисления (которая в предположении посто¬
янства среднего стехиометрического соотношения определяется количест¬
вом углерод-кислородных функциональных групп), а не концентрацией групп
-S03H. Для образцов с однородным распределением кислорода отношение
пиков О Is и О 2s (в данных экспериментальных условиях) равно пример¬
но Ю. Следуя обсуждению в разд. 9.4.1, на основании данных для отноше¬
ния пиков Ols и О 2s можно сделать следующие выводы: после аналогич¬
ной обработки толщина окисленного слоя на полипропилене значительно
меньше, чем на полиэтилене высокого давления (и для полипропилена так¬
же меньше глубины зондирования уровня О 2s, т. е. < 100 А); степень
окисления поверхности полипропилена быстро достигает равновесного зна¬
чения. Эти данные показывают, что при обработке полипропилена хромовой
кислотой происходит образование очень тонкого равновесного слоя окис¬
ленного полимера при непрерывном растворении материала поверхности,
в то время как в случае полиэтилена высокого давления степень и глубина
окисления непрерывно растут с увеличением времени экспозиции. Это сог¬
ласуется с измерениями потери веса образцов при обработке. Данные для
Д. Бриггс425отношения интенсивностей пиков О Is и С 2s в зависимости от угла при
малой степени обработки (когда шероховатости еще не развились) также
подтверждают такую интерпретацию. Увеличение шероховатости поверхнос¬
ти не сказывается на интерпретации РФЭ-спектров, но, как отмечалось, за¬
метно влияет на результаты измерений краевого угла. Данные, полученные
методом РФЭС, показывают на важность не принимавшегося ранее во вни¬
мание окисления поверхности и на неадекватность обычно используемого
метода отражательной ИК-спектроскопии для изучения модификации по¬
верхности при глубине модифицированного слоя менее 100 А.Для обработки полипропиленовых формованных изделий перед электро¬
литическим нанесением металлических покрытий (например, для декоратив¬
ной отделки или изготовления слоистых структур полимер - металл) в ка¬
честве травителя преимущественно используется Сг03 -Н20. Было пока¬
зано, что остатки хрома играют важную роль для получения оптимальной
адгезии металла к полимерной поверхности. На рис» 9.12 показаны результа¬
ты анализа методом РФЭС полиэтилена высокого давления, обработанного
этим реагентом [ 30]. Обратите внимание на наличие острого максимума
на зависимости поверхностной концентрации хрома от времени травления
и на сильную корреляцию степени окисления с адгезией поверхности к эпок
сидному клею (подобно ранее обсуждавшейся корреляции, наблюдаемой при
обработке смесью хромовой и серной кислот). Было найдено, что энергия
связи уровня Сг 2р и величина спин-орбитального расщепления уровнейРис. 9.12. Травление полиэтилена высокого давления в Сг03 - 1^о при 25°С в за¬
висимости от времени. 1 - интенсивность сигнала 01 s (Ю4 имп./с на
деление); 2 - интенсивность сигнала Сг2р3/2 (103 имп./с на деление); 3 -
прочность на сдвиг клеевого соединения (на эпоксидном клее) [зо].
4269. Применение РФЭС в технологии полимеров2^1/2 и 2р3/2 свидетельствуют о том, что хром имеет степень окисления
+ 3. Линия углерода С Is спектра высокого разрешения показана на рис. 9.13.
Поведение левого крыла линии (соответствующего большим энергиям связи)
показывает, что в начале процесса травления преобладают карбонильные
группы, затем на более поздних стадиях окисления доминирующими стано¬
вятся карбоксильные группы (это подтверждает отражательная ИК-спектро-
скопия). Момент перехода и время достижения максимальной концентрации
хрома совпадают. Данные РФЭС согласуются с вероятным механизмом
окисления, включающим образование промежуточных соединений сложного
хромового эфира» Три основных места окислительного воздействия в поли¬
этилене высокого давления — это двойные связи, третичные углеродные ато¬
мы (точки ветвления) и метиленовые группы. На основании данных, получен¬
ных с помощью спектроскопии многократного внутреннего отражения, мож¬
но сделать вывод, что Центры первого типа относительно инертны. Извест¬
ные механизмы реакций в Центрах других двух типов выглядят следующим
образом:Конечное стабильное состояние хрома есть Сгш. На поверхности он может
входить в состав комплекса, такого, какДальнейшее окисление должно приводить к разрыву полимерных цепей и,
возможно, к образованию карбоновых кислот, однако механизмы этих реак¬
ций поняты недостаточно хорошо.Расчеты показывают, что, когда концентрация хрома на поверхности
достигает максимума, на тысячу атомов углерода приходится примерно 3
атома хрома. Полиэтилены высокого давления обычно содержат около 20—30
точек ветвления на тысячу атомов углерода, однако они распределены между
аморфной и менее доступной кристаллической фазами. Поэтому небезоснова¬
тельно предполагать, что хром в результате реакции с третичным атомом уг¬
лерода связывается на поверхности в составе своего трехвалентного комплек¬
са. Дальнейшее окисление этого центра приведет к образованию карбоновой
кислоты с отрывом хрома. Эта гипотеза, следовательно, согласуется с экспе¬
риментальными наблюдениями. Более того, кислотный гидролиз, необходимый
Д. Бриггс427для образования свободной карбоновой кислоты (хром по-прежнему связан с
поверхностью подвергающегося травлению полиэтилена высокого давления),
описывается реакциейЧтобы объяснить влияние хрома, связанного на поверхности, на адге¬
зию металлов, была предложена пробная теория [ 30]. Результаты исследо¬
ваний методом РФЭС ясно указывают на причину зависимости величины ад¬
гезии от конкретных условий обработки. Такие факторы, как температура
раствора, состав и свежесть травителя (последняя определяется тем, сколь¬
ко раз травитель был использован ранее), задают время травления, за ко¬
торое достигается максимальная (оптимальная) концентрация хрома. Это
хороший пример того, какую важную роль может играть РФЭС при контроле
качества.На ранних этапах этой работы наблюдалась некоторая невоспроизводи-
мость результатов измерения адгезии для образцов, обработанных в
С г 08 -Н20. РФЭ-спектры этих поверхностей содержали небольшие пики,
соответствующие кремнию. Измерения при различных углах сбора показали,Рис. 9.13. Линия С1 s спектра высокого разрешения поверхности полиэтилена высо¬
кого давления, протравленной в Сг 03 - н20 при 25 °С в течение различ¬
ных промежутков времени: а - О с; б- 20 с; в -40 с; г-2 мин; д -
10 мин; в-1 ч; ж- 5 ч; з-16 ч. Скорость счета составляла 3*103имл./сна деление.
4289. Применение РФЭС в технологии полимеровчто атомы, отвечающие этим пикам, расположены на внешней поверхности
полимера, так как отношение интенсивностей пиков Si 2р и С Is заметно
менялось при изменении угла сбора. Например, для образца полиэтилена высокого дав¬
ления, обработанного в течение 1 мин при 20qC, данное отношение изменя¬
лось от 7,3 • 10”3 при а = 80° до 1,6 • 10~2 при а = 15°, т.е. возросло в 2,2 ра¬
за при переходе к меньшему углу сбора (по отношению к поверхности образ¬
ца). Напротив, отношение интенсивностей пиков О Is и С Is практически
не возросло. Примеси, содержащие кремний, были удалены из воды путем
многократной дистилляции до тех пор, пока значение поверхностного натя¬
жения не достигло 72,5 мН/м. В табл. 9.4 приводятся данные о влиянии этих
примесей на величину адгезии для окисленных поверхностей.Обработка электрическим (коронным) разрядом [ 50] пленок полиэтиле¬
на и полипропилена использовалась в течение многих лет для того, чтобы
сделать эти поверхности пригодными для печати, изготовления слоистых
структур и нанесения покрытий. При такой обработке (рис. 9.14) полимерная
пленка пропускается над заземленным металлическим валком, покрытым
диэлектрическим (изолирующим) материалом. На расстоянии примерно
2 мм от пленки проходит полоса высоковольтного электрода (типичное зна¬
чение подаваемого напряжения составляет 15 кВ, частота 20 кГц). В зазо¬
ре между электродом и пленкой происходят ионизация воздуха и формиро¬
вание стабильного коронного разряда, который "обрабатывает” поверхность
пленки.Обработку разрядом полиэтилена высокого давления изучали многие ис¬
следователи, особенно в связи с эффектом улучшения автоадгезии. Слипа¬
ние (сваривание) полиэтилена высокого давления происходит при контакте
двух поверхностей под давлением при температуре выше 90 °С. Однако пос¬
ле сравнительно малой степени обработки разрядом поверхности слипают¬
ся при значительно меньших температурах (~70°С). Для объяснения этого
эффекта были предложены две теории. В первой, представленной группойТаблица 9.4. Адгезия полиэтилена высокого давления к
эпоксидному клею после травления в хромовой кислоте
(напряжение сдвига) [30]. Условия травленияАдгезия, МН/м2АВ1 мин, 20 °С1,323,4520 с, 70 °С2,509*511 мин, 70 °С5,2111,065 мин, 70 °С7,8211,3815 мин, 70 °С9,4211,3830 мин, 70 °С9,9211,20Для приготовления травильного раствора и промывки поверхности
использовалась дистиллированная вода (А) и тройной дистиллят (поверхност¬
ное натяжение составляет 72,5 мН/м) (В).
Д. Бриггс429Рис. 9,14. Схематичное изображение процесса обработки поверхности пленок коронным
разрядом. 1 - металлический валок с диэлектрическим покрытием, 2 - плен¬
ка, 3 - коронный разряд, 4 - высоковольтный электрод.канадских исследователей [51], предполагалось образование электрета (ди¬
электрического поляризованного слоя). Увеличение адгезии связывалось с
электростатическими взаимодействиями. Доказательством служил тот факт,
что эффект наблюдался при обработке разрядом как в "активных” (воздух,
кислород), так и в "инертных” (азот, аргон, гелий) газах, причем величина
эффекта была связана с рассеиваемой в разряде мощностью и не зависела от
газа. Более того, максимальный эффект в окислительной атмосфере дости¬
гался прежде, чем можно было обнаружить окисление поверхности с помощью
ИК-спектроскопии (НПВО). Вторая, диаметрально противоположная теория
Оуэнса [ 52] предполагает, что эффект связан с образованием водородных
связей между полярными группами, образующимися при обработке поверх¬
ности разрядом. В этой работе изучалась, однако, только обработка полиэти¬
лена высокого давления в воздушной атмосфере.В работе автора данной главы [ 8] обработка разрядом проводилась в мо¬
дельной установке; обрабатываемые образцы пленки были неподвижны, вы¬
соковольтное напряжение прикладывалось с низкой частотой повторения
(50 Гц). Анализ методом РФЭС показал (рис0 9.15), что обработка разрядом
в воздухе, азоте и аргоне (при атмосферном давлении газов) приводит к
химической модификации поверхности, включая во всех случаях ее окисле¬
ние. Очевидно, что обработка в аргоне требует большего времени, однако
измерения рассеиваемой мощности подтвердили полученный ранее результат
[511: мощность, необходимая для достижения заданного уровня автоадгезии
(напряжения отслаивания), не зависит от используемого газа. Из рис. 9.15
следует также, что тождественным уровням автоадгезии соответствует при¬
мерно одинаковая степень окисления поверхности. Были проведены также
эксперименты с использованием разряда в водороде; при этом улучшения
автоадгезии не происходило и с помощью РФЭС окисление поверхности не наб¬
людалось. Эти результаты уверенно свидетельствуют в пользу теории Оуэнса
и отвергают электронную теорию. На рис. 9.16 показан типичный спектр
полиэтилена высокого давления, обработанного разрядом в воздухе [ 44]. Прос¬
тое разложение левого крыла линии С Is, соответствующего большим энер¬
гиям связи, дает три пика» Пик энергии связи - 286,5 эВ может быть при-
4309. Применение РФЭС в технологии полимеровРис. 9.15. Сравнение величин автоадгезии (напряжения отслаивания) и состава по¬
верхности (до данным РФЭС) для полиэтилена высокого давления, обра¬
ботанного электрическим разрядом в воздухе, азоте или аргоне (ампли¬
тудные значения напряжений 13,7; 13,7 и 2,2 кВ соответственно, часто¬
та следования высоковольтных импульсов 50 Гц). Сварка проводилась в
следующих условиях: температура 75° С, давление 1 атм, время контакта
2 с. Отношение интенсивностей пиков 01 s и С1$ является количествен¬
ной мерой степени окисления поверхности. Отношение N1 s/ds приво¬
дится для поверхностей, обработанных разрядом в азоте. Образцы, для
которых напряжение отслаивания составляет 100 г/25 мм (помечено пунк¬
тирными линиями), имеют близкие степени окисления [44].писан группе -СН20- (т. е. спиртовой и сложноэфирной, эфирной и гидро-
перекисной); пик энергии связи « 288,0 эВ - группе>С=»0 (например, альде-2гидной или кетонной) и пик энергии связи — 289,5 эВ - группе - С - О (кар¬
боновой кислоты или сложного эфира). Линия Ols еще менее информативна,
Большинство кислородсодержащих функциональных групп дают в спектре пи¬
ки О Is энергии связи * 532 эВ, исключение составляет кислород карбо¬
ксильных групп, для которого энергия связи примерно равна 533,5 эВо Сдвиг
пика О Is на рис. 9.16 вместе с возрастанием степени окисления отражает
рост относительной концентрации карбоксильных групп. Ясно, что эта инфор¬
мация недостаточно специфична для определения детальной структуры окис¬
ленного слоя и для исследования корреляций с результатами измерения ав¬
тоадгезии. Чтобы решить эту проблему, было предложено несколько реакций
образования производных соединений для пометки специфических функцио¬
нальных групп. Эти реакции приведены в табл» 9.2 [21, 22].
Д. Бриггс431Рис. 9.16. Линии С1 s и 01s спектров высокого разрешения полиэтилена высокого
давления: а - необработанного; б, в - обработанного коронным разрядом
в воздухе (амплитудное значение напряжения 13,7 кВ; 50 Гц) в течение
8 и 30 с соответственно. Скорость счета 3 • 103 имп./с на деление (С1 s)
и 103 имп./с на деление 01 s.В литературе высказывалось единое мнение, что наиболее вероятный
механизм окисления при обработке разрядом полиэтилена высокого давления
выглядит следующим образом:
4329. Применение РФЭС в технологии полимеровТаблица 9.5 Количественное определение функциональных групп на
поверхности полиэтилена высокого давления после обработки коронным
разрядом [21]РеагентОтношениеОтношение атомныхЧисло функциональныхинтенсивностейконцентрацийгрупп в расчете на однуРФЭ-пиков(элемент/углерод)группу —СН2— необрабо¬(внутреннийтанной поверхностиуровень/С 1s)PFPH®(F Is) 0,2055,5 х 10'2>С—0, 1,1 х 10~2Вг2-Н20(Вг 3d) 3,6 х 10~210,6 х 10-3СН2С=С, 5,3 х 10-3С АС б(С1 2р) 1,3 х 10'26,0 х 10'3С—ОН, 6,0 х 10“3ТААВ(Ti 2руг) 6,2 х 10-21,5 х 10'2С—ОН, 1,5 х 10~2NaOH(Na Is) 8,8 х 10~21,1 х 10-2-СООН, 1,1 х ю-2so2(S 2р) 7,6 х 10‘34.7 х 10-38.7 х 10'2С-ООН, 4,7 х 10 3-(О Is) 0,209® PFPH — пентафторфенилгидразин.САС — хлорангидрид хлоруксусной кислоты, реагирует, по-видимому, селективно с енольным
гидроксилом.ТА А — бис-ацетилацетонат диизопропилата титана; реагирует, по-видимому, селективно со спиртовым
гидроксиломПри обработке происходит как разрыв полимерных цепей, так и образо¬
вание сшивок* Основным промежуточным продуктом является группа гид¬
роперекиси* Ее стабильность и реакции разложения были предметом много¬
численных исследований.Методом РфЭС анализировались поверхности, на которых специфические
функциональные группы были помечены с помощью упомянутых выше селек¬
тивных реакций образования производных соединений. Полученные данные
допускают количественную обработку, результаты которой представлены в
табл. 9о5. Число групп - СН2=0 получено в предположении, что при броми-
ровании замещаются в среднем два атома водорода*, Поскольку эта группа
в результате таутомеризации переходит в енол, содержащий гидроксильную
группу - ОН, число групп -CH^jC^O, оцененное по реакции с бромом, и число
енольных гидроксильных групп -ОН, оцененное по реакции с хлорангидридом
хлоруксусной кислоты (CH2Q . СОС1), должны быть сравнимы, что и наб¬
людается на опыте.Линия С Is спектра углерода имеет близкие интенсивности в диапазо¬
нах энергий связи, соответствующих группам С — ОН, ^С=0 и -СООН. Ре¬
зультаты, представленные в табл. 9.5, подтверждают этот факт. Исходя из
числа групп >С*0, С-ОН, С-ООН и СООН, можно получить соотношение
числа атомов кислорода и углерода NQ/NC = 5,7 • 10~2, где Nc - число ато¬
мов углерода на необработанной поверхности. Это сравнимо с действитель¬
ной величиной 8,7 . 10~2, полученной для поверхности, обработанной разря¬
дом, из соотношения интенсивностей пиков Ols и С Is (см. табл. 9.5). На¬
ряду с группами, вступающими в перечисленные реакции образования произ¬
водных соединений, на поверхности, по-видимому, должно присутствовать
Д. Бриггс433сравнимое количество групп эфиров, перекисей и сложных эфиров. Учитывая
это, данный метод определения количества функциональных групп можно
считать вполне разумным. Явная внутренняя согласованность этих резуль¬
татов служит дополнительным доказательством надежности использованных
в работе процедур образования производных соединений. Гидроперекиси в
полиэтилене могут иметь большие времена жизни, поэтому если рассмат¬
риваемый механизм окисления является правильным, их можно обнаружить.
Реакция с S02 подтверждает это и, по-видимому, является первым прямым
доказательством данного механизма. Следовательно, среди групп, которые,
по всей вероятности, должны образовываться при разложении гидропереки¬
си, с помощью реакций образования производных соединений идентифициру¬
ются группы -С=0, С-ОН и -СООН.Эти реакции образования производных соединений также позволяют уда¬
лять с поверхности определенные функциональные группы и при использова¬
нии РФЭС для параллельного наблюдения изучать детальную картину взаимо¬
действия при адгезии к поверхности полимера. Таким образом, описанная
выше работа позволила полностью подтвердить теорию эффекта улучшения
автоадгезии после обработки поверхности коронным разрядом, предложен¬
ную Оуэнсом. Количественные исследования были проведены и на flpvrnx
системах [21]. В будущем этот метод позволит значительно расширить воз¬
можности РФЭС при изучении поведения поверхностей полимеров.Другим методом модификации поверхности полиолефинов является обра¬
ботка плазмой, пока еще не внедренная в промышленность. В этом методе
используется электрический разряд в газе низкого давления (< 1 мм рт. ст.).
Кларк и Дилкс [ 5] изучали модификацию полиэтилена высокого давления,
полипропилена и полистирола при плазменном окислении. Исследования с из¬
менением угла сбора фотоэлектронов показали, что по сравнению с обработ¬
кой коронным разрядом в этом случае модифицированный слой является
очень тонким и окислен сильнее (см. рис. 9.1).9.6.2.3. ПопиэтилентерефтапатХотя полиэтилентерефталат является полярным материалом со сравни¬
тельно большой поверхностной энергией, для получения хорошей адгезии
часто необходима обработка поверхностно При этом обработка коронным
разрядом оказывается менее успешной, чем в случае полиолефинов, посколь¬
ку обработанная поверхность сохраняет улучшенные свойства в течение ко¬
роткого интервала времени. Например, на рис. 9.17 изображена зависимость
характеристик автоадгезии для обработанных коронным разрядом пленок
от времени, прошедшего после обработки до соединения нагретых пленок
под давлением (нагрев производится до температуры, при которой необрабо¬
танные поверхности не проявляют автоадгезии). Эффект ухудшения харак¬
теристик модифицированных поверхностей со временем исследовался при
4349. Применение РФЭС в технологии полимеровРис. 9.17. Напряжение отслаивания (г/25 мм) для сварных соединений, образованных
между поверхностями полиэтилентерефталата, обработанными коронным
разрядом, в зависимости от времени между обработкой и сваркой (времени
старения). Режим сварки: 130°С, контакт в течение 2 с под давлением
1,5 кг/см2[53].помощи РФЭС [ 53]. На рис» 9.18 и 9.19 показаны спектры обработанной и
необработанной поверхностей» Ясно видно увеличение числа атомов кислоро¬
да на поверхности» Разложение контура линии С Is на отдельные пики ука¬
зывает на образование двух дополнительных функциональных групп. На осно¬
вании данных для энергий связи различных функциональных групп, полу¬
ченных из спектров и-оксибензойной кислоты и ее сложного метилового эфи¬
ра, а также других литературных данных [ 8] были идентифицированы пики
"г" и "д" на рис. 9.18. Их происхождение связано с фенольным гидроксилом
-ОН и карбоксильной группой -СООН. Согласуются также значения энергии
связи для пика Ols фенольного гидроксила и энергии связи для главного
дополнительного пика в спектре линии Ols обработанного полиэтилентере-
фтала. По аналогии со свободнорадикальным механизмом окисления, рассмот¬
ренным выше для случая обработки полиэтилена высокого давления корон¬
ным разрядом, действительно следует ожидать появления концевых групп
фенольного гидроксила и свободной карбоновой кислоты. Образование по¬
следней группы также означает, что происходит разрыв полимерных цепей
полиэтилентерефтала (т. е. уменьшение молекулярного веса).Измерения краевого угла показывают, что увеличение вклада полярных
групп в поверхностную энергию, наблюдаемое после обработки, быстро спаг
дает одновременно с ухудшением адгезионных свойств. Изменения, происхо¬
дящие на поверхности, приводят к изменению РФЭ-спектров (рис. 9.19). Бы¬
ло показано также, что большая часть окисленного материала легко смы¬
вается с поверхности водой. Этот факт служит веским доказательством то¬
го, что основная часть соединений, образующихся при обработке полиэти¬
лентерефталата коронным разрядом, имеет относительно малый молекуляр-
Д. Бриггс435Рис. 9.18. Линии С1 s и 01 s спектра поверхности полиэтилентерефталата до (верх¬
ний спектр) и после (нижний спектр) обработки разрядом (40 с, частота
следования импульсов 50 Гц, амплитудное значение напряжения 15,9 кВ;
соответствующая величина подведенной энергии 620 мДж/см2). Разложе¬
ние линии на отдельные пики проводилось с помощью прибора фирмы
Du Pont 310. Пики соответствуют углероду, входящему в следующие
группы:ный вес. Первоначальное улучшение адгезии, вероятно, большей частью
связано со способностью фенольного гидроксила, возникающего при обрат
ботке поверхности разрядом, образовывать сильную водородную связь.
После обработки, по-видимому, происходят процессы, схематично изобра¬
женные на рис. 9.20. Вслед за сравнительно быстрой миграцией низкомоле¬
кулярных компонент в толщу образца происходит более медленная пере¬
ориентация полимерных цепей большей длины. В результате появившиеся на
поверхности гидроксильные группы оказываются связаны водородными
4369. Применение РФЭС в технологии полимеровРис. 9.19. Изменения РФЭ-спектров полиэтилентерефталата, обработанного разря¬
дом, в зависимости от времени старения, а - сразу после обработки (со¬
ответствует нижнему спектру на рис. 9.18), б - через один день, в - че¬
рез 23 дня. Спектр "а" практически идентичен спектру поверхности пос¬
ле промывки ее водой. Спектр "г" получен с необработанной пленки [ 53].Рис. 9.20. Схемы молекулярных механизмов эффектов обработки поверхности разря¬
дом и старения для полиэтилентерефталата. а — до обработки, б — после
обработки, в - после некоторого времени старения. Отсутствие низкомо¬
лекулярной группы на нижнем рисунке (после некоторого времени старения)
связано с ее диффузией в объем образца.
Д. Бриггс437связями внутри образца и, таким образом, быстро теряют свою активность
на поверхности. С помощью РФЭС можно наблюдать за появлением групп
-ОН, но не за их миграцией в образец, поскольку последняя происходит
внутри зондируемой этим методом толщины образца.9.6.2.4. Другие исследованияКак отмечалось выше, в задачу данной главы не входит полный обзор
применений РФЭС в технологии полимеров. Попытка сделать это была пред¬
принята в более раннем обзоре [37]. В настоящее время литература слиш¬
ком обширна, чтобы повторять подобный опыт0 Вместо этого в нижеследую¬
щей библиографии под отдельными заголовками выделены области наиболь¬
шей активности. В частности, в этой главе не рассматривается применение
РФЭС для изучения волокон (искусственных и естественных) и тканей. В об¬
зоре [ 37] приводится список выполненных к тому времени работ, посвящен¬
ных этому предмету. Не так давно появился новый обзор по этой теме [ 43].
Много внимания сейчас уделяется углеродным волокнам, которые играют
важную роль в композиционных материалах, включающих разнообразные по¬
лимеры в качестве матриц. Основной областью исследований является опти¬
мизация свойств границы раздела волокно - матрица с помощью различных
методов обработки поверхности. Изменения, происходящие на поверхности
при старении в атмосферных условиях, исследуются для понимания механиз¬
мов фотодеградации (ранее здесь использовались только методы объемного
анализа). Предметом интенсивного изучения является также граница разде¬
ла полимер - металл как с точки зрения оптимизации адгезии, так и для пре¬
дотвращения коррозии. Один из активно исследуемых здесь вопросов рас¬
сматривался выше. Другие вопросы, относящиеся к этому же предмету, -
это нанесение декоративных покрытий на пластики, создание слоистых ком¬
позитов, конструирование и изготовление плат для электрических схем, ар¬
мирование резиновых покрышек (шин) и т. д. Совершенно иной областью при¬
менения РФЭС является исследование полиацетиленов для понимания меха¬
низмов высокой проводимости, появляющейся в системах при легировании
полимеров (например, галогенами)09.7. ЗаключениеБыстрое расширение области применения полимерных материалов свя¬
зано в основном с простотой процесса производства по сравнению с обыч¬
ными материалами (металлами, деревом, стеклом и т0 д«). Однако часто
потребителю требуется полимерный материал, поверхность которого облада¬
ет специальными свойствами. Поэтому важную роль в технологии полиме¬
ров играет анализ поверхности. Основной вклад в его развитие внесла
РФЭС. Уже сейчас этот метод неоценим при поиске и устранении различно¬
го рода дефектов» Благодаря ему быстрыми темпами углубляется наше по¬
нимание важнейших процессов модификации поверхности полимеров. В бу-
4389. Применение РФЭС в технологии полимеровдущем этот метод станет основой для создания более эффективных способовобработки.Литература1. Clark D.T., in: Photon, Electron and Ion Probes of Polymer Structure and
Properties (eds. D.W. Dwight, T. J. Fabish, H.P. Thomas), ACS Symposium
Series, 162, 255 (1981).2. Clark D.T., in: Handbook of X-ray and Ultraviolet Photoelectron Spectrosco¬
py (ed. D. Briggs), Heyden, London 1977, p. 212.3. Wheeler D.R., Pepper S.V., J. Vac. Sci. Technol., 20, 226 (1982).4. Clark D.T., in: Handbook of X-ray and Ultraviolet Photoselection Spectro¬
scopy, (ed* D. Briggs), Heyden, London 1977, p. 233.5. Clark D.T., Dilks A., J. Polym. Sci., Polym. Chem. Ed., 17, 957 (1979).6. Clark D.T., Fok C.T; J. Electron Spectrosc., 22, *173 (1981).7. Roberts R.F. et aL, Surf. Interface Anal., 2, 6 (1980).8. Clark D.T., Harrison A., J. Polym. Sci., Polym. Chem. Ed., 19, 1945 (1981).9. Dilks A., in: Electron Spectroscopy — Theory, Techniques and Applications,
(eds. C.R. Brundle, AJ)« Baker), vol. 4, Academic Press, London, 1981.10. Briggs D., Wootton A.B., Surf. Interface Anal., 4* 109 (1982).11. Clark D.T., Dilks A., J. Polym. Sci., Polym. Chem. Ed., 14, 633 (1976).12. O’Malley ].].. Thomas H.R., Lee G., Macromolecules, 12, 496 (1979).13. Chambers R.D., Clark D.T., Kilcast D., Partington S., J. Polym. Sci., Polym.
Chem. Ed., 12, 1647 (1974).14. Dilks A,, in: Photon, Electron and Ion Probes of Polymer Structure and
Properties (eds. DJVe Dwight, T.J. Fabish, H.R. Tbomas), ACS Symposium
Series, 162, 307 (1981).15. Pireaux J.J., Riga J., Caudano R., Verbist J., in: Photon, Electron and Ion
Probes of Polymer Structure and Properties, (eds. D,W« Dwight, ToJ* Fabish,
HJL Thomas), p. 169, ACS Symposium Series, 162, 169 (1981).16. Riggs W.M., Dwight D.W., J. Electron. Spectrosc., 5, 447, (1974).17. Briggs D., Brewis D.M., Konieczko M.B., J. Mater. Sci., 12, 429 (1977).18. Spell H., Christenson C., TAPPI, 1978, 283 (1978).19. Everhart D.S., Reilley C.N., Anal. Chem., 53, 665 (1981).20. Hammond J., Holubka Durisin A., Dickie R., Abstracts of Colloid and
Interfacial Science Section, ACS Miami Meeting, September 1978.21. Briggs D., Kendall C.R., Int. J. Adhesion, Adhesives, 2, 13 (1982).22. Briggs D., Kendall C.R., Polymer, 20, 1053 (1979).23. Batich C.D., Wendt R.C., in: Photon, Electron and Ion Probes of Polymer
Structure and Properties, (eds. D.W. Dwight, T.J. Fabish, H.R. Thomas),ACS Symposium Series, 162, 221 (1981).
Д. Бриггс43924. Bradley АCzuha М., Jr., Anal. Chem., 47, 1838 (1975).25. Czuha М., Jr., Riggs W.ty., Anal. Chem., 47, 1836 (1975).26. Millard M.M., Marsi М., Anal. Chem., 46, 1820 (1974).27. Everhart D.S., Reilley C.N., Surf. Interface Anal., 3, 126 (1981).28. Everhart D.S., Reilley C.N., Surf. Interface Anal., 3, 269 (1981).29. Briggs D, in: Surface Analysis and Pretreatment of Plastics and Metals,(ed. D.M. Brewis), Ch. 4, Applied Science, London, 198230. Briggs D. et al., Surf. Interface Anal., 2, 107 (1980).31. Van Ooij W., Abstracts of the Second International Conference on Quantita¬
tive Surface Analysis. Teddington, November 1981.32. Briggs D., Surf. Interface Anal., 5, 113 (1983).33. Briggs D., Brown A., van der Berg J.A., Vickerman J.C., Ion Formation from
Organic Solids, (ed. A. Benninghoven), Springer-Verlag (в печати).34. Briggs D., Surf. Interface Anal., 4, 151 (1982).35. Ratner B.D., in: Photon, Electron and Ion Probes of Polymer Structure and
Properties (eds. D.W. Dwight, T.J. Fabish, H.R. Thomas), ACS Symposium
Series, 162, 371 (1981).36. Ratner B.D. et al., J. Appl. Polym. Sci., 22, 643 (1978).37. Briggs D., in: Electron Spectroscopy: Theory, Techniques and Applications,
(eds. C.R. Bundle, A.D. Baker)., vol. 3, Academic Press, London, 1979.38. Von Brecht H., Mayer F., Binder H., Makromol. Chem., 33, 89 (1973).39. Dwight D.W.f Riggs W.M., J. Coll. Interface Sci., 47, 650 (1974).40. Collins G.C.> Lowe A.C., Nicholas D., Euro. Polym. J., 9, 1173 (1973).41. Clark D.T.f Feast W.J., Musgrave W.K.R., Ritchie J., J. Polym., Sci., Polym.
Chem. Ed., 13, 857 (1975).42. Clark D.T., Dilks A.,. J. Polym. Sci., Polym. Chem. Ed., 15, 2321 (1977).43. Millard M.M., in: Industrial Applications of Surface Analysis (eds. L.A. Casp¬
er, C.J. Powell), ACS Symposium Series, 199, 143 (1982).44. Brewis DM., Briggs D., Polymer, 22, 7, (1981).45. Briggs D., Brewis D.M; Konieczko M.B., J, Mater. Sci., 11, 1270 (1976).46. Briggs D„ Brewis DM, Konieczko M.B., Euro. Polym. J., 14, 1 (1978).47. Briggs D., Brewis D.M., Konieczko M.B., J. Mater. Sci., 14, 1344 (1979).48. Blythe A.R., Briggs D., Kendall C.R. et al., Polymer, 19, 1273 (1978).49. Briggs D., Kendall C.R.y Blythe A.R., Wootton A.B., Polymer, 24, 47, (1983).50. Briggs D., in: Surface Analysis and Pretreatment of Plastics and Metals
(ed. D.M. Brewis), Ch.9, Applied Science, London, 1982.51. Stadal М., Goring D.A.I., Can. J. Chem. Eng., 53, 427 (1975).52. Owens D.K., J. Appl. Polym. Sci., 19. 265 (1975).53. Briggs D., Ranee D.G., Kendall C.R., Blythe A.R., Polymer, 21, 895 (1980).
4409. Применение РФЭС в технологии полимеровБиблиографияМодификация поверхностиClark D.T., Dilks А.} ESCA applied to polymers(XVIII), RF glow discharge
modification of polymers in helium, neon, argon and krypton, J, Polym., Sci.,
Polym. Chem. Eds., 16, 911 (1978).Clark D.T., Dilks A., ESCA applied to polymers (XXIII), RF glow discharge mo¬
dification of polymers in pure oxygen and helium oxygen mixtures, J. Polym.
Sci., Polym. Chem. Ed., 17, 957 (1979).Huang S.•Т., Chen J.-M., Chen S.-L., Application of photoelectron spectroscopy
(ESCA) to study the surface fluorination of polypropylene, K’O Hsueh T'Ung
Pao, 24, 831 (1979).Leary H.J., Campbell D.S., ESCA studies of polvmide and modified polymide
surfaces, ACS Symp. Ser., 162, 419 (1981).Yamamoto F. et ah, Surface grafting of polyethylene by mutual irradiation inmethyl acrylate vapour (III), quantitative surface analysis by XPS, J. Polym.
Sci., Polym. Phys. Ed., 17, 1581 (1979).Фотоокисление и эрозияDilks A., Clark D.T., ESCA studies of natural weathering phenomena at selected
polymer surfaces, J. Polym. Sci., Polym. Chem. Ed., 19, 2847 (1981).Peeling J., Clark D.T,, An ESCA study of the photo-oxidation of the surface of
polystyrene film.., Polym. Degradation Stab., 3, 97 (1981).Peeling J., Clark D.T., ESCA study of the surface photo-oxidation of some nonaro-
matic polymers, Polym. Degradation Stabe, 3, 177 (1981).Peeling ]., Clark D.T*, Photo-oxidation of the surfaces of polyphenylene oxide
and polysulphone. J. Appl. Polym., Sci., 26, 3761 (1981).Взаимодействие полимеров с металламиBurkstrand J.M., Substrate effects on the electronic structure of metal
overlayers — an XPS study of polymer-metal interfaces, Phys. Rev., B20,
4853 (1979).Burkstrand J.M., Core-level spectra of chromium and nickel atoms on polystyrene,
J. Appl. Phys., 50, 1152 (1979).Burkstrand J.M., Copper-poly(vinyl alcohol) interface: a study with XPS, J. Vac.
Sci. Technol., 16, 363 (1979).Cadman P., Cossedge GM., The chemical nature of metal — PTFE tribological
interactions as studied by XPS, Wear, 54, 211 (1979).Cemia E., Ascarelli P., Spectroscopic analysis of interactions between metal¬
lic surfaces and organic polymers, Proc. Int. Conf. Org. Coat. Sci. Technol.,
1978, 103 (1978).
Д. Бриггс441Cim М., Ascarelli P., XPS applied to small metal particles and to metal-polymer
adhesion: physical insight from initial and final-state effects, NATO Adv.
Study. Inst. Ser., C73, 353 (1981).Richter К. et al., ESCA study of metal-polymer phase boundaries in composite
materials, 2, Chem., 18, 390 (1978).Roberts R.F., Schonhom М., Surface modification of polymers by deposition of
evaporated metals (I), Teflon FEP, Polym. Prepr., Am. Chem. Soc., Div.
Polym. Chem., 16, 146 (1975).Servais J.P. et al., Examination by ESCA of the constitution and effects on
lacquer adhesion of passivation films of tinplate, Br. Corros. J., 14, 126
(1979).Ootj Van W. J., Kleinhesselink A., Leyenaar S.R., Industrial applications of
XPS: study of polymer-to-metal adhesion failure, Surf. Sci., 89, 165 (1979).Van OOij W.J., Kleinhesselink A., Application of XPS to the study of polymer-
metal interface phenomena, Appl. Surf. Sci., 4, 324 (1980).Волокна и тканиBenerito R.R. et al., Modifications of cotton cellulose surfaces by use of radio
frequency cold plasmas and characterisation of surface changes by ESCA,
Text. Res. J., 51, 224 (1981).Chand N., Surface studies of poly aery lonitrite yarn by XPS (ESCA), Indian J.
Text. Res., 6, 133 (1981).Chand N., XPS study of drawn poly(ethylene terephthalate) fibres, Pop. Plast.
Rubber, 26, 6 (1981).Hammer G.E., Graphite fibre surface analysis by XPS and polar dispersive free
energy analysis. Appl. Surf. Sci., 4, 340 (1980).Hopfgarten F., ESCA studies of carbon and oxygen in carbon fibres, Fibre Sci.
Technol., 12, 283 (1979).Ishitoni A., Application of XPS to surface analysis of carbon fibre, Carbon, 19,
269 (1981).Proctor A., Sherwood M.A., XPS studies of carbon fibre surfaces (I). Carbon
fibres surfaces and the effect of heat treatment, J. Electron Spectrosc., 27,
39 (1982).Proctor A., Sherwood P.M.A., XPS studies of carbon fibre surfaces (III). The
effect of electrochemical treatment, Carbon, 21, 53 (1983).Proctor A., Sherwood P.M.A., XPS studies of carbon fibre surfaces (III).
Industrially treated fibres and the effect of heat and exposure to oxygen,
Surf. Interface Anal., 4, 212 (1982).Takeshige F.J. et al., Fundamental studies on water repellency of textile
fabrics by ESCA, Teikoku Gakuen Kiyo, 1981, 83 (1981).
4429. Применение РФЭС в технологии полимеровПопиацвтипен и его модификацииHsuS.K. et al*, Highly conducting iodine derivatives of polyacetylene: Raman,
XPS and XRD studies, J. Chem. Phys., 69# 106 (1978).Ikemoto l*C* et al*, XPS study of highly conductive bromine-doped poly acetylene,
Bull. Chem. Soc. Jpn., 5Б, 721 (1982).Ikemoto J*C* et alXPS study of highly conductive iodine-doped poly acetylene,
Chem. Lett., 10, 1189 (1979).Salanek W*R* et al*, Photoelectron spectra of arsenic (V) fluoride-doped
polyacetylenes, J, Chem. Phys., 71, 2044 (1979).Stevens G»C,, Bloor D*, Williams P.M., Photoelectron valence band spectra of
diacetylene polymers, Chem. Phys., 28, 399 (1978).Thomas H*R* et al*, Photoelectron spectra of conducting polymers. Molecularly
doped polyacetylenes, Polymer, 21, 1238 (1980).РазноеBigelow R*W* et ah, Structure variation of sulphonated polystyrene surfaces, Adv.
Chem. Ser., 187, 295 (1980).Brion D*, Mavel G„ A study of the photoelectron spectroscopy (XPS) of mouldings
based on PVC, Eur. Polym. J., 16, 159 (1980).Casarini A* et aL, XPS and the problems of film producing industry, Poliplasti
Plast. Rinf., 27, 80 (1979).Dilks A*, The Identification of peroxy-features at polymer surfaces by ESCA, J.
Polym. Sci., Polym. Chem. Ed., 19, 1319 (1981).Dwight D*W* et al*, ESCA analysis of polyphosphazene and poly (siloxane/car¬
bonate) surfaces, Polym. Prepr. An. Chem., Soc., Div. Polym. Chem., 20,
702 (1979).Gianelos ]*, Grulke E*A*> Some studies of chlorinated PVC using XPS, Adv.X-ray Anal., 22, 473 (1979).Knecht J., Baessler H*, An ESCA study of solid 2,4-hexadiyne 1,6-diol bis
(toluene sulphomate) and its constituents before and after polymerisation,
Chem. Phys., 33, 179 (1978).Knutson K,f Lyman D./., Morphology of black copolyurethanes (II). FTIR and
ESCA techniques for studying surface morpholy, Org. Coat Plast. Chem.,42, 621 (1981).Lacaze P*C*f Tourillon G., Spectroscopic study (XPS-SIMS) of the aging ofpolyacetonitrile thin films electrochemically deposited on a platinum elect¬
rode, J. Chim. Phys., Phys. Chim. Biol., 76, 371 (1979).Leary H.J., Campbell D.SSurface analysis of aromatic polymide films using
ESCA, Surf. Interface Anal., 1, 75 (1979).
Д. Бриггс443Nagarajan S., Stachurshi Z.HHughes M,%,, Larkins F,P., A study of the
PE-PTFE system (II). ESCA measurements, J.Polym. Sci. Phys. Ed., 20,
1001 (1982).O'Malley /.J., Thomas H.R., Surface studies of multicomponent polymer solids,
Contemp. Top. Poly. Sci., 3, 215 (1979).Shuttleworth D. et aL, XPS study of low molecular weight polystyrene-poly-
dimethylsiloxane black copolymers, Polym. Prepr., Am. Chem. Soc., Div.
Polym. Chem., 20, 499 (1979).Stone-Masui J*H»> Stone W,E*E*, Characterisation of polystyrene latexes by
photoelectron and infrared spectroscopy, Polym. Colloids, 2, 331 (1980).Sung С., Paik S., Ни С*В*. ESCA studies on surface chemical composition of
segmented polyrethanes, J, Biomed. Mater. Res., 13, 161 (1979).Takai У. eU ah, Photoelectron spectroscopy of poly-p-xylyene polymerised
from the vapour phase, Polym. Photochem. 2, 33 (1982).Thomas R.H., 0*Malley /./. Surface studies on multicomponent polymer studies
by XPS: polystyrene/poly(ethyleoxide) homopolymer blends, Macromolecu¬
les, 14, 1316 (1981)*
Глава 10ПРИМЕНЕНИЕ ОЖЕ-ЭЛЕКТРОННОЙ
И ФОТОЭЛЕКТРОННОЙ СПЕКТРОСКОПИИ
В ИССЛЕДОВАНИЯХ КОРРОЗИИН.С. Макинтайрп10.1. ВведениеПроцессы коррозии локализованы в очень тонких поверхностных слоях.
Металл начинает подвергаться коррозии в тех местах, где нарушена защит¬
ная или пассивирующая оксидная пленка, в результате чего становится воз¬
можным контакт химически активных атомов металла с атомами и молекула¬
ми, атакующими поверхность. Однако достаточно в местах повреждения за¬
щитной пленки вновь образоваться химически инертному покрытию толщиной
всего в несколько атомных слоев, как происходит репассивирование поверх¬
ности. Задача науки о коррозии состоит как в выяснении причин химического
разрушения металлов, так и в разработке методов защиты их поверхности.
Следовательно, анализ тонких поверхностных пленок всеми доступными мето¬
дами должен был бы стать одной из первых задач ученых, исследующих по¬
верхность.Однако этого не произошло и главным образом из-за того, что исследо¬
ватели коррозии были заняты решением проблем, связанных с применяемыми
на практике материалами, а физики и химики предпочитали исследовать ме¬
нее сложные процессы на поверхности. Контакты между двумя группами уче¬
ных в основном имели место тогда, когда анализировался состав поверхности
активно корродирующего образца. Такие исследования часто давали информа¬
цию, касающуюся в большей степени самой исследуемой поверхности и в мень¬
шей - истинных причин коррозии. Лишь в последние несколько лет были пред¬
приняты согласованные действия ученых в попытке исследовать методами
анализа поверхности процессы начальной стадии коррозии.Область химии и физики поверхности, изучающая процессы коррозии, весь¬
ма обширна. Если основную задачу определить как достижение понимания про¬
цессов, инициирующих и предотвращающих разрушение металлов в различных
средах, то эта область будет существенно шире традиционного испытания
стойкости металлов в водной среде. Электрохимические методы стали однимиA/.S. McIntyre, Surface Science Western, University of Western, Ontario
London, Ontario, Canada N6A 5B7.
Н.С. Макинтайр445из наиболее важных в числе методов, используемых для контроля видов и ско¬
рости различных процессов, протекающих на границе металла и внешней сре¬
ды. Особенно многообещающим представляется объединение методов электро¬
химии и анализа поверхности. Ряд примеров такого синтеза обсуждается в
этой главе. Многие проблемы коррозии столь же тесно, как с химией, связа¬
ны с металлургией. В случае металлов коррозии часто подвержены преиму¬
щественно границы зерен; на межкристаллитную коррозию сильное влияние
оказывают нагрев и механическая обработка. Сами металлурги широко ис¬
пользуют методы анализа поверхности, особенно оже-электронную спектро¬
скопию для исследования сегрегации зерен и сенсибилизации сплавов (см.
гл. 7). Тем не менее методы анализа поверхности до сих пор лишь в немно¬
гих случаях использовались для исследования процессов проникновения кор¬
розии в микроструктуру металла и в еще меньшей степени для исследования
коррозионного растрескивания поверхности при механических нагрузках. Яв¬
ления, связанные с микроструктурой изъязвления поверхности, также только
недавно стали привлекать внимание исследователей. Возможное значение та¬
кой работы будет обсуждаться в этой главе. Областью, где аналитические
методы должны будут более широко применяться, является разработка но¬
вых методов и материалов для защиты поверхности. В настоящей главе будут
описаны некоторые исследования органических и неорганических ингибиторов
коррозии.Самым первым инструментом, который специалисты по коррозии исполь¬
зовали для исследований поверхности, был оптический микроскоп. Этот при¬
бор в сочетании с методами селективного химического травления позволял
исследовать поперечные срезы пленок коррозии на металлических подложках.
Такая техника дает возможность изучать поверхностное распределение и од¬
нородность пленок коррозии толщиной не менее 5-10 мкм. Кристаллографи¬
ческую структуру этих пленок, если она вообще существовала, иногда удава¬
лось определить методом дифракции рентгеновских лучей. Существенный прог¬
ресс в анализе пленок был достигнут в 60-е годы, когда стали широко доступ¬
ными методы микроаналитического электронного зондирования. Пространствен¬
ное разрешение рентгеновского флюоресцентного элементного анализа с ис¬
пользованием электронного микрозонда достигло 2 мкм, что позволило иссле¬
довать элементный состав пленок в поперечном сечении.Очевидно, эти методы применимы только для исследований достаточно
толстых, однородных коррозийных пленок и не могут дать информацию о на¬
чальной стадии коррозии, о микроструктуре поверхности или о химии тонких
пассивирующих пленок. Появление в начале 70-х годов методов электронной
спектроскопии послужило важной вехой на пути развития новых подходов к
изучению процессов коррозии.
44610. Применение ЭОС и РФЭС в исследованиях коррозииИз всего многообразия методов исследования поверхности в этой главе
мы ограничимся рассмотрением рентгеновской фотоэлектронной спектроско¬
пии (РФЭС) и оже-электронной спектроскопии (ЭОС), что соответствует теме
настоящей книги. Безусловно, в исследованиях поверхности, связанных с воп¬
росами коррозии, эти методы используются очень широко. В тех случаях, ког¬
да мы встретимся с ограничением применимости данных методов, будут так¬
же описаны альтернативные подходы, использующие другие хорошо известные
методы анализа поверхности.Чтобы сделать настоящую главу полезной как для специалистов по корро¬
зии, так и для специалистов в области анализа поверхности, в разд. 10,2 обсуж¬
даются те аспекты электронной спектроскопии, которые особенно существен¬
ны для исследований коррозии. В разд. 10.3 дан обзор последних работ, посвя¬
щенных исследованию процессов коррозии, в которых использовались методы
РФЭС и ЭОС. Дополнительную информацию по обоим разделам можно найти
в более ранних обзорах Джоши [ 1] и Кастла [2, 3].10.2. Особенности применения РФЭС и ЭОС для исследования
коррозии10.2.1. Мувствитепьность анализа поверхностиЭлектроны с энергией 100- 1000 эВ, анализируемые в обычных РФЭС- и
ЭОС-измерениях, имеют среднюю длину пробега в диапазоне 1 - 3 нм [ 4]. Та¬
ким образом, при характерной чувствительности обоих методов обычно не со¬
ставляет труда зарегистрировать монослой пассивирующей пленки на подлож¬
ке с другим элементным составом. В определенных случаях, когда имеет мес¬
то заметный химический сдвиг, методом РФЭС можно детектировать верх¬
ний монослой, отличающийся от подложки только по химической структуре.В случае слоистого характера распределения продуктов коррозии толщи¬
ну верхнего слоя на бесконечно толстой подложке можно определить из экс¬
поненциальной зависимости, описываемой уравнением (5.27) в гл. 5. Изме¬
ряя угловое [5] или энергетическое распределение оже- или фотоэлектронов
от верхнего слоя и подложки, можно определить характер верхнего слоя:
островковый или слоистый. Кастл [3], используя систему двух переключае¬
мых источников рентгеновского излучения с различной энергией, определял
отношение интенсивностей фотоэлектронов и сравнивал полученное значение
с результатами расчета, проведенного для модели слоистого распределения.В случае слоистого характера распределения измерению могут быть доступ¬
ны 10-20 монослоев. В будущем с помощью сканирующей оже-электронной
микроскопии (СОМ) можно будет проводить количественные исследования из¬
менений более сложных поверхностных структур.
Н.С. Макинтайр447Часто пленки на начальной стадии окисления состоят из нескольких очень
тонких слоев, различающихся по составу. В этих случаях даже очень тщатель¬
ные измерения зависимости энергии электронов от угла вылета не могут дать
однозначной картины оксидной пленки. Так, например, РФЭС-исследования ок¬
сидированной поверхности сплава Alloy 600 показали, что вблизи поверхности
слои состоят из оксидов никеля и железа, а оксиды хрома расположены ближе
к границе между металлом и оксидной пленкой. Использование же более чувст¬
вительного метода рассеяния ионов низких энергий позволило показать, что
верхний монослой пленки образован исключительно оксидом железа [ 6].10.2.2. Чувствительность к элементному составуКак РФЭС, так и ЭОС чувствительны ко всем элементам, за исключени¬
ем водорода, представляющим интерес при исследовании процессов коррозии.
Возможность анализа легких элементов (Z < 11) представляет особый инте¬
рес в связи с анализом кислорода в пленках коррозии. Такая информация, как
интенсивность линии О Is и энергия связи, очень важна при определении харак¬
теристик пассивирующих пленок.Чувствительность метода РФЭС ограничена высоким уровнем фона, свя¬
занного с преобладанием в спектре электронов, потерявших энергию при рас¬
сеянии в образце. При использовании большинства коммерческих источников
рентгеновского излучения и практически приемлемого времени счета (пример¬
но 1 ч) предел относительной чувствительности для различных элементов ле¬
жит в пределах 1,0-0,1% (при отношении сигнал/шум, равном 3). Это означа¬
ет, что такие предшественники коррозии, как хлориды и фосфаты, детектиру¬
ются с трудом или вообще не детектируются методом РФЭС. До тех пор пока
не удастся существенно увеличить скорость счета электронов (примерно на
порядок), метод РФЭС будет оставаться инструментом исследования только
основных фаз коррозии.За редким исключением эквивалентный предел чувствительности ЭОС не¬
сколько ниже, чем у РФЭС. Это объясняется несколькими причинами (см. гл. 3),
но для большинства образцов основной причиной, по-видимому, является элект¬
рический заряд, накапливаемый на диэлектрических оксидах и гидроксидах.
Накопление заряда можно уменьшить путем использования очень малого тока
первичных электронов, но при этом существенно возрастает время счета.При детектировании малых и следовых количеств таких соединений, как хло¬
риды, возможности ЭОС больше, чем РФЭС, поскольку эти соединения часто
имеют локальный характер распределения.Определенные ограничения возможностей РФЭС и ЭОС в исследованиях
процессов коррозии связаны с нечувствительностью этих методов к водороду.
Водород в составе гидратов и гидроксидов представляется важной компонен-
44810. Применение ЭОС и РФЭС в исследованиях коррозиитой пленок. Кроме того, гидриды металлов, образующиеся в некоторых систе¬
мах, являются предшественниками образования хрупкого разрушения. Альтер¬
нативными методами анализа водорода служат масс-спектрометрия вторич¬
ных ионов (ВИМС) и ядерный микроанализ.10.2.3. Количественный анализВ гл. 5 обсуждались некоторые модели РФЭС и ЭОС с использованием и
без использования стандартов. Для случая анализа пленок коррозии эти мо¬
дели были несколько видоизменены. Асами, Хасимото и Симодаира [7, 8], ис¬
ходя из первых принципов, развили наиболее полную модель РФЭС, особенно
пригодную для анализа тонких пассивных оксидных пленок на поверхности
сплавов. Уравнение (5.27) в гл. 5 было модифицировано так, чтобы учесть
эффект ослабления, вносимый загрязнением поверхности углеродом, и разли¬
чие плотностей оксидной пленки и металлической подложки. Таким образом,
было определено относительное содержание элементов в различных образцах.
Удалось даже оценить содержание водорода по измерениям - ОН и связанной
воды (см. ниже). Эта модель успешно применялась для анализа оксидов на по¬
верхности сплавов с совершенно различным химическим составом [9]. На
рис. 10.1 приведены зависимости относительной концентрации оксидов метал¬
лов на поверхности хромомолибденовой стали от анодного поляризационного
потенциала в соляной кислоте. Линии Fe 2Р3/2, Сг 2Ръ/2 и Мо Ы фотоэлект¬
ронного спектра записывались как в режиме интегрирования, так и в режиме
спектрального разрешения (см. приложение 3), чтобы разделить линии, соот¬
ветствующие металлическим компонентам с меньшей энергией связи и окси¬
дам. Суммы интенсивностей, определенные раздельно для металла и оксида,
использовались для вычисления толщины оксидной пленки, а эта величина в
свою очередь использовалась для вычисления состава оксидной пленки и спла¬
ва (рис. 10,1 б). Несмотря на существенное изменение состава оксидной плен¬
ки в ходе эксперимента было получено хорошее согласие результатов РФЭС-
анализа состава металлической подложки с известным объемным составом
сплава, что подтверждает справедливость количественной модели.Используя ту же модель РФЭС, Асами и Хасимото [10] провели расчеты
состава известных оксидны к структур. При этом для всех фотоэлектронных
линий использовалось одно и то же значение средней длины пробега электро¬
нов независимо от химического состояния элемента. На рис. 10.2 приведено
сравнение известного для данного оксида отношения оксид/катион и отноше¬
ния интенсивности линий О ls/M 2р, исправленного с учетом средней длины
свободного пробега электронов и загрязняющего слоя. Очень хорошее согла¬
сие результатов измерений с известным составом большинства оксидов сви¬
детельствует о справедливости использования в таких исследованиях одного
Н.С. Макинтайр449Рис. 10.1. Количественные измерения составов сплавов и оксидных пленок на их по¬
верхностях. Анализ электрода из хромо-молибденовой стали, поляризован¬
ного в 1М растворе НС1 в течение 3600 с. а - зависимость относитель¬
ной весовой концентрации катионов в оксидной пленке от приложенного по¬
тенциала; б- зависимость относительной весовой концентрации металлов,
входящих в состав нержавеющей стали, от приложенного потенциала. Сплош¬
ные линии соответствуют составу в объеме образца [э].значения средней длины пробега электронов. Более того, из углов наклона пря¬
мых на рис. 10.2 для оксидов хрома и железа можно определить среднее отно¬
шение сечений фотоэлектронной эмиссии О/М.Важное значение имеет определение количества кислорода, содержащего¬
ся в пленках коррозии в определенных соединениях, особенно в тех случаях,
когда гидрирование или гидроксилирование поверхности может влиять на ее
химическую инертность. Асами и сотр. [11] удалось количественно оценить со-
45010. Применение ЭОС и РФЭС в исследованиях коррозииРис. 10.2. Количественный РФЭС-анализ оксидов металлов с известным составом.Приведена зависимость скорректированного отношения 01s/M2p для не¬
скольких оксидов хрома и железа от отношения кислород/металл в объе¬
ме образца.держание кислорода, входящего в три различных соединения - оксид металла,
гидроксид металла и связанную воду. Кислород, связанный с металлом или в
гидрооксиде металла, детектировался по химическому сдвигу; количество
связанной воды определялось по разности полной интегральной интенсивности
линии О Is и интенсивности, соответствующей линиям кислорода, связанного
с катионами, записанными во всем РФЭС-спектре.В другом подходе количественного исследования пленок коррозии мето¬
дом РФЭС для разделения компонент оксида и металла используются стандар¬
ты [6]. Хотя этот подход является менее гибким по сравнению с описанным
выше, однако, используя метод анализа спектров, который к тому же мэжно
автоматизировать, удается достичь разумного количественного разделения
основных фаз оксида и металла.Количественный ЭОС-анализ, по-видимому, осуществлялся главным обра¬
зом с использованием стандартов, хотя общий подход, развитый "из первых
принципов", хорошо описан в литературе (см. гл. 5). Проблему вклада в оже-
электронный спектр низколежащих атомных слоев рассматривали на количест¬
венном уровне Понс, Лехериси и Лонжерон [12], используя многослойную мо¬
дель поверхности. ЭОС-измерения профиля градиента концентрации по глуби¬
не пленки коррозии на 5 - 6 монослоев существенно осложняются из-за вкла¬
да в сигнал от несколько отличающихся по составу более низколежащих слоев.
В дифференциальном методе при послойном анализе результаты измерения
состава верхнего слоя корректируются с учетом информации о составе после¬
дующего слоя. Для максимальной эффективности данного метода необходимо
Н.С. Макинтайр 451
45210. Применение ЭОС и РФЭС в исследованиях коррозиипри послойном анализе проводить измерения состава каждого монослоя
(~0,3 нм). Этот метод использовался для повышения разрешения по глубине
при ЭОС послойном анализе состава пассивирующих пленок на поверхности
сплавов Alloy 600 и 800 [13]. На рис. 10.3, а приведены исходные результаты
измерений интенсивности линий ЭОС при послойном анализе, а на рис. 10.3, б
показаны профили относительных концентраций атомов, полученных после об¬
работки данных дифференциальным методом. Благодаря устранению вклада
от низколежащих слоев на рис. 10.3, б стали отчетливо видны максимумы кон¬
центраций хрома и железа внутри поверхностной оксидной пленки. Митчел [14]
расширил модель Понса, Лехериси и Лонжерона, включив в нее упрощенное вы¬
ражение, учитывающее эффект поглощения оже-электронов каждым предшест¬
вующим слоем. Кроме того, при использовании модифицированного пуассонов-
ского распределения был учтен статистический характер процесса распыления.10.2.4. Пространственное разрешениеВ РФЭС минимальная площадь измеряемой поверхности составляет вели¬
чину порядка нескольких квадратных миллиметров* что определяется угловой
апертурой приемной электронной оптики. Пятно рентгеновского излучения
покрывает большую площадь на поверхности. В современных же микроанали¬
тических сканирующих оже-спектрометрах минимальная анализируемая пло¬
щадь поверхности достигает 0,01 мкм2.Низкое пространственное разрешение метода РФЭС является серьезным
препятствием при исследовании некоторых вопросов коррозии. Процессы кор¬
розии по своей природе очень часто носят локальный характер: многие реак¬
ции инициируются на изгибах поверхности, на границах зерен или в трещинах.
До тех пор пока эти явления не будут воссоздаваться искусственно, изолиро*
ванно от общих процессов коррозии поверхности, трудно себе представить воз¬
можность использования стандартных РФЭС-приборов для исследований локаль¬
ных химических процессов коррозии. Недавно появилось сообщение о создании
прототипа РФЭС-прибора с рентгеновским пучком малого диаметра1*. Это тех-Рис. 10.3. Обработка данных ЭОС-анализа по глубине дифференциальным методом,
повышающим разрешение по глубине, а - профиль по глубине интенсив¬
ностей линий ЭОС-спектра поверхности сплава Alloy 600 пассивированной
в 0t5NU2soA при статическом потенциале 0,8 В (нормальный водородный
электрод) в течение 30 мин, скорость распыления, 1,5 А/мин; б- полу¬
ченные в результате обработки данных рис. 10.3, а) дифференциальные про¬
фили состава, выраженные в относительных атомных долях для Ni, Сг,Fe и О. [13].DSurface Science Laboratories, Inc., Sunnyvale, California.
Н.С. Макинтайр453ническое решение, а также использование мощных источников синхротронного
излучения с малым диаметром пучка могут открыть новые, очень интересные
направления исследований в области химии коррозии.Главным преимуществом метода ЭОС является возможность анализа лока¬
лизованных продуктов коррозии. Локальный анализ областей (например, впадин
и трещин), отстоящих друг от друга на расстояние 1 мкм, является обычным
при исследовании пленок коррозии. Особенно важная информация получается
при строчном сканировании вдоль области с переменным составом. Посколь¬
ку при этом одновременно регистрируется несколько элементов, можно прове¬
сти калибровку по интенсивности. На рис. 10.4 приведена запись линий хрома,
марганца и титана при строчном сканировании поверхности сплава Alloy 800,
оксидированной при 600 °С в потоке кислорода [15]. Распределение марганца
и титана носит локальный характер и, возможно, связано с пересечением гра¬
ниц зерен с поверхностью.. Более того, распределение титана сильно зависит
от температуры, которая меняется вдоль поверхности из-за потока газа в на¬
правлении сканирования. При исследовании коррозии полезную качественную
информацию дает восстановление пространственных распределений в виде
оже-изображений. Так, в работе [16] для нержавеющей стали марки 304, на-Рис. Ю.4. Полученное методом строчного ЭОС-сканирования распределение хрома,
марганца и титана на поверхности сплава Alloy 800, оксидированной при
600 °С в течение 1 мин.
45410. Применение ЭОС и РФЭС в исследованиях коррозиигретой до 1000 °С, были получены оже-карты, показывающие обогащение хро¬
мом вдоль границ зерен. В литературе можно найти много других примеров
получения оже-изображений методом сканирования.10.2.5. Профилирование по глубинеАнализ пленки коррозии на глубину более 2 нм обычно включает ионную
бомбардировку с одновременным анализом методом ЭОС или с последующим
анализом методом РФЭС. О структурных изменениях, вызванных ионной бом¬
бардировкой окисленной поверхности, написано довольно много (см. гл. 4).
Информация об этих изменениях представляется очень важной, если при по¬
слойном анализе необходимо получить качественные химические или количест¬
венные характеристики оксидных слоев. Так было обнаружено, что иногда лу¬
чевое повреждение выражается в изменении химического состава оксидов ме¬
таллов - сильного в одних случаях и достаточно слабого в других. Сведения
о степени химических изменений для одной и той же системы оксидов весьма
различны, что заставляет предположить важность геометрии системы. Напри¬
мер, многие исследователи сообщают, что при бомбардировке ионами аргона
низких энергий происходит разложение оксидов железа до FeO. В некоторых
работах [17, 18] обнаружено дальнейшее восстановление FeO до металла, в то
время как в других работах [19, 20] найдено, что FeO является стабильным
продуктом. Если в режиме стационарного распыления оксидной структуры не
происходит ее восстановления до металла, то при послойном анализе оксид¬
ная и металлическая фазы будут количественно различаться. Макинтайр и др.
[6, 21] обнаружили, что в условиях их эксперимента хром, кобальт, никель и
железо, типичные представители первого ряда элементов, подверженных кор¬
розии, при ионной бомбардировке не восстанавливаются до металла.Детальный анализ соотношения оксидной и металлической фаз возможен
только методом РФЭС, в спектрах которой вклад от оксидов и металла хоро¬
шо разделяется. На рис. 10.5 представлены диаграммы относительного содер¬
жания оксидов и металлической фазы на поверхности сплава Alloy 600, окси¬
дированной при 300 °С при 5 и 0,01%-ном содержании кислорода в газовой сме¬
си. Глубокое проникновение в подложку оксида никеля в случае оксидирования
при более высоком парциальном давлении кислорода является основным отли¬
чием от оксидирования в атмосфере, бедной кислородом. Этот эффект едва ли
можно было зарегистрировать методом ЭОС.Безусловно, определить химическую структуру оксидов после ионной бом¬
бардировки обычно невозможно, потому что более высокие состояния окисле¬
ния восстанавливаются, а также происходит дегидратация. В этом случае дру¬
гие неразрушающие методы анализа по глубине оказываются полезными, хотя
и несколько утомительными. При определении химических изменений в оксиди-
Н.С. Макинтайр455Рис. 10.5. РФЭС-профили по глубине пленок на поверхности сплава Alloy 600, по¬
лученных при 300 °С и двух различных концентрациях кислорода: а -
0,01% 02, 5 мин, б- 5% 02, 5 мин.рованных поверхностях может быть полезной механическая обдирка корроди¬
рующих поверхностей с использованием in situ шаровой мельницы. В настоя¬
щее время эта техника вместе с ЭОС используется для быстрого профилиро¬
вания очень толстых слоев [22]. Химическое травление, используемое при
химическом послойном анализе поверхности полупроводников [23], также мо¬
жет быть полезным при анализе некоторых оксидных пленок.10.2.6. Экспериментальные методы
10.2.6.1. Исследование коррозииВ литературе, посвященной анализу поверхности применительно к проб¬
леме коррозии, в большинстве случаев рассматривается анализ образцов, по-
45610. Применение ЭОС и РФЭС в исследованиях коррозиилученных в специальных экспериментах по коррозии, а не материалов, корро¬
дирующих в реальных условиях. Описан широкий круг экспериментов, в кото¬
рых искусственно воспроизводятся условия питтинга, щелевой коррозии, кор¬
розии под напряжением или условия общей атаки поверхности. Испытания
материалов проводились в средах, воспроизводящих рабочие условия (напри¬
мер, в автоклаве с высоким давлением пара, в автоклаве, заполненном кисло¬
той, in situ при растяжении и т.д.). Некоторые исследования коррозии, глав¬
ным образом посвященные фундаментальным вопросам, выполнены в кювете
с электрохимическим контролем. Во всех случаях, когда не ожидается обра¬
зование очень толстых пленок, вопрос приготовления поверхности образца
является очень важным. Так, при механической полировке (холодная обработ¬
ка) свойства поверхности по отношению к окислению зависят от ее шерохова¬
тости [6], а при электрохимической полировке может происходить обеднение
или обогащение поверхности некоторыми компонентами [3]. Для фундаменталь¬
ных исследований процесса окисления сплавов, по-видимому, лучше всего при¬
менять вакуумный отжиг образцов. Хотя в случае изготовления реальных изде¬
лий холодная обработка неизбежна, однако часто желательно исследовать сам
эффект окисления.10.2.6.2. Перемещения образцаВо многих случаях, и особенно при электрохимических исследованиях,
очень важно защитить образец от дальнейшего химического изменения при
его перемещении из области реакции в аналитическую камеру. Когда актив¬
ный металл или сплав с непассивирующей пленкой на поверхности извлекается
из кюветы, воздействие воздуха на образец, естественно, является основным
фактором. В случае некоторых относительно пассивных пленок образец при
перемещениях можно хранить в жидком азоте [13]. В более совершенном мето¬
де манипуляций с образцом экспериментальная кювета помещается в бокс с
инертной атмосферой, и образец переносится из бокса непосредственно в спект¬
рометр через шлюз. Как показано в работе [24], этих мер достаточно, чтобы
обеспечить сохранность восстановленной поверхности U02 Q. В наиболее со¬
вершенных системах при перемещениях образца контакт с какой-либо атмос¬
ферой вообще отсутствует. Одна из таких систем для ЭОС-исследований была
описана в [25], а в работах [26, 271 описаны камеры для манипуляций с образ¬
цом при комбинированных электрохимических - РФЭС-исследованиях.10.2.6.3. Влияние вакуумаОчевидно, что в условиях вакуума в камере спектрометра происходит де¬
сорбция некоторых гидратов и разложение гидроксидов. Асами и Хасимото
[10] обнаружили, что a-FeOOH в вакууме при температуре 50 °С медленно
разлагается, теряя 3% гидроксильных групп за каждые 100 мин. Кроме того,
Н.С. Макинтайр457все большее число исследований показывает, что даже молекулы воды, находя¬
щиеся внутри кристаллической решетки оксида, воспроизводимо детектируют¬
ся в РФЭС-0 Is спектре [28, 29]. Молекулы воды, физически или химически
адсорбированные на поверхности, конечно, значительно легче десорбируются.
На десорбированных молекулах происходит рассеяние О ls-электронов, что
оказывает влияние на результаты измерений интенсивности линии О Is связан¬
ной воды в РФЭС-спектре [9]. Поскольку полагают, что деградация пассиви¬
рующего слоя зависит от присутствия связанной воды [30], развитие методов
ее детектирования представляется многообещающим для определения парамет¬
ров пленок коррозии. Измерения методом ядерного микрозонда до и после
РФЭС-анализа позволят более точно установить корреляцию между содержа¬
нием воды и изучаемыми процессами.10.2.6.4. Влияние облученияРентгеновское излучение, воздействующее на образец при РФЭС-анализе,
вызывает значительно меньшее повреждение поверхности, чем эквивалентный
поток электронов. Однако имеются сообщения о частичном восстановлении не¬
которых оксидов металлов под действием рентгеновского излучения (напри¬
мер, СиО ч. Си20, Со ООН -* СоО), и по мере увеличения мощности использу¬
емых источников рентгеновского излучения этот эффект проявляется более
отчетливо. Под действием потока электронов происходят химическое разло¬
жение [31], десорбция воды и адсорбированных ионов, например хлора [32], а
при больших потоках электронов - восстановление оксидов [33].10.2.7. Химические эффектыХимический сдвиг линий спектра РФЭС , соответствующих фотоэмиссии
внутренних электронов, связан с изменением электростатического потенциала
внутренних оболочек при увеличении или уменьшении вблизи атома плотности
заряда валентных электронов (см. гл. 3). Таким образом, существует соотно¬
шение между энергией связи электронов в атоме и его химическим состояни¬
ем. Благодаря химическому сдвигу продукты коррозии в виде оксидов элемен¬
тов можно легко отличить от самих элементов, присутствующих в восстанов¬
ленной форме. Часто возможно также отличить оксиды металла различной ва¬
лентности (например, СгО*~ и Сг203) или оксид и гидроксид [например, NiO
от Ni (ОН) 2]. Химическая структура влияет также и на другие особенности
фотоэлектронных пиков и обусловливает ряд дополнительных изменений в со¬
стояниях окисления. Многие спектроскопические особенности химических сдви¬
гов и формы линий, характерные для различных продуктов коррозии, пока не
представлены в литературе в форме, удобной для использования учеными, ис¬
следующими процессы коррозии. Характерные изменения в спектрах, связан-
45810. Применение ЭОС и РФЭС в исследованиях коррозииные с химической структурой, обычно невелики, а приводимые в литературе
данные либо противоречивы, либо имеют столь большие пределы ошибок, что
практически становятся бесполезными. Положение можно улучшить, если бо¬
лее тщательно стандартизировать процедуру калибровки энергии (см. прило¬
жение 1) и развить более точный метод определения формы и положения спек¬
тральных линий. В приложении 4 наиболее полно представлены данные о хими¬
ческих сдвигах для различных соединений, представляющих интерес для ис¬
следователей коррозии - оксидов основных элементов сплавов: железа, нике¬
ля, хрома, кобальта, меди и марганца, что существенно дополняет более ран¬
ние данные, собранные в работе [34].В ЭОС по химическому сдвигу хорошо различаются лишь металл и его
оксид. Вполне возможно, что в будущем с увеличением числа используемых
в ЭОС-спектрометрах анализаторов с высоким энергетическим разрешением
ситуация изменится.Общий обзор, посвященный химическим сдвигам в РФЭС и ЭОС, приве¬
ден в других главах настоящей книги. Однако имеется несколько элементов,
представляющих особый интерес для специалистов по коррозии. Для оксидов
этих элементов полезно привести возможно полный объем данных о химиче¬
ских сдвигах, что и будет сделано ниже.10.2.7.1. КислородПри анализе различных оксидов на поверхности часто используется ли¬
ния О Is (примерно 530 эВ). Она имеет относительно малую ширину и сим¬
метричную форму, что позволяет осуществлять более точную подгонку слож¬
ной комбинации полос. Обширный обзор, посвященный энергиям связи О ls-
электрона в оксидах металлов, был опубликован Джонсоном [35]. И хотя, ка¬
залось бы, нельзя установить простого соответствия между характером свя¬
зи в оксиде металла и энергией связи О ls-электрона, однако эксперимен¬
тальные значения этой энергии для некоторых оксидов довольно значительно
различаются, что позволяет идентифицировать эти оксиды. Одним из приме¬
ров может служить различие химических сдвигов у СиО и Си20 [ 36, 37].Обычно кислород в гидроксильной группе (ОН~) и в оксиде (02“) отлича¬
ют по химическому сдвигу О Is-линии, который составляет величину от 1 до
1,5 эВ. Например, линия кислорода, связанная с NiO, имеет энергию
529,6 ± 0,2 эВ, в то время как для Ni(OH)2 эта линия имеет заметно большую
энергию связи 531,2 ±0,2 эВ [37]. Более того, в смешанном окси дно-гидро -
ксидном соединении FeO(OH) вклад в спектр линии О 1 s от каждого кисло¬
рода можно отчетливо выделить [19]. Более сложной задачей является иден¬
тификация воды на поверхности по спектру О Is. Величина химического
сдвига лежит в широхом диапазоне, превышающем 3 эВ, поскольку молекулы
Н.С. Макинтайр459воды на поверхности могут занимать различноэ положение. Нортон [38] обна¬
ружил, что для замороженной воды энергия связи составляет около 533 эВ,
в то время как Асами и сотр. [7, 8] нашли, что энергия связи О Is у гидрок¬
сида и у воды, связанной внутри кристаллической решетки, различаются не¬
значительно (531 эВ). Часто детектируемая в диапазоне 531 г-534 эВ широкая
огибающая линии О Is связана с большим количеством различных форм хими¬
чески и физически связанной воды на поверхности и внутри кристаллической
решетки. К сожалению, линии хемосорбированных и физисорбированных кис¬
лорода и гидроксильных групп лежат в этой же области энергий.Экспериментально получаемые линии кислорода в оже-электронной спект¬
роскопии имеют слишком большую ширину, чтобы с их помощью можно было
проводить идентификацию химического состояния кислорода. Кислород в кри¬
сталлической решетке оксида и хемосорбированный кислород, одновременно
присутствующие в образце, были обнаружены в работе [39].10.2.7.2. ТитанКак показали исследования методом РФЭС, ТЮ2 можно уверенно отли¬
чить от TiO по положению линии Ti 2р [34, 40, 41]. TiN - соединение,
предположительно образующееся в некоторых коррозийно-стойких пленках на
поверхности, - также можно отличить от ТЮ и ТЮ2 по химическому сдвигу
линии Ti 2р [40].Методом ЭОС соединения Ti О и ТЮ2 легко идентифицируются благода¬
ря тому, что их L3M23-линии сдвинуты относительно друг друга на 2 эВ [42].
По форме линии этого перехода можно также отличить металлический титан
от TiH2 [43].10.2.7.3. ВанадийВ спектре РФЭС линии оксидов V205 и V02 различаются примерно на
1 эВ. Однако ЭОС-спектры в этом случае дают больше информации, чем РФЭС.
По оже-переходам VL2 3М2 3V и L2M2 3М2 3 можно идентифицировать це¬
лый ряд оксидов с различным стехиометрическим соотношением ванадий -
кислород [44-46].10.2.7.4. ХромПри РФЭС-исследованиях содержание оксидов хрома обычно определяется
по линии Сг 2Р2/2. Оксиды трех- и четырехвалентного хрома довольно легко
разделяются благодаря большому (~2 эВ) химическому сдвигу [ 47, 48]. В слу¬
чае трехвалентного хрома оксид Сг203 и гидроксиды Сг(ОН)3 и СгООН также
можно различить по разнице в энергиях связи, хотя и несколько меньшей
(~ 0,5 эВ) [ 10, 49]. При идентификации различных структур хромитов может
быть полезной информация о мультиплетном расщеплении линии Сг 2Ръ/г [10].
46010. Применение ЭОС и РФЭС в исследованиях коррозии10.2.7.5. МарганецМетодом РФЭС было исследовано несколько оксидов марганца с различ¬
ным стехиометрическим соотношением [50]. МпО и Мп02, по-видимому, осо¬
бенно просто различить по положению в спектре линии Мп 2р^/2 и ее форме
[51110.2.7.6. ЖелезоИсследованию оксидов железа методом РФЭС было посвящено несколько
работ [19, 52, 53]. Вследствие сильной связи дырки, образовавшейся в резуль¬
тате фотоэмиссии электрона из внутренней оболочки, с валентной оболочкой
в состояниях с большим значением спина, Fe 2^-фотоэлектронные спектры
оксида и субоксида железа имеют довольно сложный вид. Некоторые особен¬
ности этого сложного спектра могут быть использованы для аналитики, по¬
скольку форма линии Fe 2р довольно чувствительна к химическим измене¬
ниям. Это можно видеть из рис. 10.6, где для сравнения приведены спектрыРис. 10.6. Рентгеновские фотоэлектронные спектры некоторых характерных продук¬
тов коррозии. Показан спектр линии Fe2ръ/2 для a- FeOOH (a),a-Fe2°3(6), Fe304 (e), Fe(COOH)2 (г).
Н.С. Макинтайр461oc-FeOOH, a-Fe203, Fe304 и Fe(COOH)2 в области линии Fe 2р3/Г Центр
линии гидроксида трехвалентного железа (рис. 10.6, а) сдвинут по отношению к
a- Fe203 примерно на 1 эВ в область больших энергий авязи, а a - Fe203 и у- Fe208
(не показан) можно различить по расщеплению основного пика. В случае магнетита
(Fe304), содержащего как Fe11 так и Fe111, спектр состоит из двух перекры¬
вающихся компонент (рис. 10.6, в). К сожалению, в случае реальных образцов
поверхность Fe304 четко идентифицировать часто бывает довольно сложно,
поскольку воздействие воздуха приводит к окислению Fe304 до Fe203. И на¬
конец, некоторые органометаллические соединения, представляющие интерес
при исследовании коррозии, имеют спектр, отличающийся от спектров окси¬
дов. Как видно из рис. 10.6, г, оксалат железа можно идентифицировать по
четко выраженному сателлиту (отмечен стрелкой).Для исследователей коррозии также представляют интерес некоторые ок¬
сиды двухвалентного железа. В спектре FeO на крыле линии Fe 2Р3/2 имеет¬
ся четко выраженный горб [18]. Аналогичный спектр наблюдается у смешан¬
ного оксида FeMoO, [54].Большое внимание при исследовании коррозии уделяется сульфидам же¬
леза. Сульфиды железа на поверхности в присутствии воздуха часто подвер¬
гаются гидролизу с образованием оксидов, что существенно усложняет спектр.
Однако FeS и FeS2 имеют заметно различающиеся спектры линии Fe 2р3 /2,
что опять-таки является следствием различного мультиплетного взаимодей¬
ствия. Форма и положение этой лийии в спектрах FeS2 и металлического же¬
леза очень близки [ 55], а в спектре FeS эта линия представлена широким пи¬
ком, сдвинутым почти на 2 эВ в сторону больших энергий по сравнению с FeS2.Для ряда оксидов железа в спектре ЭОС характерно наличие пиков в об¬
ласти малых кинетических энергий. Эклунд и Лейграф [56] обнаружили два не¬
зависимых Fe LFF-пика с энергиями ~ 43 эВ и 51 эВ и предположили, что
они связаны с Fe2+ и Fe3+ соотаетственно. Сео и сотр. [57] обнаружили тре¬
тий пик с энергией 46 эВ и высказали предположение, что пики с энергией
46 эВ и 51 эВ относятся к двухвалентному железу, в то время как трехвалент¬
ное железо имеет пики с энергией 43 и 51 эВ.10.2.7.7. КобальтОксиды кобальта И и кобальта III в спектре РФЭС можно различить по
их магнитным свойствам. Парамагнитные оксиды кобальта И в своих спект¬
рах имеют интенсивный сателлит, на 6 эВ превышающий энергию линии Со
%Р3/2> в то время как диамагнитные оксиды кобальта III такой особенности
в спектрах не имеют. Гидроксид и оксид кобальта II различаются по химиче-
46210. Применение ЭОС и РФЭС в исследованиях коррозиискому сдвигу ^ 1,0 ±0,2 эВ [18, 37j. Смешанные оксиды кобальта СоМоОд и
Со А1204 имеют химический сдвиг 0,5 эВ в области больших энергий [58].10.2.7.8. НикельНа оксидированной поверхности никеля в отличие от кобальта во всех
практически интересных случаях, по-видимому, все соединения содержат ни-
каль с одинаковой степенью окисления. Положение линии Ni %Р3/2 в спектрах
NiO и Ni (ОН) 2 различается на величину химического сдвига (около 2 эВ),
что позволяет идентифицировать поверхность Ni(OH) 2 [37].Положение линии Ni 2рз/2 в спектре ионов Ni2+, содержащихся в различ¬
ных решетках оксидов, может существенно изменяться в зависимости от хи¬
мического окружения, что, возможно, связано с мультиплетным взаимодейст¬
вием [6]. Из рис. 10.7, а и б видно, что в спектре NiO и шпинели (NiFe204)
максимумы пиков Ni 2р3/2 смещены относительно друг друга на 1,5 эВ. Кро¬
ме того, в спектре NiO главный пик расщеплен, а в спектре NiFe204 расщеп¬
ление пика отсутствует.Рис. 10.7. Фотоэлектронные спектры простых и смешанных оксидов никеля. Показан
спектр линии Ni2р /2 для NiO (a); NiFe204 (б); смешанного оксида
железа — никеля (в) (Ni /Fe = 1).
Н.С. Макинтайр46310.2.7.9. МедьОдним из элементов, наиболее полно исследованных методом РФЭС, яв¬
ляется медь. В ряду переходных металлов структура спектров оксидов меди
наиболее просто интерпретируется. Однако эти оксиды более сильно подвер¬
жены изменениям при нагреве или облучении.При исследовании методом РФЭС в твердых пленках были обнаружены
два устойчивых оксида меди Си20 и СиО [59]. Для гидроксида меди был
также получен характерный спектр РФЭС [37].Все три соединения образуются скорей всего на поверхности в результа¬
те коррозии металлической меди. Поэтому важно иметь возможность отли¬
чать эти соединения от металлической подложки. Фотоэлектронные спектры
закиси меди и металлической меди совпадают с точностью до 0,1 эВ. В то же
время индуцированные рентгеновским излучением оже-спектры металлической
меди и закиси меди (СиЦМ4 gM4 5), впервые описанные Шёном [60], сущест¬
венно различны, что позволяет дать количественную характеристику для ок¬
сида и металла [24]. Вклад в спектр от Си20 и от металлической меди мож¬
но определить по отношению интенсивности пиков при двух различных энер¬
гиях, если вклад от оксидов меди более высокой валентности отсутствует. В
оже-спектрах СиО и Си(0Ю2 линия меди ЦМ4 5М4 5 сдвинута на различную
величину кинетической энергии по сравнению с Сд^О. Однако эта линия в обо¬
их спектрах имеет большую ширину и под ее огибающей содержится большое
число дискретных оже-линий, что несколько затрудняет ее идентификацию.Идентификацию СиО и Си(ОН)2 обычно осуществляют по линии Си 2р3/ 2.
По сравнению с Си20 максимум главного пика в спектре СиО сдвинут на
1,3 ± 0,2 эВ в сторону больших энергий. Однако этот максимум трудно опре¬
делить по отношению к линии Си20 из-за большого уширения пика, возможно
связанного с мультиплетным расщеплением. Пик Си 2рз/2 в спектре Си(ОН)
сдвинут в сторону больших энергий на 2,5 ±0,15 эВ по сравнению с Си20.
Соединения меди можно также характеризовать с помощью двух линий, распо¬
ложенных на 6 и 8 эВ выше основной линии Си 2Р3/2 [37]. Единственным от¬
личием для СиО и Си(ОН)2 в этой области спектра служит разное соотноше¬
ние интенсивностей двух пиков огибающей "встряски".10.2.7.10. МолибденКоррозионные свойства молибдена, входящего в состав многих нержавею¬
щих сталей, исследовались методом РФЭС. В отличие от первого ряда пере¬
ходных металлов для элементов второго ряда, таких, как молибден, измене¬
ния спектров, вызванные окислением, более предсказуемы, и в спектрах не
46410. Применение ЭОС и РФЭС в исследованиях коррозиисодержится сателлитная структура. Таким образом, оксиды МоIV и Мо VI
легко идентифицируются по химическому сдвигу линии Мо 3d [58]. ДляМо03
химический сдвиг линии Мо Ы относительно Со Мо 04 составляет приблизи¬
тельно 0,8 эВ.10.3. Обзор работ по применению РФЭС и ЭОС
в исследованиях коррозииВ настоящем разделе будет дан обзор работ, в которых использовались
методы РФЭС и ЭОС для решения проблемы коррозии путем нанесения корро-
зионно-стойких покрытий из одного материала на другой. Здесь мы не будем
рассматривать вопросы, связанные с химическими реакциями на поверхности
полупроводников, а также с реакциями, относящимися больше к вопросам ме¬
таллургии, чем к коррозии. Будут рассмотрены реакции на поверхности, в
которых участвуют вода, кислород и другие элементы. Однако многие фунда¬
ментальные исследования реакций на поверхности металлов, не имеющие пря¬
мого отношения к процессам коррозии, не включены в настоящий обзор.10.3.1. Легкие металлыЗенер, Барбулеско и Дженкинс [61], используя ЭОС, исследовали процесс
окисления поверхности бериллия. По пику в спектре энергетических потерь,
расположенному ниже линии BeKLL, им удалось обнаружить сегрегацию сле¬
дов кремния. Метод ЭОС использовался также в работе [62], где было обнару¬
жено, что анодное окисление монокристалла алюминия приводит к образова¬
нию слоев со стехиометрией А1203.Однако в исследованиях методом РФЭС алюминиевых фольг, пассивиро¬
ванных в естественных условиях, наряду с А1203 был обнаружен целый ряд
гидроксидов [63]. Как показали исследования методом РФЭС, пленки алюми¬
ния, образующиеся при анодировании в серной кислоте, содержат, помимо
А1203, также сульфид и сульфат [64]. Исследования цинкмарганцевых спла¬
вов алюминия с использованием комбинации сканирующего оже-микрозонда
с методикой испытаний на ударное растяжение показали, что средняя зерно¬
граничная концентрация цинка, меди и марганца может косвенно коррелиро¬
вать с плато на графике скорости коррозионного растрескивания в напряжен¬
ном состоянии [65].10.3.2. Металлы первого ряца и их сплавыПочти вся современная литература относительно поверхности посвящена
исследованиям сплавов на основе хрома, железа, кобальта, никеля и меди.
Ниже приводится обзор этих исследований.
Н.С. Макинтайр46510.3.2.1. ХромВ работе Коннера [66] исследовался процесс твердофазного окисления
хрома методами РФЭС и ЭОС. В нескольких работах [67, 68] исследовался
вопрос стабильности хромовых защитных покрытий на других металлах с ис¬
пользованием метода РФЭС; в частности, контролировалась степень окисле¬
ния хрома. Природа пассивирующих пленок, образующихся на поверхности
хрома в нейтральных и кислых растворах, исследовалась методом ЭОС в ра¬
боте [69].10.3.2.2. Железо и низколегированные сталиОкисление чистого железа на воздухе исследовалось методами РФЭС и
ЭОС в ряде работ [70, 71]. Имеются сообщения [57, 72, 73] об электрохимиче¬
ских исследованиях водной коррозии чистого железа в присутствии анионов
борной кислоты в качестве буфера. Стабильное присутствие бора во внешней
части пассивирующей пленки позволяет предположить, что внутренняя часть
оксида образуется в результате окисления металла, а внешняя - в результа¬
те осаждения субоксида железа из раствора [57]. Измерение отношения О/М
при профилировании по глубине подтверждает наличие двух оксидных струк¬
тур, однако для окончательного суждения необходимо быгь уверенным, что
под действием ионного пучка не происходит восстановление оксидов железа.
Другие ингибиторы, присутствующие в растворах, такие, как СгО2", МоО*~
и As20|“, внедряются в пассивирующие пленки в большей степени, чем бор
[74]. Эффект проникновения ионов хлора в пассивирующие оксидные пленки же¬
леза хорошо известен. Однако современный уровень развития методов ЭОС и
РФЭС не позволяет обнаружить хлор в этих пленках [75, 76]. В работе [76]
было сделано предположение, что с увеличением воздействия ионов хлора на
поверхность происходит увеличение содержания углеводородов в поверхност¬
ном слое оксида железа. Этот эффект приписывается потере поверхностью
гидроксильных групп и связанной воды лри депассивации. Кроме того, воздей¬
ствие хлора, по-видимому, приводит к увеличению площади поверхности, до¬
казательством чему является большая величина сигнала, соответствующего
О Is-энергии связи (большее количество физисорбированного газа).В работе [77] исследовалось внедрение анионов иода в деформированную
пленку из мягкой стали в зависимости от поляризационного потенциала. Изме¬
рения показали, что с изменением потенциала содержание иода в пленке меня¬
ется (рис. 10.8). В отсутствие иода пленка становится неустойчивой к корро¬
зии под напряжением (область I).Исследования методом ЭОС процесса коррозии железа в растворах нитра¬
тов показали, что наблюдаемая межкристаллитная коррозия, возможно, связа¬
на с тем, что карбидная фаза в этих растворах не пассивируется [78].
46610. Применение ЭОС и РФЭС в исследованиях коррозииРис. 10.8. Связь между образованием пассивной пленки на поверхности железа с по¬
верхностной концентрацией иода [ 77].10.3.2.3. Двойные сплавы железа и нержавеющие сталиГазофазное окисление железохромовых сплавов и нержавеющих сталей
интенсивно исследовалось методами РФЭС и ЭОС в работах [79-38]. Для
большинства температур, при которых проводились исследования, вначале
наблюдается образование тонкого слоя оксида хрома, поверх которого затем
формируется слой оксида железа. Бэр [87] исследовал окисление нержавею¬
щей стали марки 304 при температуре 800 °С в атмосфере с различным содер¬
жанием кислорода. С помощью сканирующего оже-микроскопа он показал, что
низкое парциальное давление кислорода благоприятствует начальному росту
пленки чистого оксида хрома, равномерно распределенной по зернистой по¬
верхности сплава. При высоких давлениях кислорода оксид хрома концентри¬
руется вблизи границ зерен, а смешанный оксид, как было обнаружено, рас¬
пределен по самому зерну. В другой работе [88] использовались ЭОС и другие
методы для определения состава пленки, образующейся на гранулированной
поверхности сплава Fe - 26% Сг при окислении в атмосфере, содержащей
Ю“3 мм рт. ст. 02 при 600 °С. После нескольких секунд окисления на поверх¬
ности формируется двойная пленка. По отношению интенсивностей линий спект¬
ра ЭОС O/Fe внешний слой идентифицируется как a-Fe203 [88]. Исполь¬
зуя ЭОС и другие методы, удалось установить, что нижележащий слой состо¬
ит из а-Сг203, а лежащая под этими слоями подложка обеднена хромом. По¬
лученная информация может быть полезной при разработке методов создания
защитных пленок на сплавах, являющихся частью технологии обработки дета¬
лей перед их эксплуатацией [89].
Н.С. Макинтайр467Рис. 10.9. Зависимость поверхностной концентрации О2 , 0Н“ и связанной воды на
поверхности стали от поляризационного потенциала в 1MHCL. Для сплавов
Fe - 30%С и Fe - 30%Сг - 2%Мо пассивация начинается при 0,2 эВ
(относительно насыщенного коломельного электрода, HK3J. Результаты,
представленные здесь для Fe - 30%Сг, можно сравнить с зависимостями
концентраций катионов для того же сплава, показанными на рис. 10.1, а.
Лучшая воспроизводимость аналитических результатов для железохроммо-
либденового сплава, возможно, связана с большей однородностью пассиви¬
рующего слоя [91].Пассивирование нержавеющих сталей в процессе водной коррозии и со¬
став образующихся при этом пассивирующих пленок являются одними из на¬
иболее изученных вопросов в науке о коррозии. Как ЭОС-, так и РФЭС-иссле*
дования показали, что по мере приближения к пассивирующему потенциалу
концентрация хрома в поверхностном слое Fe - Сг-сталей увеличивается
[5, 8, 30, 89-93]. По-видимому, обогащение хромом связано с селективным
растворением железа из оксида; в некоторых случаях наблюдается также
обеднение железом металла [91]. Подробно природу пассивирующей пленки ис¬
следовали Кастл и Клейтон [5], а также Асами, Хасимото и Симодаира [91].
Кастл и Клейтон обнаружили двойной слой: внутренний слой богат хромом, а
внешний содержит большое количество гидроксида или связанной воды вместе
46810. Применение ЭОС и РФЭС в исследованиях коррозиис органическими молекулами. Асами, Хасимото и Симодаира нашли, что при
пассивирующем потенциале концентрация в пленке гидроксида и связанной во¬
ды, образующих гидратированный оксигидроксид хрома, достигает максиму¬
ма (рис. 10.9).Добавление в нержавеющую сталь молибдена увеличивает коррозионную
стойкость, особенно при питтинговой атаке. Исследования методом РФЭС [9,
94, 95] помогли выяснить роль молибдена. Предполагается, что шестивалент¬
ный молибден взаимодействует с активными центрами на растворяющейся по¬
верхности, где оксигидроксид не может образоваться. Это приводит к умень¬
шению активности реакционных центров и формированию более однородного
пассивирующего слоя [9] (рис. 10.9). Аналогичный механизм пассивации пред¬
полагается и в случае железомолибденовых сплавов. В двух работах [54, 96],
в которых использовался метод РФЭС, было показано, что большая часть пас¬
сивирующего оксида представляет собой оксигидроксид железа и только в ак¬
тивных областях (т.е. в коррозионных язвах) был обнаружен молибден. В од¬
ной из этих работ [54] молибден был обнаружен в составе соединения FeMo04.
В работе [ 97] при исследовании коррозии молибденовой нержавеющей стали
в растворе Рингера в пассивирующей пленке не бы/ю обнаружено хоть сколько-
нибудь заметного количества молибдена.Реакции сплава Fe - Сг с раствором хлоридов также исследовались, при¬
чем идентификация хлора в поверхностном слое в этом случае осуществля¬
ется легче, чем в случае железа. Саниман [98], используя метод РФЭС, ис¬
следовал влияние концентрации раствора хлорида на проникновение его в по
верхность. Анализ концентрации и толщины пленки показал, что проникнове¬
ние хлорида связано с реакцией обмена, а не с реакцией присоединения. В
работе [99] исследовалась также устойчивость оксидной пленки на поверхно¬
сти нержавеющей стали к щелевой коррозии в растворе хлоридов.В работе [ 100] проводилось сравнение анодных покрытий на железонике¬
левых бинарных сплавах и на железоникелевых сплавах, содержащих молиб¬
ден и бор или фосфор и бор. В последнем случае в пассивирующей пленке на
поверхности сплава содержались оксиды бора и фосфора. По форме пика в
спектре ЭОС было экспериментально установлено, что фосфор присутствует
в виде фосфита. Предполагают, что в этой форме он ответствен за уменьше¬
ние потенциала никеля и, следовательно, за уменьшение скорости его раст¬
ворения. Авторы работы [101], используя сочетание методов ЭОС - РФЭС,
исследовали естественные оксидные пленки на поверхности сплавов
Fe — Ni — Сг _Р_В. Было показано, что пассивирующие пленки действи¬
тельно обогащены бором и фосфором, причем большая часть фосфора рас¬
пределена в слое, следующем за слоем, обогащенным оксидом хрома.Исследования методом ЭОС межкристаллитной коррозии нержавеющей
стали 304 [102] позволили установить, что многие примеси на границах зерен
Н.С. Макинтайр469играют определенную роль в увеличении восприимчивости этих границ к ата¬
ке активными частицами. Например, сера в несенсебилизированных сталях
способствует межкристаллитной коррозии в растворе нитрат-бихромата. С по¬
мощью метода ЭОС было показано, что в процессе коррозии образцов в раство¬
ре H2S04 - CuS04 происходит обеднение границ зерен хромом.10.3.2.4. Кобальт и его сплавыИсследование методом РФЭС процесса окисления кобальта во влажном
воздухе показало, что первичным продуктом реакции на поверхности металла
является Со(ОН)2 [18]. Анодное окисление чистого кобальта исследовалось
методом ЭОС [ ЮЗ]; для пассивирующей пленки были получены характерные
спектры.В работе [67] обнаружено, что коррозия кобальт-хромовых сплавов в нейт¬
ральных и кислых растворах связана с селективным растворением кобальта.
Макинтайр, Мерфи и Зетарук, используя метод РФЭС, также показали, что в
щелочных растворах при 300 °С происходит преимущественное растворение ко¬
бальта из кобальт-хромвольфрамового сплава. Проведенные одновременно ис¬
следования газофазного окисления показали, что на воздухе формируется
слой, богатый кобальтом (Со304); такая миграция кобальта наружу будет
увеличивать его преимущественное растворение в водной среде.10.3.2.5. Нинель и его сплавыОкисление поликристаллического никеля исследовалось методом ЭОС в
работах [104-106].. Мюллеру и сотр. [106] удалось обнаружить на ранней ста¬
дии окисления образование островков Ni02.Со сплавами Ni - Fe был проведен ряд исследований. Методом ЭОС анали¬
зировалась оксидированная поверхность пермаллоя (80% Ni - 20% Fe) [107],
а метод РФЭС использовался при анализе поверхности сплава Ni - 20% Fe,
оксидированного при 500 °С [108]. Богатый железом оксид, формирующийся
на начальной стадии на отожженной поверхности, в конечном счете приводил
к образованию шпинели NiFe204-Смит и Шмидт [109] исследовали процесс окисления сплава Ni — 20% Сг
при 800°С. Было обнаружено, что двойной оксид Сг203 - NiO формируется
везде, за исключением мест вблизи пересечения границ зерен. Аналогичные
результаты были получены с использованием м тода РФЭС для кратковремен¬
ного газофазного окисления сплава Alloy 600 при 500°С [6]. Кроме того, бы¬
ло обнаружено, что железо, будучи минимальной добавкой в объеме сплава,
концентрируется во внешних слоях оксидной пленки. Было показано, что парци¬
альное давление кислорода влияет на содержание в пленке оксида никеля.
47010. Применение ЭОС и РФЭС в исследованиях коррозииВ работе [НО] методом РФЭС исследовались поверхности нескольких бо¬
гатых никелем сложных сплавов, оксидированных при Ю00°С. Было обнаруже¬
но, что поверхности обогащаются ТЮ2, являющимся незначительной добавкой
во всех исследованных сплавах. Реакция при высоких температурах с Na SO_ VT 2 4приводит к окислению почти всего хрома на поверхности до Сг , чт о, очевид¬
но, ведет к ускоренной коррозии.Методом РФЭС исследовали процесс коррозии никелевого электрода, нахо¬
дящегося в контакте с твердым электролитом [27],, в кислом [111-113] и ней¬
тральном [113] растворах и в жидком фтористом водороде [114]. В двух рабо¬
тах [111, 112] с использованием метода РФЭС было показано, что пассивирую¬
щая пленка состоит из двух слоев: верхний слой образован Ni(OH)2, а сле¬
дующий за ним - из NiO. В работе [ 115] метод ЭОС использовался для изме¬
рения толщины пленки NiO в процессе растворения никеля [115], но никакой ин¬
формации о стехиометрическом составе пленки получено не было. Мак-Доугалл,
Митчел и Грехем [113] анализировали пассивирующие анодные пленки на поли-
кристаллическом никеле. Используя метод ЭОС для получения картины распре¬
деления кислорода на поверхности, они показали, что на некоторых зернах
пассивирующая пленка на одну треть тоньше, чем на других зернах. Исследо¬
вание профиля пленки по глубине обнаружило протяженную границу между ок¬
сидом и металлом. Авторы считают это доказательством того, что пленка
имеет большое количество дефектов; это играет существенную роль в про¬
цессе пассивации.В работе [116] с использованием метода РФЭС было показано, что анод¬
ное окисление сплава Ni - 25% Fe в растворе кислоты приводит к образова¬
нию пленки из NiO непосредственно на границе с металлом, а внешний слой
покрытия состоит из гидроксидов железа и никеля. В противоположность этим
результатам двойные пассивирующие пленки на сплаве Ni - Мо [117] и ха-
стеллой [31] имеют внутренний слой, состоящий из смешанного никель-молиб-денового оксида, а на внешней поверхности присутствует только оксид или
гидроксид никеля.Пленки на поверхности сплавов Alloy 600 и Alloy 800, образованные в
NaOH, исследовали Хасимото и Асами [118], используя метод РФЭС. Первич¬
ной пассивирующей пленке была приписана формула оксигидроксида хрома, а
вторичная пленка состояла из Ni(QH)2. Макинтайр, Зетарук и Оуэн [119] ис¬
следовали поверхность сплава Alloy 600 после окисления образца в условиях
водяного реактора под давлением. В условиях нормального восстановления
был обнаружен оксигидроксид эдэома. В эксперименте варьировались окисли¬
тельные свойства раствора; тем самым было показано, что в условиях силь¬
ного окисления образуется пленка из гидроксида, богатого никелем, а хромо¬
никелевые гидроксиды образуются в промежуточном случае. Профили по глу-
Н.С. Макинтайр471бине пассивирующих пленок на сплавах Alloy 600 и 800 исследовались в работе
[13] с помощью метода ЭОС. Было обнаружено, что с увеличением pH раство¬
ра относительный термодинамический избыток хрома на поверхности умень¬
шается.Водная коррозия сплава Ni - Си (66% Ni) в условиях парового котла ис¬
следовалась методом РФЭС в работе [120], где было показано, что Ni(OH)2
должен быть основной поверхностной компонентой в случае активной атаки
поверхности при 300°С. В условиях восстановления в соответствии с пред¬
сказаниями термодинамики на поверхности обнаружено небольшое количест¬
во оксида или он вообще отсутствовал.10.3.2.6. Медь и ее сплавыМедь и процесс ее окисления исследовались методам4 РФЭС и ЭОС в те¬
чение почти десяти лет [36, 121]. Как было установлено методом РФЭС в ра¬
боте [122], окисление сплава 70/30 Си - Ni приводит к образованию на по¬
верхности пленки, богатой NiO.Анодное окисление металлической меди в слабом щелочном или кислом
растворе, как было показано методом РФЭС, приводит к образованию двой¬
ной пленки; внешний слой образован Си(ОН)2, а к металлу примыкает слой из
Си20 [123].Недавно Шусмит и сотр. [24] исследовали методом РФЭС начальные стадии
роста аналогичной пленки в гидроксиде калия. Процессы формирования пленок на по¬
верхности медно-никелевых сплавов в солевых растворах исследовались методами
РФЭС [124] и ЭОС [125]. В последней работе было обнаружено образование двойной
пленки, состоящей из верхнего слоя - смешанного медно-никелевого оксида
и нижнего, более толстого слоя оксида, богатого никелем. Окисление Си -2% Be
в аммиачных растворах приводит к образованию на поверхности слоя, богатого
бериллием [126].Кастл, Эплер и Пеплоу [127] исследовали методом РФЭС химические про¬
цессы на поверхности труб конденсора из алюминиевой бронзы (медь 76%,
цинк 22%, алюминий 2%) в морской воде. На поверхности труб был обнаружен
слой с большой концентрацией магния и алюминия. Было сделано предположе¬
ние, что происходит осаждение из морской воды на поверхность либо чистого
магния, либо в комбинации с алюминием, присутствующим в сплаве. В рабо¬
те [128] методом РФЭС исследовалось осаждение Mg(OH)2 на алюминиевую
бронзу в лабораторных условиях в зависимости от pH раствора. Для рациональ¬
ного объяснения измеренной методом РФЭС толщины пленки Mg(OH)2 следует
предположить, что значение pH у поверхности больше, чем в объеме раствора.Анализ защитных органических покрытий на меди проводился несколькими
исследовательскими группами. В работах [129, 130] методом РФЭС исследова-
47210. Применение ЭОС и РФЭС в исследованиях коррозиилось взаимодействие меди с бензотриазолом; было подтверждено, что бензо-
триазол реагирует с закисью меди на поверхности. Процесс разрушительного
окисления комплекса бензотриазола с медью с образованием оксида меди ис¬
следовался в работе [131]. В работе [132] методом РФЭС исследовались реак¬
ции меркаптобензотриазола с поверхностями меди (I), которые приводят к ин¬
гибированию процессов коррозии.70.3.3. Тяжелые металлыИсследования поверхности применительно к проблеме коррозии элементов
более тяжелых, чем элементы первого ряда, не столь обширны. В связи с этим
вследствие малого количества полученных к настоящему времени результатов
трудно дать последовательный обзор исследований этих элементов.Электрохимию и процесс окисления молибдена исследовали методом
РФЭС с использованием специальной камеры с инертной атмосферой Ансель
и сотр. [26]. В работе [133] с помощью метода РФЭС было показано, что катод¬
ное разложение оксида ниобия приводит к образованию гидратированного окси¬
да с более низкой степенью окисления. Естественные оксидные пленки на по¬
верхности редкоземельноподобных элементов - церия9 иттрия и лантана - ис¬
следовались методом РФЭС [134], а на поверхности гадолиния - методом ЭОС
[135].Несколько работ было посвящено исследованию процессов окисления и
коррозии олова« Ансель и сотр. [136] методом РФЭС исследовали электрохи¬
мическое окисление олова в щелочном растворе. Было обнаружено, что в пас¬
сивной области оксида на поверхности содержится SnIV, а не Sn11, который
образуется на начальной стадии. Об исследовании методом РФЭС твердофаз¬
ного окисления олова сообщалось в работе [137]. Анализ методом РФЭС пас¬
сивирующей пленки на поверхности сплава Sn - Ni приводился в работе
[138]. Серве и сотр. [139] исследовали пассивирующие пленки хрома на поверх¬
ности олова и пришли к заключению, что отсутствие адгезии этих пленок свя¬
зано с образованием оксида олова.Оксиды на поверхности сплавов индий ~ золото и индий — свинец - золо¬
то анализировались методом РФЭС [140]. Анодное окисление золота в серной
кислоте, как было показано в [141], приводит к образованию на поверхности
пленки Au203. Окисление свинца исследовалось методом РФЭС [142]. Сандер
и сотр. [143] также методом РФЭС исследовали электрохимическое окисление
субоксида урана.10.3.4. Органические покрытияДля создания барьера между атакующими активными частицами и метал¬
лом на поверхности металлов наносятся органические защитные покрытия.
Н.С. Макинтайр473Более того, если проникновение через защитное покрытие все же происходит,
коррозийная атака будет ограничена только непосредственной областью про¬
никновения и не будет распространяться в стороны, как это имеет место при
отсутствии защитного покрытия. Потеря адгезии таких покрытий на поверх¬
ности железа и стали изучалась методом РФЭС.Проводились исследования коррозии поверхности стали, покрытой эпок¬
сидными пленками на основе сложных эфиров [144], уретана и аминов [145], в
результате нарушения адгезии в солевых растворах. Обе стороны пленки в ме¬
сте прорыва исследовались методом РФЭС и затем в дополнительных химиче¬
ских испытаниях поверхности пленки контроль осуществлялся также методом
РФЭС. Со стороны как органического слоя, так и подложки, в местах проник¬
новения присутствуют следы омыления поверхности, приводящего к образова¬
нию карбоксилата натрия. Предполагается, что этот процесс происходит бла¬
годаря присутствию гидроксильных групп, образующихся в результате корро¬
зии.Высокочувствительный метод детектирования карбоксилатов включает в
себя их "прикрепление” к серебру - иону, обладающему высокой селективно¬
стью по отношению к карбоксигруппе и большим сечением фотоэмиссии элект¬
ронов. Используя этот метод, можно детектировать карбоксилаты с поверхно¬
стной концентрацией около 1% (см. гл. 9).Исследовался также эффект использования "конверсионного" покрытия
между сталью и органическим слоем. Было обнаружено, что в местах наруше¬
ния адгезии конверсионное покрытие, фосфат цинка, сохраняет адгезию к ме¬
таллической подложке и содержит следы органики. Отсюда следует, что нару¬
шение адгезии происходит вблизи границы с металлом, но в органической
фазе.Хаммонд и сотр. [146] анализировали спектры линий С Is, N Is и О Is по¬
верхностей раздела покрытий, содержащих несколько органических слоев. Они
провели детальное измерение энергий связи и отношений интенсивностей для
ряда карбоксильных анионов и простых полимеров.Эффекты механического и химического нарушения адгезии полибутадие-
новых пленок сравнивались путем использования метода РФЭС в [147]. Было
показано, что механическое нарушение адгезии обусловлено нарушением ко¬
гезии. Химическое же нарушение адгезии связано с гидролизом сложных эфи¬
ров на границе с металлом, что подтверждает результаты более ранних работ.10.4. ЗаключениеОчевидно, что методы РФЭС и ЭОС становятся все более эффективными
в исследованиях химических процессов и определении состава пленок корро¬
зии. Особенно следует отметить, в какой степени за последние несколько лет
47410. Применение ЭОС и РФЭС в исследованиях коррозииоба метода стали действительно количественными - не только со стороны оп¬
ределения элементного состава, но часто и применительно к установлению
присутствующих химических соединений. Кроме того, мы все больше убежда¬
емся, что даже весьма неустойчивые структуры, например гидраты, можно
сохранить в вакууме спектрометра. Прогресс в этих двух направлениях поз¬
волил сделать важные открытия, касающиеся природы пассивирующих пленок.Начинает накапливаться значительное количество данных относительно
коррозии металлов и сплавов ряда железо- хром - никель; так, в некоторых
случаях в различных экспериментах получены данные, согласующиеся между
собой. Это признак зрелой области исследований!Особенно важны электрохимические эксперименты, поскольку в них име¬
ется возможность существенно изменять условия коррозии в пределах прием¬
лемого интервала времени. Многие ситуации нельзя смоделировать в ячейке.В связи с этим предполагается, что аналитические данные о поверхности по¬
зволят связать результаты, полученные в электрохимических экспериментах,
с результатами исследований процесса коррозии в условиях "разомкнутой це¬
пи", сделав, таким образом, исследование коррозии в электрохимической ячей¬
ке более информативным по отношению к коррозии в поле (в естественных ус¬
ловиях).По мнению автора этой главы, метод РФЭС в настоящее время дает не¬
сколько большую информацию, чем метод ЭОС. Это связано с более мягкими
условиями возбуждения, а также с тем, что для РФЭС в настоящее время луч¬
ше известны химические сдвиги. Однако коррозия является существенно мик¬
роскопическим процессом, поэтому особое внимание следует вновь уделить
методам анализа поверхности с высоким пространственным разрешением, та¬
ким, как ЭОС, поскольку возможности метода РФЭС в этом плане исчерпаны.В связи с этим особую важность могут приобрести попытки понять эффекты,
связанные с воздействием на образец электронного пучка в методе ЭОС, а
также исследования оже-химических сдвигов. Микроскопические исследования
поверхности будут иметь особое значение для понимания роли органических и
неорганических ингибирующих слоев, а также процессов щелевой и питтинго-
вой коррозии.Можно надеяться, что прогресс в проведенных за последние пять лет ис¬
следованиях, связанный с использованием методов ЭОС и РФЭС, в будущем
приведет к заслуженному более широкому применению этих методов в экспе*
риментах по коррозии.БлагодарностиАвтор выражает благодарность мисс Т. Чен за квалифицированную по¬
мощь в подготовке настоящей главы.
Н.С. Макинтайр475^итература1. Joshi A., Investigation of passivity, corrosion and stress corrosion cracking
phenomena by AES and ESCA, in Corrosion 77, Paper 16, National Associa¬
tion of Corrosion Engineers, Houston, Texas (1977).2. Castle Surf. Sci., 68, 583 (1977).3. Castle J.E., Application of XPS analysis to research into the causes of cor¬
rosion, in: Applied Surface Analysis (Eds. T.L. Bair and L.E. Davis), ASTM
STP 699, p. 182, American Society for Testing and Materials, Philadelphia,
Pennsylvania (1980).4. Seah M.P., Dench W.A.? Surf. Interface Anal., 1, 2 (1979).5. Castle Clayton C.R., Corrosion Sci., 17, 7 (1977).6. McIntyre N.S., Owen D.G., Zetaruk D.C., Appl. Surf. Sci., 2, 55 (1978).7. Asami K., Hashimoto КShimodaira S., J. Jpn Inst. Metals, 40, 438 (1976).8. Asami КHashimoto K;. Shimodaira S., Corrosion Sci., 17, 713 (1977).9. Hashimoto K., Asami K., Teramoto K., Corrosion Sci., 19, 3 (1979).10. Asami K., Hashimoto K.9 Corrosion Sci., 17., 559 (1977).11. Hashimoto КNaka М., Asami K., Masamoto Т., Corrosion Sci., 19, 165 (1979).12. Pons F., Le Hericy Longeron J.PSurf. Sci., 69, 547 (1977); 69, 565
(1977).13. Seo М., Sato N; Corrosion, 36, 334 (1980).14. Mitchell D.f\, Appl. Surf. Sci., 9, 131 (1981).15. McIntyre Applications of surface analysis in the nuclear industry, in:
Industrial Applications of Surface Analysis (Eds. Powell C.J., Casper L.),ACS Symposium Series No. 99, p. 345, American Chemical Society, Washing¬
ton, DC (1982).16. Baer D»H*f Appl. Surf. Sci., 7, 69 (1981).17. Kim K.S.. Baitinger W.£., Amy /.Jf., Winograd N., J. Elect. Spectrosc., 5,351 (1975).18. Chuang T.J., Brundle С.Я., Wandelt К•, Thin Solid Films, 63, 19 (1978).19. McIntyre N.S., Zetaruk D.G., Anal. Chem., 49, 1521 (1977).20. Mitchell D.F., Sproule G.L, Graham M»J., J. Vac. Sci. Technol., 18, 690
(1981).21. McIntyre N*St; Murphy E.V., Zetaruk D.G., Surf. Interface Anal., 2, 151 (1979).22. Walls J.M., Hall D.D., Sykes D.E., Surf. Interface Anal., 1, 204 (1979).23. Schmidt C.J., Lenzo P.F., Spencer E.Q*, J. Appl. Phys., 46, 4080 (1975).24. McIntyre N.S., Sunder S., Shoesmith D.W., Stanchell F.W., J. Vac. Sci.Technol., 18, 714 (1981).25. Revie R.W., Baker B.C., Bockris L’M.» Surf. Sci., 52, 664 (1975).26. Ansell R.O., Dickinson Т., Povey A.R., Sherwood M.A., J. Electroanal.Chem., 98, 69 (1979).
47610. Применение ЭОС и РФЭС в исследованиях коррозии27. Yang C.Y., O’Grady W.E** J. Vac. Sci. Technol., 20. 925 (1982).28. Kudo К*, Shibata F*,t Okamoto G*, Sato N., Corrosion Sci., 18, 809 (1978).29. Castle J*E*, Proc. of the Conference on Metal Coatings, p. 435, Lehigh Uni»
versity (1978).30. Okamoto G*, Corrosion Sci., 13, 471 (1973).31. Burstein G*T; Materials Science and Engineering, 42, 207 (1980).32. f!zklarska-Smialowska Z., Viefhaus H*, Hanik-Czachor М., Corrosion Sci.,16, 649 П976).33. Smith T*, Surf. Sci., 55, 601 (1976).34. Wagner C.D. et aL, Muilenberg G*E*, Handbook of X-ray Photoelectron
Spectroscopy, Physical Electronics Division, Perkin-Elmer Corporation, Eden
Prairie, Minnesota (1979).35. Johnson 0.» Chem. Scripta, 8, 162 (1975).36. Schoen G, J. Electron. Spectrosc., 1, 377 (1972).37. McIntyre N*S*, Cook M*G», Anal. Chem., 47, 2208 (1975).38. Norton P.R* J. Catalysis, 36, 211 (1975).39. Matsushima T*, Almy D*B*, White J*M*, Surf. Sci,, 67, 89 (1977).40. Ramqvist L* eU ah, J. Phys. Chem. Solids, 30, 1835 (1969).41. Franzen J*F*, Umana M*X*, McCreary J*R*, Thom R.J* J. Solid State Chem.
18, 363 (1976).42. Solomon J*S*, Baun W*L; Surf. Sci., 51, 228 (1975).43. Knotek M*L; Houston J*, Phys. Rev., В15, 4580 (1977).44. Sawatsky G*A*, Post D*, Phys. Rev., В 20, 1546 (1979).45. Fiermans L*yyennik ]*, Surf. Sci., 35, 42 (1973).46. F./. Szalkowski, Somorjai G.A., J. Chem. Phys., 61, 2064 (1974).47. Allen G.Q., Curtis M.J., Hooper A.J., Tucker Р.Ц., J. Chem. Soc. (Dalton),
1973, 1675 (1973).48. Allen G.C., Tucker P.ty., Inorg. Chim. Acta, 16, 41 (1976).49. Ikemoto /. et aL, J. Solid State Chem., 17, 425 (1976).50. Oku М., Hirokawa К., Ikeda S., J. Electron Spectrosc., 7, 465 (1975).51. Aoki A•, Jpn J. Appl. Phys., 15, 305 (1976).52. Allen G*C«, Curtis M.T., Hooper A.J., Tucker P.M., J. Chem. Soc. (Dalton)
1976, 1526 (1976).53. Brundle C.R., Chuang T.J., Wandelt КSurf. Sci., 68, 459 (1977).54. Stout D.A., Lumsden J.B»» Staehle R.W., Corrosion, 35, 141 (1979).55. Binder H., Z. fur Naturforsch., В 28, 256 (1973).56. Ekelund S., Leygraf C., Surf. Sci., 40, 179 (1973).57. Seo М., Sato MLumsden J.B; Staehle R.W., Corrosion Sci., 17, 209 (1977).58. Patterson T.A., Carver J.C., Leyden D.£., Hercules D.M*, J. Phys. Chem.,
80, 1702 (1976).
Н.С. Макинтайр47759. Gaarenstroom S.W., Winograd N., J. Chem. Phys., 67, 3500 (1977).60. Schoen G., J. Electron Spectrosc., 7, 377 (1972).61. Zehner D.M», Barbulesco М., Jenkins L.H., Surf. Sci., 34, 385 (1973).62. Matsuzawa S., Baba N., Tajima S., Electrochim, Acta, 24, 1199 (1979).63. Barr T.U J. Vac. Sci. Technol., 14, 660 (1977).64. Treverton J.A., Davies N.С» Electrochim. Acta, 25, 1571 (1980).65. Shastry C.R., Levy М., Joshi A., Corrosion Sci., 21, 673 (1981).66. Conner G.ft., J. Vac. Sci. Technol., 15, 843 (1978).67. Storp S., Holm R., Surf. Sci., 10, 68 (1977).68. Baer D.R., Use of surface analytical techniques to examine metal corrosion
problems, in: ACS Symposium Series, Industrial Application of Surface
Analysis, American Chemical Society, Washington, DC (1982).69. Seo М., Saito R., Sato N., J. Electrochem. Soc., 127, 1909 (1980).70. Brundle С.Ц., Surf. Sci., 66, 581 (1977).71. Grimzewski J.K., Surf. Sci., 62, 386 (1977).72. Revie R.W., Baker B.G., Bokris O'M., J. Electrochem. Soc., 122, 1460 (1975).73. Davies й.Ц., Burstein G.T., Corrosion, 36, 416 (1980).74. Lumsden J.B., Szklorska-Smialowska ZStaehle R.W., Corrosion, 34, 169
(1977).75. M. da Cunha Belo, Rondot В., Pons F., Lanyeron J.P., J. Electrochem. Soc.,
124, 1317 (1977).76o Cocke D.L*, Nilsson Pof Murphy O.J., Bokris J. Q*M., Surf. Interface Anal.,4, 94 (1982).77. Asami K., Totsuka NTakano М., Corrosion, 35, 208 (1979).78. Tauber G*, Grabke H.J., Corrosion Sci., 19, 793 (1979).79. Leygraf СHutqvist GEkelund SSurf,, Sci., 51, 409 (197).80. Coad J.Pb, Cunningham J.G., J. Electron Spectrosc., 3, 435 (1974).81. Betz G, Wehner G.%, Toth L., Joshi A., J. App. Phys., 45, 5312 (1974).82. Ole fjord I., Corrosion Sci., 15, 687 (1975) >83. Leygraf С., Hultqvist G., Surf. Sci., 61, 69 (1976).84. Wild ft./i., Corrosion Sci., 17, 87 (1977).85. Smith Т., Crane L.Vf., Oxid. Met., 10, >135 (1976).86. Driscoll T.J.» Needham P.$.9 Oxid. Met., 13, 283 (1979).87. Baer D.R., Appl, Surf. Scio, 7, 69 (1981).88. Jensen C.P., Mitchell D.F., Graham M.J., Corrosion Sci. (в печати).89. Chattopodhyaу B.9 Wood G.C., J. Electrochem. Soc., 117, 1163 (1970).90* Olefjord I., Fischmeister H.9 Corrosion Sci., 16, 387 (1976).91. Asami К.» Hashimoto К., Shimodaira S., Corrosion Sci., 16, 387 (1976)*92., Asami K., Hashimoto K., Shimodaira S., Corrosion Sci ., 18, 125 (1978).
47810. Применение ЭОС и РФЭС в исследованиях коррозии93. Leygraf С. tt ah9 Corrosion Sci„, 19, 343 (1979)-94. Sugimoto K.9 Sawada YCorrosion, 32, 347 (1976).95. Hashimoto K*9 Asami КCorrosion Sci., 19, 251 (1979).96. Hashimoto K.9 Naha M*9 Asami K.9 Masumoto Г., Corrosion Sci., 19, 165
(1979).97 , Cahoon J*R*9 Bandy R.9j Corrosion, 38, 299 (1982).98. Saniman MSc Thesis, University of Surrey, Guildford, Surrey (1977).99" Hultqvist G., Leygraf C*9 J. Vac,, Sci„ Technol., 17, 85 (1980).100., Burstein G*J.9 Corrosion, 37, 549 (1981)o101. Baer D./?., Petersen D.4*> Pederson L.R*9 Thomas J. Vac. Sci.
Technol., 20, 957 (1982).102. Joshi AStein D.F., Corrosion, 28, 321 (1972).103. Davies D.#., Burstein G.T,, Corrosion Sci., 20, 989 (1980).104. Kulpa S.H., Frankenthal R.P., J. Electrochem. Soc., 124, 1588 (1977).105. Sharma S.P., J. Electrochem. Soc., 125, 2005 (1978).106. Miiller FL-H., Beckmann P., Schemmer М.,Benninghoven A.f Surf. Sci., 80, 325( 1979).107. Lee W.Y>, Eldridge J. Electrochem. Soc., 124, 1747 (1977).108. Olefjord L, Marcus P., Surf. Interfacial Anal., 4, 23 (1982).109. Smith K.L., Schmidt L.Q., J. Vac. Sci. Technol., 20, 364 (1982).110. Smith S*R. et aL, Oxidation of Metals, 14, 415 (1980).111. Dickenson Т., Povey A.F., Sherwood M.A*, J. Chem. Soc., Faraday
Trans. I, 73, 327 (1977).112. Marcus P., Oudar Olefjord /., J. Microsc. Spectr. Electron, 4, 63
(1979).113. MacDougall B., Mitchell D.G., Graham M.J., Corrosion, 38, 85 (1982).114. Watanabe N>, Haruta MElectrochim. Acta, 25, 461 (1980).115. Datta М., Mathieu H.J., Landolt D., Electrochim. Acta, 24, 843 (1979).116. Olefjord /., Marcus P., Surf. Interface Anal., 4, 29 (1982).117. Burstein G.T., Hoar T.P., Corrosion Sci., 17, 939 (1977).118. Hashimoto К•, Asami K., Corrosion Sci., 19, 427 (1979).119. McIntyre N.S., Zetaruk D.G., Owen DJ. Electrohem. Soc., 126, 750 (1979).120. McIntyre N*$., Rummery T.$., Cook M.G., Owen D., J. Electrochem. Soc.,123, 1164 (1976).121. Robert T.f Bartel М., Offergeld G., Surf. Sci., 33, 128 (1972).122. Castle J»%*, Nature Physical Sciences, 234, 93 (1971)»123. Strehblow H*mfl, Titze B., Electrochim. Acta, 25, 839 (1980).124. Hulett L.D., Bacarella A*L*, Lidonnici L., Griess J.CJ. Electron
Spectrosc., 1, 169 (1972).125. McGuire G.£. et al., J. Electrochem. Soc., 125, 1801 (1978).126. Newman R.C., Brustein G.T., Corrosion Sci., 20, 375 (1980).
Н.С. Макинтайр479127. Castle J.E, Epler D.C„ Peplow D*B,, Corrosion Sci., 19, 457 (1979).128. Castle /.E., Tanner°Tremain R., Surf. Interface Anal. 1, 49 (1979).129. Roberts R.F., J. Electron. Spectroscop., 4, 273 (1974).130. Fox P.G; Lewis GBoden P.]., Corrosion Sci,, 19, 457 (1979).131. Chadwick D., Hashami Т., Corrosion Sci., 18, 39 (1978).132. Ohsawa M„ Suetaka W., Corrosion Sci., 19, 709 (1979).133. Sugimoto КBelanger G., Piron D.L., J. Electrochem. Soc., 126, 535 (1979).134. Barr T.L*, in: Quantitative Surface Analysis of Materials (ed. N.S.,McIntyre),
ASTM STP 643, p. 83, American Society for Testing and Materials, Phila¬
delphia, Pensylvania (1978).135. Bevolo A,J., Beaudry B.J.- Gschneidner K.A., J. Electrochem. Soc., 127,
2556 (1980).136. Ansell R*Q; Dickinson T., Povey A.F., Sherwood M.A., J, Electrochem.Soc., 124, 1360(1977).137. Lau C»L*, Wertheim G.K., J. Vac. Sci. Technol., 15, 622 (1978).138. Thomas J.H.> Sharma S.P., J. Vac. Sci. Technol., 14, 1168 (1977).139. Servais J.P., Lempereur Renaud LLeroy V., Br. Corros. J., 14, 126
(1979).140. Baker /.Ajf., Johnson R.W., Poliak R.A., J. Vac. Sci. Technol., 16, 1534
(1979).141. Dickinson Т., Povey Л.Е., Sherwood PM; J. Chem. Soc., Faraday Trans.I,
71, 298 (1975).142. Hewitt R.W., Winograd NSurf. Sci., 78, 1 (1978).143. Sunder S. et ah, J. Electroanal. Chem., 130, 163 (1981).144. Hammond /.5., Holubka /.Vj., Dickie R.A., J, Coatings Technol., 51,655 (1979).145. Holubka J.W., Hammond J,$., DeVries Dickie Я.Д., J. Coatings
Technol., 52, 63 (1980).146. Hammond /.S., Holubka J.W., DeVries J.E., Dickie R,A., Corrosion Sci.,21, 239 (1981).147. Dickie R.AHammond J.S., Holubka J.W., Ind. Eng. Chem., Prod. Res.
and Dev., 20, 339 (1981).
Приложение 1
КАЛИБРОВКА СПЕКТРОМЕТРОВМ.Т. Энтони 1>П.1.1. Калибровка РФЭС-установокТочная калибровка рентгеновских фотоэлектронных спектрометров явля¬
лась важной задачей спектроскопистов в течение последних десяти лет. Воз-
можнрсти достоверной интерпретации спектров, полученных на различных
установках, без точно определенной спектральной шкалы энергий сильно огра¬
ничены. Некоторые исследователи [1-9] сообщали о калибровке энергии,
приводя таблицы данных для меди, серебра и золота. Эти элементы имеют то
преимущество, что они легко очищаются, химически инертны, а также явля¬
ются хорошими проводниками» С целью калибровки можно с максимальной
точностью определить энергии связи, если принять за нуль энергии уровень
Ферми проводящих образцов, так как это позволяет избежать ошибок, возни¬
кающих в связи с различием работ выхода спектрометров. </-зоны палладия и
никеля дают достаточно интенсивные и резкие границы Ферми, что позволяет
точно определить этот нуль. Литературные калибровочные значения энергий
связи меди, серебра и золота, определенные таким способом, представлены
в табл. П. 1.1. Видно, что данные, приведенные в этой таблице, различаются
на величину порядка 0,3 эВ, которая, как это ни удивительно, с годами не
уменьшается.Комитет ASTM Е-42 организовал круговое сличение (a round-robin) [10] с
целью опубликовать энергии связи, полученные на спектрометрах различных
типов. Эго пролило свет на величину ошибок, возникающих яри калибровке энер¬
гий. Анализ с помощью графиков Иодена позволяет отделить вклады случайных
ошибок, сдвига нуля и погрешности отсчета напряжений. Наиболее существен¬
ная погрешность порядка 0,3 эВ возникает при определении нулевой точки.
Случайная ошибка порядка 0,1 эВ демонстрирует воспроизводимость измере¬
ний. Наконец, отсчет напряжения в спектрометрах приводит к ошибкам, рав¬
ным приблизительно 5*10“4, или 0,5 эВ, при энергии связи 1000 эВ.Т. Anthony, Division of Materials Applications, National Physical Laboratory,
Teddington, Middlesex, UK.
М.Т. Энтони481Таблица П.1.1. Калибровочные энергии связи РФЭС (эВ) по
литературным даннымШон, Йохансон Асами, Рихтер и1972 [2] и др., 1976 [4] Пеплин-
1973 [3] ски,1978 [5]СиЗр 75,2 ± ,1Au4f7/284,083,8 ±,284,0784,0Ag3of5/2368,2368,2 ±,2368,23Cu2р3/2932,2 ±,1932,8 ±,2932,53932,7CuLMM, КЭ 919,0 ±,1918,3 ± ,2918,65918,35ЭС 567,6 ±,1568,35 ±,2567,96568,25Отн. УФPdPdPdPdСпектро¬
метр AEIES100 Магнитный AEIES200 AEIES200Вагнер, Пауэл, Берд и Фагл и ЛибаглГейл и Эриксон Свифт, Мортен- и др.,Реймонд, и Мейди, 1980 [7] сон, 1981 [9]1978 [6] 1979 [10] 1980 [8]СиЗр75,175,175,13Аи4^7/283,884,083,98 ± ,02 83,784,0Ag3d5/2367,9368,21 ± ,03367,9368,2Cu2p3/2932,4932,6932,66 ± ,06932,5932,57CuLMM, КЭ918,6918,7918,64 ± ,04ЭС567,9567,9567,97 ± ,04Отн УФАи4/?/2PdСпектро¬метрVarianIEE-15ОбзорЕ-42VGAEIES200B ESCA3MkHP
I 5950AС учетом как предыдущих литературных данных [1-9], так и данных,
приведенных в обзоре комитета ASTM [10], стало ясно, что точная калибров¬
ка энергий будет возможна только в том случае, если все измерения в про¬
цессе калибровки полностью прослеживаются. Работа, проделанная в Нацио¬
нальной физической лаборатории (НФЛ) [ 11] на спектрометре ESC АЗ Мк И
фирмы VG Scientific, заставила обратить особое внимание на следующие мо-
482Приложение 1. Калибровка спектрометровменты: а) воспроизводимость калибровки, б) корректное определение точки,
соответствующей нулевой энергии связи, и в) точное определение шкалы энер¬
гии. Эта работа полностью выявила влияние рабочих параметров на воспроиз¬
водимость и, кроме того, позволила сделать вывод, что для нулевой точки
лучше всего подходит край уровня Ферми в никеле при использовании в качест¬
ве источника излучения ЩКа. Определение шкалы энергии было получено пу¬
тем предварительной проверки тождественности энергии и сканируемого напря¬
жения с последующим максимально точным измерением сканируемого напря¬
жения. Измерительная цепь калибровалась по стандартному элементу, кото¬
рый сам был откалиброван по эталону вольта НФЛ. Таким образом, шкала
энергии НФЛ-спектрометра полностью просдеживается до эталона вольта. Для
энергий электронов в диапазоне 0 - 1550 эВ была достигнута точность изме¬
рений порядка 2 *10~б с суммарной погрешностью порядка 11 • 10 “б.Энергии связи меди, серебра и золота относительно уровня Ферми нике¬
ля были измерены на НФЛ-спектрометре и представляют собой впервые полу¬
ченные по полностью прослеживаемой методике стандартные энергии связи.
Энергии связи для излучения как А1 Ка, так и MgKa представлены в
табл. П.1.2 [ 11].Сравнение предыдущих литературных данных из табл. П. 1.1. с табличными
данными НФЛ показывает, что недавно выполненная тщательная работа Бер¬
да и Свифта [7], в которой приводится самая низкая погрешность из всех
указанных в литературе, дает очень близкие значения. Другие сопоставления
внутри лаборатории [11] показывают, что при использовании правильной ме-Таблица П.1.2. РФЭС стандартные энергии связи
(эВ) (за нуль отсчета принят край уровня Ферми
Ni)A\Ka Mg KaСиЗр75,14 ± 0,0275,13 ± 0,02Аи4'7Й83,98 ± 0,0284,00 ± 0,01АдЗо^368,27 ± 0,02368,29 ± 0,01Си L3MM567,97 ± 0,02334,95 ± 0,01Си2рж932,67 ± 0,02932,67 ± 0,02Ад MtNN1128,79 =ь 0,02895,76 ± 0,02Примечания.1. AIKa-MgKa = 233,02 эВ.2. ЭС Аи4Л|^AlqKa занижена из-за вклада от хвоста линии
Аи4 ^5/2-3 ЭС Ад3^5 + 2МдКа увеличена из-за присутствия рентге¬
новского сателлита АдЗс^.
М.Т. Энтони483тодологии калибровка с некоторыми усилиями воспроизводима с точностью
± 0,02 эВ, а с точностью ± 0,05 эВ достигается довольно легко.П.1.2. Калибровка ЭОС-установокПодавляющее большинство электронных спектрометров, используемых в
настоящее время в электронной оже-спектроскопии, представляют собой ана¬
лизаторы типа цилиндрическое зеркало (АЦЗ) в отличие от анализаторов ти¬
па сферического сектора (ССА), которые обычно используются в РФЭС-уста-
новках высокого разрешения. Более низкое разрешение, достигаемое при
использовании одиночного АЦЗ-анализатора, наряду с трудностью точной
установки образца ограничивает точность калибровки энергии по сравнению с
точностью такой калибровки в РФЭС. Тем не менее для получения методом
ЭОС информации о химическом состоянии необходима точная калибровка
энергетической шкалы.В отличие от РФЭС, когда кинетическая энергия Е отсчитывается от
уровня Ферми, в ЭОС положению пиков сопоставляется кинетическая энергия
Ек, отсчитываемая от уровня вакуума. В результате абсолютная шкала энер¬
гий чувствительна к разности работ выхода спектрометров, хотя разности
энергий различаются не так сильно. Кроме того, для АЦЗ-установок критич¬
ным является положение образца в отличие от спектрометров со сферичес¬
ким анализатором, используемых в РФЭС-установках. Испытания в НФЛ с
использованием АЦЗ-анализаторов производства фирмы Varian показали, что
в случае пика Cu L^MM положение медного образца необходимо фиксировать
с точностью 15 мкм для получения экспериментальной воспроизводимости
0,1 эВ. Этот эффект возникает из-За фокусирующих свойств АЦЗ-анализато¬
ров, для которых приращение Д/ фокального расстояния I анализатора про¬
является в сдвиге Д£ энергии Еа Как правило, I имеет величину порядка
150 мм, вследствие чего указанная точность не может быть достигнута.Комитет ASTM Е-42, проведя недавнее круговое сличение, представил
положение оже-пиков для меди и золота, используемых в качестве металлов
сравнения, пики этих металлов регистрировались на самых разнообразных
установках. Энергии пиков имеют значительный разброс относительно сред¬
него значения, который варьируется от 1,9 эВ для низкоэнергетичных пиков
до 7,4 эВ для высокоэнергетичного перехода М N /V. _ в золоте* Явное раз-5 6,7 6* •личие этих величин возникает вследствие проблем калибровки, что приводит
к ненадежности информации о химическом состоянии.Поскольку большинство электронных оже-спектров регистрируются в
виде производных (d{ En(E)\/dE) с модуляцией напряжения, прикладываемого
к анализатору, то энергиям пиков обычно ставится в соответствие положение
484Приложение 1. Калибровка спектрометровотрицательного смещенного пика, расположенного в высокоэнергетичной
части. К сожалению, энергии пиков, определенные таким способом, сильно
зависят от амплитуды модуляции, что можно видеть из рис. П0Ы для слу¬
чая упругого пика с энергией 100 эВ, а также зависят от разрешения анали¬
затора, Можно было бы установить точную калибровку для пиков меди, се¬
ребра и золота при заданной модуляции при разных значениях разрешения,
однако для большинства анализаторов влияние реальной модуляции на энер¬
гетический спектр не известно с достаточной точностью. Во многих анали¬
заторах модуляция подается в фазе и строго пропорциональна приложенно¬
му постоянному напряжению не для всех электродов. Этот вопрос подробно
обсуждается в работе Сиха (15], который показал, почему в этом случае
трудно осуществить калибровку модуляции эффективной энергии анализатора.
Чаще всего эта проблема возникает в том случае, когда РФЭС-анализатор
используется для ЭОС0 Испытания на двух АЦЗ фирмы Varian показали, что
эффективная модуляция энергии спектрометра на 20 ± 1% ниже, чем пред¬
сказываемая по величине произведения измеренной модуляции напряжения
зеркального электрода и постоянной спектрометра. В дополнение к этомуРис. П.1.1. Упругий пик с энергией
100 эВ. Дифференциаль¬
ный режим регистрации
{d\En{E)\/E) для моду¬
ляции с максимальным
размахом 1, 2, 5 и 10 В
и прямой Еп(Е) режим.
М.Т. Энтони485эффекту, обусловленному конструкцией анализатора, необходимо обеспечить
калибровочную стабильность генератора напряжения. Эти проблемы являют¬
ся общими для большинства приборов ЭОС, а не присущи какой-то конкретной
модели.Во избежание трудностей, связанных с неточностью модуляции и необ¬
ходимостью корректировок разрешения анализатора, можно провести замену
положения каждого пика в дифференциальном режиме энергией, при которой име¬
ет место пересечение линии нулевого сигнала между положительным и отрицатель¬
ным выбросами. Эта точка пересечения нечувствительна к использо¬
ванным мэдуляции и разрешению анализатора и поэтому может быть опреде¬
лена с высокой точностью. На рис. П .1.1 показан ряд различных мо¬
дуляций для упругого пика 100 эВ и продемонстрировано, что точка
пересечения нулевого уровня смещается менее чем на 1% сдвига, наблюдаемого
для обычных отрицательных пиков при амплитуде модуляции до 2В. Для малых моду¬
ляций точка пересечения нулевого уровня соответствует положениям пиков, реги¬
стрируемым в прямом £п(£)-спектре (показан в правом углу на рис. П.1.1). В дей¬
ствительности для достижения наивысшей возможной точности калибровки энергии
рекомендуется использовать этот Еп(Е) -режим, так как только в этом
случае энергия пика полностью не зависит от модуляции и связанных с ней
ошибок, влияющих на ее величину» В таблв Пв103 приведены положения пиков
Еп(Е) для калибровочных металлов - меди, серебра и золота - при исполь¬
зовании электронного пучка 5 кВ„ Положения этих пиков те же, что и поло¬
жения точек пересечения с нулевым уровнем в дифференциальном режиме при
малой амплитуде модуляции. Приведенные данные получены в НФЛ на спектро¬
метре ESCA3 фирмы VG Scientific, модифицированном для относительныхТаблица П.1.3. Кинетические энергии пиков оже-
электронов (эВ)Ек ЕКСим2>3м4>5м4р559,2 ± 0,163,5 ± 0,1АиА/6704504568,2 ± 0,172,5 ± 0,1А9М4А/45А/45353,6 ± 0,1357,9 ± 0,10uL3M4,SMA,5914,4 ± 0,1918,7 ± 0,1Примечания: 1. Энергии пиков, измеренные в Еп(Е)-режиме,
соответствуют точкам пересечения с нулевым уровнем в диффе¬
ренциальном режиме при малой амплитуде модуляции. 2. Кинетиче¬
ские энергии содержат соответствующие релятивистские поправ¬
ки, Ек — кинетическая энергия относительно уровня вакуума, Ец —
кинетическая энергия относительно уровня Ферми
486Приложение 1. Калибровка спектрометровизмерений, при которых его энергетическая шкала прослеживается до этало¬
на Вольта [11]. Таким образом, положения пиков точно связаны с первичным
стандартом, однако с учетом того, что кинетические энергии, отсчитываемые
от уровня вакуума (как это обычно принято), зависят от "работы выхода”
спектрометра, в табл. П.ЬЗ добавлен последний столбец, содержащий значе¬
ния кинетических энергий относительно уровня Ферми» Еще один момент, ко¬
торый следует отметить, состоит в том, что при изменении энергии в откло¬
няющем поле следует учитывать эффективную массу электрона, а не его мас¬
су покоя, т. е. в этом случае возникают релятивистские эффекты, при кото¬
рых истинная энергия электрона Е^ связана с энергией анализатора исполь¬
зуемой в низкоэнергетичном приближении, следующими соотношениями [ 16,17]:где R - отношение между кинетической энергией электрона в поле сферичес¬
кого дефлектора и Таким образом, АЦЗ-анализатор, откалиброванный по
истинному значению пика золота при 2020 эВ, будет, если предположить
линейность энергетической шкалы, давать для пика меди 914,4 эВ значение
энергии на 0,43 эВ больше, т. е„ 914,8 э„ отличие от РФЭС использование табл. П0103 для калибровки ЭОС-анали-
заторов не всегда является тривиальным. На РФЭС-установках, на которых
также есть возможность для ЭОС, спектрометр уже тщательно прокалибро¬
ван методом РФЭС» Для анализаторов, применяемых в РФЭС и используемых
для ЭОС, эту таблицу можно непосредственно использовать и без наличия
РФЭС, так как положение образца не критично. Однако в установках, основан¬
ных на АЦЗ-анализаторах, в случае, когда положение образца не воспроизво¬
дится с точностью ±15 мкм, необходимо привлекать дополнительную методи¬
ку. Большинство АЦЗ-анализаторов имеет стандартную энергию пучка 2000 эВ,
так что положение образца можно подстраивать до тех пор, пока на энергети¬
ческой шкале не появится пересечение нулевого уровня для упругого пика при
2000 эВ0 Как правило, для стабилизации прибор необходимо выдержать при
такой установке образца в течение порядка 10 мин, однако даже после этого
энергия, вообще говоря, не будет равна точно 2000 эВ» Спектрометр необходи¬
мо настроить по меди или золоту и аккуратно смещать образец до тех пор,
пока разности энергий пиков, измеряемых цифровым вольтметром, не будут
М.Т. Энтони487соответствовать значениям, указанным в табл. П. 1.3. Образец теперь распо¬
ложен правильно и можно тщательно определить истинное значение номиналь¬
ной (2000 эВ) энергии пучка» Эго значение энергии затем используется при
всех последующих установках образца, и прибор калибруется согласно данным
табло ПЛ.З.Литература1. Siegbahn К., Nordling С., Fahlman А. et al., ESCA-Atomic Molecular and
Solid State Structure Studied by Means of Electron Spectroscopy, Almqvist,
Uppsala, 1967.2. Schon G., J. Electron Spectrosc., 1, 377 (1972).3. Johansson G., Hedman /., Berndtsson A., et al., J. Electron Spectrosc., 2,295 (1973).4. Asami K., J. Electron Spectrosc., 9, 469 (1976).5. Richter K., Peplinski B., J. Electron Spectrosc., 13, 69 (1978).6. Wagner C.D., Gale L.H., Raymond R.H., Anal. Chem., 51, 466 (1979).7. Bird R.J., Swift P., J. Electron Spectrosc., 21, 277 (1980).8. Fuggle J.C., Martensson /V., J. Electron Spectrosc., 21, 275 (1980).9. Lebugle A., Axelsson U., Nyholm R., Martensson N., Phys. Scr., 23, 825
(1981).10. Powell C.J., Erickson N.E., Madey T.E., J. Electron Spectrosc., 17, 361
(1979).11. Anthony M.T., Seah M.P. (в печати),12. Seah M.P., Anthony М.Т. (в печати).13. Seah M.P., Anthony М.Т. (в печати).14. Powell C.J., Erickson N,E., Madey T.E., J. Electron Spectrosc., 25, 87 (1982).15. Seah M.P., Surf. Interface Anal., 1, 91 (1979).16. Keski-RahkonenO., Krause M.O., J. Electron Spectrosc., 13, 107 (1978).17. Keski-Rahkonen 0., J. Electron Spectrosc., 13, 113 (1978).
Приложение 2
МЕТОДЫ УЧЕТА СТАТИЧЕСКОЙ ЗАРЯДКИ77. Свифт, Д. Шатлуорс,1)М. П. Сихг)П.2.1. ВведениеУчет статической зарядки представляет собой проблему, которая обычно
возникает при интерпретации РФЭС-спектров* Статическая зарядка появляет¬
ся как следствие создаваемого на поверхности непроводящих образцов поло¬
жительного заряда, когда атомы теряют электроны в процессе фотоэмиссии.
Этот положительный заряд создает тормозящее поле перед образцом, в резуль¬
тате чего фотоэлектроны имеют кинетическую энергию более низкую, чем ки¬
нетическая энергия, получаемая из простого соотношениягде hv - энергия фотона данного рентгеновского источника, - энергия
связи соответствующего внутреннего уровня, а Е отсчитывается от уровня
Ферми спектрометра. Проводники, находящиеся в электрическом контакте
со спектрометром, не испытывают этих эффектов зарядки, так как фотоэлек¬
троны в этом случае замещаются электронами, проходящими через образец.
Однако для электрически изолированных проводников и диэлектриков скорость
оттока фотоэлектронов больше скорости, с которой они замещаются в образце,
и в результате происходит зарядка поверхности.Как правило, в спектрометрах с ахроматическим рентгеновским источни¬
ком этот заряд частично нейтрализуется фоновыми электронами низкой энер
гии (т. е. < 5 зВ), которые возникают при облучении рентгеновским тормоз¬
ным излучением окна рентгеновской пушки и внутренних частей установки.
Если поверхность заряжена положительно, то эти низкоэнергетичные электро¬
ны притягиваются к образцу, и таким образом устанавливается равновесный
статический заряд, и появляющиеся в спектре фотоэлектронные пики теперь
имеют энергию, определяемую соотношением^ P.Swift, D.Shuttleworth, Shell Research Ltd., Thornton Research Centre, PO Box 1,
Chester CHI 3SH.2 M.P. Seah, National Physical Laboratory, Teddington, Middlesex, UK.(П2.1)(П2.2)
П. Свифт, Д. Шатлуорс, М.П. Сих489где С - число, определяемое установившимся статическим зарядом, которое
обычно имеет положительное значение.Установившийся статический заряд, обсуждавшийся выше, можно рас¬
сматривать как равновесный статический заряд С, который обычно устанав¬
ливается в течение нескольких секунд после облучения образца рентгеновски¬
ми лучами от ахроматического источника, если только образец не обладает
изменяющейся электропроводностью,, Последнее явление может возникать из-
за повреждений образца рентгеновскими лучами, что тем самым изменяет его
физическую природу (например, восстановление непроводящего соединения
с образованием металлических участков)»Если рентгеновский источник монохроматичен, то проблема зарядки по¬
верхности усложняется вследствие более низкой интенсивности потока электро¬
нов и полного устранения тормозного рентгеновского излучения» Это приво¬
дит к значительному уменьшению числа рассеянных низкоэнергетичных фото¬
нов в области образца, вследствие чего установление стационарного заряда
редко достигается» Если используется монохроматический рентгеновский ис¬
точник, то необходимо направить на поверхность образца, как это описано в
разд» 2о6, управляемый пучок низкоэнергетичных электронов от электронной
пушки, что приведет к образованию статического заряда С^9 управляемого
этой пушкой»Дополнительно к изменению кинетически.« энергии фотоэлектронов, воз¬
никающему из-за равновесного статического заряда, может наблюдаться
уширение спектральных пиков непроводящих материалов» Это явление возни¬
кает потому, что не все участки поверхности имеют одинаковые значения С,
хотя на всей поверхности при заданном потоке фотонов может устанавливать¬
ся равновесие статического заряда» Этот эффект особенно заметен для гете¬
рогенных образцов, когда компоненты образцов различаются по изоляцион¬
ным свойствам (например, минералов) и в худших случаях спектральные пики
от фотоэлектронов, эмитируемых с одинаковых атомных уровней, но с раз¬
личных участков образца, могут различаться на несколько электронвольт»Хотя статическая зарядка может значительно сдвигать кинетическую
энергию фотоэлектронных пиков, из этого совсем не следует невозможность
определения их истинной энергии связи, конечно, при условии, что значение С
измерено* Ниже обсуждается несколько методов, которые можно рассматри¬
вать как дополнение или как альтернативу использованию оже-параметров, ко¬
торые обсуждались в гл» 3»П.2.2. Примесные поверхностные слоиЕсли образцы не подготавливать для анализа при тщательном контроле
газовой среды, то на их поверхности, как правило, наносятся примеси. Как
только эти примеси появились в спектрометре, дальнейшее загрязнение мо¬
жет происходить вследствие адсорбции остаточных газов, особенно в уста-
490Приложение 2. Методы учета статической зарядкиновках с диффузионными масляными насосами» Эти примесные слои можно
использовать для калибровки, если известно, что они точно отражают уста¬
новившийся статический заряд на поверхности образца и дают пики элементов
с известной энергией связи. Углерод является элементом, который наиболее
часто обнаруживается в примесных слоях, и именно фотоэлектроны с атом¬
ного уровня С Ь наиболее часто используются для целей калибровки» Для
уровня С 1« в этих примесях энергия связи часто принимается равной
284,8 эВ и разность между измеренным положением этого пика в энергети¬
ческом спектре и приведенным выше значением энергии связи дает обуслов¬
ленное зарядкой значение С в уравнении (П»2»2)»Зигбан и др. [ 1] первыми сообщили о наличии примесного углеродного
слоя и пришли к заключению, что его источником является масло насоса,
так как интенсивность пика С Ь возрастала с увеличением времени на¬
хождения образца в вакууме спектрометра» Впоследствии стало общеприня¬
тым, что С Ь -электроны в этом примесном слое дают насыщенные углево¬
дороды или атомы углерода в химическом состоянии с электроотрицатель¬
ностью, аналогичной электроотрицательности в насыщенном углеводороде»
Обсуждение экспериментальных проблем и вопросов надежности при исполь¬
зовании примесного углерода для калибровки энергии содержится в обзоре
Свифта [ 2]»Основной недостаток этого метода состоит в неопределенности, связан¬
ной с разбросом от 284,6 до 285,2 эВ опубликованных в литературе [2] зна¬
чений энергии С Ь -электронов» Поэтому если С 1« -электроны от примесного
слоя будут использоваться для калибровки, то экспериментатору рекоменду¬
ется на своем спектрометре измерить энергию связи этих электронов» В
идеальном случае это измерение следует проводить на поверхности, кото¬
рая по своим химическим и физическим свойствам аналогична веществу, ко¬
торое будет исследоваться и которая однородна покрыта исключительно тон*
ким (т» е» порядка монослоя) примесным слоем»На измеряемую энергию связи в примесных слоях углерода и на ее по¬
следующее использование для калибровки отсчета энергии могут влиять
следующие факторы:а) химическое состояние углерода в примесном слое [ 3],б) толщина примесного слоя [4-6],в) химическая и физическая природа подложки, на которой производятся
измерения [ 2, 4],г) процедура приготовления образца И' эффекты обработки поверхности
[4,7,8],д) точность калибровки спектрометра, на котором проводятся измере¬
ния»Несмотря на кажущиеся ограничения и неопределенности, связанные
П. Свифт, Д. Шатлуорс, М.П. Сих491с использованием примесного углерода для учета статической зарядки, этот
метод является самым удобным и широко используемым. Однако по причи¬
нам, которые были перечислены выше и обсуждались в обзоре Свифта [ 2], к
интерпретации энергий связи необходимо подходить с осторожностью» Более
того, если необходимо сравнить результаты, полученные в разных лабора¬
ториях, то следует осуществить абсолютную калибровку энергий спектромет¬
ров и привести надежную эталонную энергию связи (например, Au 4/^), Эта
информация совместно с используемой энергией связи С Ь -электрона и ин¬
формацией о подложке, на которой она была измерена, будет служить надеж¬
ной основой для обеспечения надежного сравнения результатов»П.2.Э. Напыленные поверхностные слоиВ тех случаях, когда отсутствует постоянный примесный поверхностный
слой, необходимо ввести калибратор путем его преднамеренного осаждения0
Как правило, в качестве эталонных соединений используются органические
соединения и металлы, так как их можно надежно наносить йа поверхности в
сверхвысоком вакууме. Можно использовать также калибраторы с неизвест¬
ной структурой, такие, как масло для насосов, если их повторное испарение
в сверхвысоком вакууме происходит без разложения,,Следует убедиться, что калибратор не разлагается и не реагирует с
подложкой после конденсации и что наблюдаемая энергия связи эталонного
вещества, которое может осаждаться в виде тонкого поверхностного слоя
или в виде островков, является объемной характеристикой. Это можно осу¬
ществить путем регистрации спектра в зависимости от времени и количест¬
ва осажденного эталонного вещества. Целесообразно также определить с
помощью угловых измерений вид конденсата (т, е» равномерный поверхност¬
ный слой или островки). Это, конечно, неприменимо к порошковым образцам.Преднамеренное осаждение паров масла в системах с диффузионной
откачкой часто используется для целей калибровки, так как его можно осу¬
ществить частичным размораживанием охлаждаемых жидким азотом лову¬
шек,, Другой способ [ 10] состоит в том, что в спектрометрах некоторого
типа можно отогреть охлаждаемое окно рентгеновского источника и таким
образом осуществить перенос осажденных примесей непосредственно на об¬
разец, В обоих случаях процесс конденсации можно ускорить путем охла¬
ждения образца» При использовании этого метода необходимо проявлять
осторожность, чтобы избежать "эффекта памяти”, связанного с ранее
введенным летучим веществом, которое не предполагалось использовать в
качестве эталона и которое может конденсироваться на образце и приводить
к неправильной калибровке.Еще один метод, использующий для калибровки органические вещества,
состоит во введении этих веществ извне вакуумной камеры, как описано
492Приложение 2. Методы учета статической зарядкиКоннором [111 Этот редко используемый метод позволяет так подобрать
органическое соединение, чтобы оно было совместимо с используемым
образцом, например в отношении смачиваемости и отсутствия перекрытия
линий остовных уровней.Металлы также часто применяются для калибровки в РФЭС, причем ча¬
ще используются драгоценные металлы из-за их инертности и простоты
испаренияо На практике напыляется обычно in situ около одного монослоя
(например, золота)» Этот метод является одним из немногих детально раз¬
работанных методов коррекции зарядки, для которого было показано [ 12 -
15], что условия, необходимые для достижения электрического равновесия
и, таким образом, для точного учета зарядки, зависят от степени покрьь
тия поверхности» Существует оптимальная толщина золота для эффектив¬
ной коррекции зарядки, которая для полиэтилена и ПТФЭ приблизительно
равна 0,6 нм [15]» Эту толщину следует считать эквивалентной толщиной,
так как известно, что при такой низкой степени покрытия золото осажда¬
ется в виде островков, Здесь необходимо проявлять осторожность, так как
имеются данные о наличии небольших сдвигов (^ 0,25 эВ) энергии связи пи¬
ка Аи 4/ при низких степенях покрытия [ 17]. Были опубликованы также
данные [ 18 — 21] о реакции золота с подложками некоторого типа, особенно
с галогенидами и цианидами.» Тем не менее, если соблюдать некоторые
предосторожности, метод декорирования золотом можно использовать для
получения энергий связи, скорректированных на зарядку, используя эталон¬
ную энергию связи линии Au 4/ , равную 84,0 эВвЕсли приходится иметь дело с образцами, которые легко испаряются
без разложения в сверхвысоком вакууме, то калибровка становится относи¬
тельно простой» В этом случае первоначальные данные о соответствующем
образце в виде тонкого слоя получаются путем его конденсации на чистую
металлическую подложку (например, золото) и привязки к известной энер¬
гии связи остовного уровня этой подложки, Необходимо убедиться в надеж¬
ности электрического контакта путем регистрации спектра при различной
толщине образца и проверки равного смещения всех пиков спектра при пода¬
че электрического смещения на образец.П.2.4. СмесиБыла исследована возможность осуществления коррекции на статичес¬
кую зарядку энергии изолирующих материалов путем изготовления мелко¬
зернистых смесей из порошков, содержащих компоненту с известной энер¬
гией связи» В этом методе наиболее часто используемым эталонным ве¬
ществом является порошковый графят [4, 22 - 24], хотя рассматривались
также варианты с фтористым литием [25], солями калия [26, 27], свинцовым
суриком, трехокисью молибдена [29, 30] и золотом [30]. Обычно считают,
П. Свифт, Д. Шатлуорс, М.П. Сих493что основным ограничением этого метода является сложность практичес¬
кого получения мелкодисперсных гомогенных смесей (т. е. в пределе на
атомном уровне)0 При интерпретации данных необходимо учитывать следую¬
щие факторы:а) размер частиц в порошках, при котором они могут влиять на степень
плотности смеси,б) взаимодействие частица - граница раздела между различными компо¬
нентами,в) разная зарядка частиц различных составляющих смеси»Для смеси порошков обычно получаются очень плохие результаты, и
Вагнер [ 31] показал, что для некоторых систем могут возникать ошибки
вплоть до 10 эВ,Смешивания на молекулярном уровне можно добиться для ограниченного
количества веществ путем сплавления эталонного вещества с неизвестным
соединением, однако получаемые данные редко будут характеризовать исход¬
ные соединения» Совместная конденсация летучего эталонного вещества с
неизвестным соединением также является одним из методов калибровки энер¬
гии, широко применявшимся Коннором [11], который достиг хорошей самосо-
гласованности результатов с воспроизводимостью энергий связи, равной
± 0,2 эВо Как оказалось, эти результаты не зависят от относительного содер¬
жания совместно сконденсированных эталонного вещества и неизвестного
соединения, а также от конкретной молекулы, используемой для калибровки»Из трех методов калибровки на основе смесей, обсуждавшихся выше,
последний, как было показано, является самым надежным. Однако этот ме¬
тод применим только для стабильных летучих соединений,, Как правило, при¬
менять методы смешивания в качестве средства калибровки энергии не ре¬
комендуется независимо от того, используются они для коррекции статичес¬
кой зарядки или нетоП.2.5. Внутренние стандартыВозможно, наиболее надежный метод калибровки спектров состоит в
использовании внутренних стандартов [32]; он базируется на инвариантности
энергии связи выбранной химической группы в различных молекулах. Этот
метод привлекателен тем, что эталон теперь зафиксирован в неизвестном
веществе и может нести информацию о статической зарядке системы при его
равномерном распределении по поверхности.Как было показано в работе [ 33], широкое распространение этот метод
получил при исследовании углеродсодержащих веществ и, в частности, поли¬
мерных систем, для которых, как было обнаружено, этот метод дает надежные
результаты при изучении химических сдвигов. Для использования этого мето¬
да, конечно, требуется знать молекулярную формулу исследуемого вещества.
494Приложение 2. Методы учета статической зарядкиНеобходимо с помощью некоторого независимого метода получить информа¬
цию об инвариантности энергии связи используемой химической группы при
различном окружении (например, создавая тонкие проводящие пленки).Новый вариант этого метода состоит в использовании в качестве стан¬
дарта объемных растворителей в быстро замороженных растворах [34]. Хотя
этот метод имеет ограниченное приложение, он тем не менее ценен»П.2.6ь Низковольтная компенсирующая электронная пушкаНизковольтные компенсирующие электронные пушки можно использовать
для стабилизации электростатической зарядки диэлектриков, анализируемых
методом РФЭС [35], особенно при использовании монохроматичных рентгенов¬
ских лучей.В ряде публикаций [36, 37], в которых рассматривались конструкции и
режимы работы компенсирующих пушек, было показано, что наилучшие резуль¬
таты получаются для электронного пучка с низкой энергией (< 1 эВ) по отноше¬
нию к вакуумной камере, находящейся при потенциале земли, при источнике
пучка, расположенном очень близко к образцу» Оптимальные рабочие условия,
например положение образца, существуют для каждой конкретной конфигура¬
ции, и они, как правило, должны быть определены экспериментатором. Для
достижения максимальной эффективности нейтрализации и уменьшения коли¬
чества индуцированных бомбардирующими электронами реакций необходимо
использовать электроны с низкой энергией»Низкоэнергетичную компенсирующую электронную пушку в принципе мож¬
но использовать для следующих трех типов РФЭС-экспериментов:а. Исследования с использованием монохроматических рентгеновских лу¬
чей, когда сдвиги, обусловленные зарядкой, для диэлектриков обычно очень
велики и не устраняются обсуждавшимися ранее методами. Компенсирующая
пушка заменяет вторичные электроны, создаваемые тормозным излучением,
что происходит при замене ахроматического источника рентгеновских лучей
на монохроматический источник. Этот способ обычно позволяет преодолеть
основную проблему, однако при этом остаются небольшие поправки, которые
необходимо учесть, используя методы, в общих чертах описанные в преды¬
дущих разделах.б. Абсолютной калибровки спектра можно добиться с помощью компенси¬
рующей пушки, откалиброванной должным образом на конкретной эксперимен¬
тальной установке [ 36]. Этим способом можно уменьшить или даже сделать
отрицательной величину С, входящую в уравнение (П»2.2). Величина CQ9 кото¬
рая была введена ранее в связи с уравнением (П„2»2), равна напряжению като¬
да за вычетом постоянной пушки» Используя откалиброванную пушку, в случае
гладких образцов потенциал поверхности можно измерить с погрешностью
П. Свифт, Д. Шатлуорс, М.П. Сих495± 0,01 эВ, что позволяет точно определять энергии связи» В настоящее время
конструкционные особенности компенсирующих пушек не позволяют достигнуть
такой точности для порошковых образцов, которые чаще всего исследуются методом
РФЭС» В этом случае абсолютная точность уменьшается до ± 0,50 эВ»в» В тех случаях, когда статическая зарядка неоднородна по поверхности
образца, может возникать уширение пика и с помощью компенсирующей пушки
можно уменьшить или даже полностью устранить это уширение [ 38]»Из пяти методов калибровки энергии, изложенных в этом приложении, ме¬
тод низковольтной компенсирующей электронной пушки имеет наибольшие по¬
тенциальные возможности, так как он применим для образцов всех типов,
встречающихся в РФЭС»Литература1. Siegbahn К., Nordling С., Fahlman A. et al.9 ESCA, Nova Acta Regiae, Soc.Sci. Ups., IV, 20, 1967.2. Swift P., Surf. Interface AnaL, 4, 47 (1982).3e Brandt E.S., Untereker D.F., Reilley С./V., Murray R.W., J0 Electron Spectrosc.,14, 113 (1978),4. Johansson GHedman J., Bendtsson Aet al.9 J. Electron Spectrosc», 2,295 (1973).50 Kinoshita S., Ohta T.9 Kuroda H.9 Bull. Chem. Soc. Jpn., 49, 149 (1976).6. Clark D.T., Dilks A.9 Thomas H.R,, J. Polym. Sci., Polym. Chem. Ed., 16,1461 (1978).7. Wagner C.D., Appl. Surface Analysis, ASTM STP, American Society for Testing
and Materials, Philadelphia, 1980o8. Jaegle A., Kait A.f Nanse G.9 Peruchetti J.C., Analysis, 9, 252 (1981).9o Bird R.J., Swift P., J. Electron Spectrosc., 21, 227 (1980).10. Clark D.T., Thomas H.R., Dilks A.9 Shuttleworth D., J. Electron Spectrosc.,40, 455 (1977).11. Connor J.A.9 Handbook of X-ray and Ultrayiolet Photoelectron Spectroscopy,(ed. D. Briggs), Heyden, London, 1977, Ch. 5, p. 183012. Hnatowich D.J., Hudis J., Perlman M.L., Ragnini R.C., J. Appl. Phys., 42
4883 (1971).13. Ebel M.F., Ebel H.9 J. Electron Spectrosc., 3, 169 (1974).14. Grinnard C.R.f Riggs H.M.9 Anal. Chem., 46, 1306 (1974).15° Uwamine У., Ishizuka T.9 Yamatera H.9 J. Electron Spectrosc., 23, 55 (1981).16. Pashley D.W.9 Adv« Phys., 14, 327 (1965).17. Adams /., Thesis Ph.D., University College of Wales, Aberystwyth, 1972,
p. 137.18. Betteridge D.9 Connor J.C., Hercules D.M., Electron Spectrosc., 2, 327 (1973).19o Urch D.S., Webber M.t J. Electron Spectrosc., 5, 791 (1974).
496Приложение 2. Методы учета статической зарядки20о Matiengo L.I., Grim S.О., Anal. Chem., 46 , 2052 (1974).21„ Nefedov V.L, Salyn Va.V., Leonhardt G., Scheibe R., J. Electron Spectrosc.,
10, 121 (1977).22. Nordberg R., Brecht HAlbridge R.G., et al., J.R., Inorg. Chem., 9 , 2469
(1970).23. Nefedov V.L, Kakhana M.M., Zh. Anal. Kim., 27 , 2049 (1972).24. Kumar G., Blackburn J.R., Albridge R.G., et al., Inorg. Chem., 11, 296 (1972).25. Bremser W., Linnemann F., Chem., 1971, Fig. 95, 1011 (1971).26. Jack J.J., Hercules D.M., Anal. Chem., 43, 729 (1971).27. Hulett L.D., Carlson T.A., Appl. Spectrosc., 25, 33 (1971).28. Stec W.J., Morgan W.E., Albridge R.G. et al., Inorg. Chem., 11, 219 (1972).29* Schwartz W.E., Watt P.H., Watts J .P. et al., Anal. Chem., 44, 2001 (1972).30* Dianis W.P., Lester J.E., AnaL Chem., 45, 1416 (1973).31. Wagner C.D., J. Electron Spectrosc., 18, 345 (1980).32. Ogilvie J.J., Wolburg A., Appl. Spectrosc., 26, 401 (1972).33. Clark D.T., Thomas H.R., J. Polym. Sci. Chem. Ed., 14, 1671 (1976).34. Burger K., J. Electron Spectrosc., 14, 405 (1978).35. Huchital D.A., McKeon R.T., Appl. Phys. Lett., 20, 158(1972).36. Hunt C.P., Stoddart C.T.H., Seah M.P., Surf. Interface AnaL, 3, 157 (1981).37 „ Hunt C.P., Antony M.T., Stoddart C.T.H., Seah M.P., NPL Chem. Report 108,March 1980c
38« Windawi H., J. Electron Spectrosc., 22, 373 (1981).
Приложение 3
ОБРАБОТКА ДАННЫХ В РЕНТГЕНОВСКОЙ
ФОТОЭЛЕКТРОННОЙ СПЕКТРОСКОПИИП. М. А. Шервуд ”П.3.1. ВведениеВ настоящее время проявляется все возрастающий интерес к сбору высо¬
кокачественных данных, которые можно обрабатывать строгими методами
анализа. Это вызвано пониманием важности выявления малых изменений в фо¬
тоэлектронных спектрах, а также наличием дешевых микрокомпьютеров, по¬
зволяющих автоматизировать сбор данных, и многочисленных цифровых мето¬
дов их обработки, которые можно использовать, не прибегая к помощи боль¬
ших и дорогостоящих ЭВМ. В этом приложении будет сделана попытка дать
обзор методов сбора и обработки данных, а также рассмотрен ряд примеров
из работ автора, иллюстрирующих эти методы.П.3.2. Системы сбора данныхПодавляющее большинство данных в рентгеновской фотоэлектронной
спектроскопии все еще регистрируется при помощи аналоговых измерителей
скорости счета и самописцев. Аналоговые данные подвержены погрешностям
в определении положения и площади пиков, что связано с зависимостью этих
величин от постоянной времени регистрации и скорости сканирования.
Аналоговая регистрация не дает практически никакого представления о
качестве данных, что обусловлено их сглаживанием при выборе больших по¬
стоянных времени. Если имеется необходимость в сглаживании данных, то
гораздо лучше осуществить его контролируемым способом в цифровом виде
(об этом речь пойдет ниже), чем полагаться на правильный выбор оператором
постоянной времени и скорости сканирования. Собранные цифровые данные
можно обрабатывать различными методами, а аналоговые данные, как только
они зарегистрированы, больше нельзя изменить, так как они уже были изме¬
нены аналоговой системой регистрации. Кроме того, обработка данных час¬
то проводится такими аналоговыми методами, как аппроксимация кривых сSherwood, Department of Inorganic Chemistry, The University, Newcastle-
upon-Tyne NE1 7RU, UK.
498Приложение 3. Обработка данных в РФЭСиспользованием графического решающего устройства Дюпона. Такой подход
очень субъективен и поэтому часто приводит к ошибкам. Одним из наиболее
серьезных ограничений аналоговых данных является невозможность их на¬
копления с целью увеличения в спектре отношения сигнал/шум (S/N).Большинство коммерческих спектрометров можно приобрести с систе¬
мой, позволяющей контролировать работу спектрометра и проводить опреде¬
ленного типа обработку данных. Такие системы обработки данных могут
быть довольно дорогими, но наличие дешевых микрокомпьютеров позволяет
реализовать их и по значительно более низкой цене. Микрокомпьютеры имеют
то преимущество, что, несмотря на их назначение решать в основном одну
задачу, низкая их стоимость позволяет иметь несколько микропроцессоров,
каждый из которых будет решать свою конкретную задачу. Кроме того, про¬
граммы всегда можно модифицировать с учетом последних улучшений и до¬
стижений в используемой системе сбора и обработки данных, что было бы
невозможно при использовании приборов типа многоканальных анализаторов.Способ построения системы обработки данных, основанный на микро¬
компьютерах, проиллюстрирован на рис. П.3.1, где изображен используемый
в лаборатории автора способ связи спектрометра с микро- и основным компь¬
ютерами. В этой системе используются два микрокомпьютера и пять перифе¬
рийных устройств, связанных с основной ЭВМ. Все печатающие устройства и
графопостроители связаны между собой через распределительную плату, кото-Рис. П.3.1. Схематическое изображение системы обработки данных, в которой исполь¬
зуются микрокомпьютер и основной компьютер. 1 — телетайп, 2 - графо¬
построитель, 3 - принтер, 4 — цветной дисплей, 5 — тектроникс, 6 - цифро¬
вой градопостроитель, 7 - принтер/графопостроитель, 8 — дисплей,9 — скоростной перфосчитыватель.
П.М.А. Шервуд499рая обеспечивает возможность независимого обращения к ним как микрокомпьюте¬
ров, так и через терминалы основной ЭВМ, а также передачу по шести линиям инфор¬
мации от микрокомпьютеров и периферийных устройств к основной ЭВМ.Данные регистрируются одновременно аналоговым (А) и цифровым (D) из¬
мерителями скорости счета. Аналоговый измеритель выдает графики в коор¬
динатах X - У, которые полезны при регистрации обзорных спектров и для
контроля любых их изменений во времени (эти изменения могут быть вызва¬
ны разложением образцов или же дрейфом их потенциала). Для контроля из¬
менений спектров во времени можно использовать также цифровой измери¬
тель скорости счета. Для этого наряду с основным выходом, через который
информация поступает на микрокомпьютер Apple II, он имеет дополнительный
выход, через который данные в случае необходимости можно зарегистриро¬
вать на перфоленте.Микрокомпьютер Apple II имеет оперативную память объемом 64 кбайт,
которая достаточна для записи сложных программ сбора данных с полным
графическим обеспечением. Этот микрокомпьютер запрограммирован на пря¬
мое общение с оператором, который с его помощью может убедиться, что вся
система работает правильно, а также на сбор всей спектральной информации.
Его программа составлена таким образом, что неопытный оператор получа¬
ет всю необходимую информацию на цифро-буквенном дисплее размером
80 х 24. Опытный оператор может не обращать никакого внимания на эту ин¬
формацию, так как она выводится с очень большой скоростью. Затем одно¬
временно с информацией на текстовом дисплее на дополнительном графиче¬
ском дисплее изображается спектр в процессе его накопления. Одновремен¬
ное изображение программных инструкций и графиков позволяет отказаться
от инструкций по эксплуатации и легко создавать самообучающиеся програм¬
мы. Такой подход представляет особую ценность при использовании микро¬
компьютеров для анализа данных. Данные записываются на гибких дисках,
которые затем можно вставить в другой микрокомпьютер (Apple И) и про¬
вести анализ, при этом первый микрокомпьютер используется для регистра¬
ции следующего спектра, что полностью ликвидирует холостое время систе¬
мы.Слектры непрерывно записываются на гибком диске (на диске диамет¬
ром 128 мм можно записать приблизительно семьдесят достаточно длинных
спектров). Эти спектры можно также передать на основную ЭВМ, где они за¬
писываются на магнитной ленте. Основная ЭВМ используется для обработки
данных и для сложных расчетов с целью интерпретации результатов (напри¬
мер, расчетов по методу молекулярных орбиталей). Мощная ЭВМ необходима
для некоторых методов обработки данных, однако в лаборатории автора и для
этих целей все чаще используются микрокомпьютеры.
500Приложение 3. Обработка данных в РФЭСП.3.3. Простые операции с даннымиБольшинство систем регистрации данных контролируют основные ха¬
рактеристики спектрометров, такие, как начальная кинетическая энергия,
диапазон, число сканирований, остановка сканирования после достижения за¬
данного отношения сигнал/шум, ток и напряжение пушки рентгеновского ис¬
точника, а также контролируют работу аргоновой ионной пушки. Данные мо¬
гут изображаться в процессе их регистрации или же после записи всего спект¬
ра. Выбирая способ накопления данных, необходимо учитывать, что в некото¬
рых выпускаемых системах автоматически осуществляется предварительная
обработка данных (сглаживание и вычитание фона).Это очень неудачная прак¬
тика. Как будет ясно из этого приложения, не существует "корректного" спо¬
соба проведения подобных операций и такой подход может приводить к невер¬
ным результатам. Поэтому, как правило, ко всем методам обработки данных
необходимо относиться с осторожностью, а первоначальные необработанные
данные всегда следует хранить в неизмененном виде.Некоторые очень простые операции все же можно проводить, и об этом
речь пойдет ниже. Все эти операции можно легко осуществить с помощью
микроЭВМ. Преимущество микроЭВМ заключается в том, что спектр после по¬
добной обработки практически мгновенно можно изобразить на дисплее.П.3.3.1. Изображение спектра и изменение масштабаИзображение спектра, как правило, представляет собой набор точек,
каждая из которых соответствует цифровой информации, полученной за фик¬
сированное время при конкретной кинетической энергии или же, что иногда
практикуется, при кинетической энергии, линейно изменяющейся в малом
диапазоне. Такое точечное изображение экспериментальных данных, по мне¬
нию автора, более предпочтительно по сравнению с изображением, на кото¬
ром с целью получения непрерывной кривой экспериментальные точки соеди¬
нены прямыми линиями. Часто на спектр накладывают перемещаемые перпен¬
дикулярные линии, что позволяет определить для конкретной точки в спектре
координаты х (кинетическую энергию или энергию связи) и у (число отсчетов).
Эта информация, как правило, изображается в верхней части спектра. Обычно
также предусмотрено простое увеличение масштаба по осям х и у. Полезной
является также возможность нормировать спектр таким образом, чтобы точ¬
ка с максимальным числом отсчетов соответствовала максимуму дисплея.
Особенно это удобно в том случае, если имеется возможность убрать часть
спектра для выделения его характерных особенностей.
П.М.А. Шервуд501П.3.3.2. Интегрирование и измерение площадейПолезной является также возможность измерения площадей. Как прави¬
ло, при этом вычисляется площадь между прямой, проходящей через две точ¬
ки, положение которых определяется пересекающимися перпендикулярными
линиями, и спектром. Иногда проводится интегрирование всего спектра пу¬
тем суммирования (после вычитания некоторого горизонтального фонового
уровня) всех точек, и каждая точка проинтегрированной кривой представляет
собой сумму всех точек, расположенных слева от нее.П.3.3.3. Устранение выбросовИногда в спектре вследствие колебаний питания или включения приборов
могут появляться выбросы. В этом случае полезно предусмотреть возмож¬
ность стирания любой выбранной точки и ее замены на другую точку. Очевид¬
но, что такую процедуру можно применять только для ограниченного числа
точек!Возможность устранения выбросов перед проведением дальнейшей обра¬
ботки данных очень существенна, так как наличие двух или даже одного выб¬
роса может серьезно повлиять на результаты таких операций над данными,
как сглаживание и обработка кривых по нелинейному методу наименьших
квадратов.П.3.3.4. Вычитание сателлитовВ некоторых коммерческих системах обработки данных предусмотрено
устранение сателлитов рентгеновского излучения. При этом даже заявляют,
что такой подход не хуже использования рентгеновских монохроматоров, ко¬
торые, конечно, полностью устраняют сателлиты. Вычитание сателлитов не¬
обходимо проводить с большой предосторожностью. Во-Первых, необходимо
аккуратно вычесть фон в рассматриваемой области и из последующего изло¬
жения будет видно, что эта операция отнюдь не является операцией, которую
можно осуществить с абсолютной точностью. Во-вторых, необходимо иден¬
тифицировать все оже-пики в этой области (так как они, конечно, не связаны
с рентгеновскими сателлитами) и аккуратно рассчитать их спектральный
вклад, чтобы соответствующую им интенсивность можно было вычесть из ин¬
тенсивности сателлитов рентгеновского излучения. В-третьих, необходимо
идентифицировать все сателлиты, связанные с пиками, расположенными вне
этой области,т.е. с сателлитами AlKc^ 2-излучения из магниевого анода с
алюминиевыми рентгеновскими окнами и кроссовера двуханодных систем, а
также сателлиты от фотоэлектронных пиков, расположенных при энергиях
выше рассматриваемой области и связанных с дискретными потерями энер¬
гии (см. гл. 3).
502Приложение 3. Обработка данных в РФЭСП.3.3.5. Вычитание постоянной составляющейВычитание фона спектра является нетривиальной операцией, которую
мы рассмотрим ниже. Вычитание же горизонтального постоянного уровня
является простой операцией, которая представляет собой удобный метод улуч¬
шения внешнего вида спектра, имеющего небольшие спектральные особенно¬
сти, наложенные на большой фон.П.3.3.6. Сложение и вычитание спектровС целью сравнения спектры можно складывать и вычитать друг из дру¬
га. Во многих системах сбора данных предусмотрена возможность непосред-
ственнного сложения и вычитания. Если же операцию вычитания необходимо
провести более тщательно, то процедуру вычитания необходимо осуществить
нетривиальным способом, такие методы будут обсуждаться ниже.П.3.3.7. Определение положения максимума пикаПроцедура определения положения точки в спектре с максимальной ско¬
ростью счета, т.е. положения максимума пика, осуществляется достаточно
легко. Такая процедура, как правило, позволяет отделить истинный пик от
случайных выбросов.П.3.4. СглаживаниеПочти во всех системах сбора данных заложена возможность сглажива¬
ния данных. Такую операцию необходимо проводить осторожно, и в этом раз¬
деле рассматриваются те изменения, которые процесс сглаживания оказыва¬
ет на данные, а также предположения о том, каким способом необходимо осу¬
ществлять сглаживание для достижения максимальной эффективности и ми¬
нимальных искажений. Как правило, лучше всего использовать несглаженные
данные, так как любая процедура сглаживания в конце концов приводит к не¬
которым искажениям спектра. Тем не менее существует много ситуаций, ког¬
да сглаживание необходимо; к таким ситуациям относится регистрация с хо¬
рошим отношением сигнал/шум очень слабых сигналов с недопустимо боль¬
шим временем регистрации спектра или же случай исследования нестабиль¬
ных образцов, когда необходимо записать спектр за очень малый промежу¬
ток времени. Кроме того, часто необходимо перед дальнейшей обработкой
сгладить спектр с довольно хорошим отношением сигнал/шум; это имеет мес¬
то при дифференцировании или вычислении разностного спектра.Сглаживание является операцией, направленной на улучшение корреляции
между точками и на подавление некоррелированного шума. Сглаживание осу¬
ществляется путем вычисления свертки данных с некоторой сглаживающей
П.М.А. Шервуд503функцией. Используются два типа сглаживающих операций, а именно метод
наименьших квадратов относительно центральной точки, предложенный Са-
витским и Голеем [1], и связанные с ним методы, а также метод преобразо¬
вания Фурье. Чаще всего применяется первый метод и простейшие его моди¬
фикации используются в большинстве систем обработки данных.П.3.4.1. Метод наименьших квадратовВ этом методе используется свертка данных с функцией типа(П.3.1)где у(0) - сглаженная точка, соответствующая центру интервала, содержа¬
щего нечетное число точек Р(р = 2т + 1); f(t) - экспериментальные данные
в точках рассматриваемого интервала, которые суммируются с целочислен¬
ными коэффициентами С(; NORM есть нормирующий фактор.В простейшем случае вычисления среднего по интервалу С = 1 для всех
t и NORM = Р. Обработку данных на интервале Р методом наименьших квад¬
ратов с использованием полиномов степени п можно осуществить, используя
приведенную выше формулу при соответствующем выборе целочисленных
коэффициентов для различных Ри п, В работах Гильденбранда[2], Проктора
и Шервуда [3] приводится общая формула для вычисления необходимого набо¬
ра полиномов р.В общем случае выбор количества точек, используемых при сглаживании
этим методом, определяется следующими соображениями [31:а. Степень сглаживания увеличивается с увеличением числа точек в ин¬
тервале сглаживания. Оптимальная величина этого интервала равна 0,7 от ши¬
рины пика на половине его высоты.б. Степень сглаживания уменьшается при увеличении степени сглаживаю¬
щего полинома. В общем случае применение квадратичных и кубических поли¬
номов более эффективно, чем применение полиномов более высокой степени.в. Обычные методы сглаживания приводят к потере точек у обоих концов
спектра. Если интервал сглаживания содержит N точек, то число точек, теря¬
емых у каждого конца спектра, равно (N - 1)/2; и, таким образом, этот эф¬
фект становится все более значительным по мере увеличения интервала сгла¬
живания. Этот нежелательный эффект можно ликвидировать путем устране¬
ния потери точек при использовании соответствующего уравнения (см. уравне¬
ние (8) в [3]). Это позволяет повторять операцию сглаживания любое число раз.7. Для увеличения степени сглаживания процедуру сглаживания мэжно
повторить (особенно если при этом не возникает потерь точек, как это описа-
504Приложение 3. Обработка данных в РФЭСно в п. "в"), хотя наибольшая степень сглаживания имеет место после перво¬
го цикла процедуры сглаживания. При повторном сглаживании переходят к но¬
вому сглаживающему полиному, в котором положительные точки имеют боль¬
ший вес и который шире первоначального полинома, имеет высоту, уменьшаю¬
щуюся по мере уширения сглаживаемой функции, и который приводит к появ¬
лению пульсаций на концах этих функций. Такое широкое варьирование сгла¬
живающих функций делает процесс повторного сглаживания весьма полезным
[3]. Было обнаружено, что хорошие результаты такого повторного сглажива¬
ния получаются при выборе минимально возможного интервала сглаживания
и повторении процесса сглаживания максимально возможное число раз, хотя
размер интервала и количество циклов сглаживания не влияют на результат,
если ширина интервала не слишком превышает ширину пика.Иллюстрация успешного приложения описанных методов к типичному фо¬
тоэлектронному спектру атомного остова представлена на рис. П.3.2 и в
табл. П.3.1. Спектр с высоким уровнем шума Is-оболочки атома О, который
представляет собой сумму трех пиков, стократно сглажен но интервалу, со¬
держащему 21 точку (21 точка соответствует ширине пика на полувысоте, ум¬
ноженной на 0,7), и результат такого сглаживания сравнивается с тем же
спектром, накопленным за большой промежуток времени. Конечно, сглажен¬
ный спектр не содержит никакой новой информации, так как сглаживание
есть просто косметическая операция, однако в сглаженном спектре некото¬
рые его особенности проявляются более четко, и, таким образом, он более
информативен, чем несглаженный спектр. Так, наименьший из трех пиков в
сглаженном спектре более четко представлен как визуально, так и с точки
зрения параметров, указанных в табл. П.3.1. ЗначениеX уменьшается до
42% своего начального значения, когда третий пик просто добавляется к
сглаженному спектру, а для несглаженного спектра эта же величина состав¬
ляет только 97% начального значения.П.3.4.2. Метод преобразования ФурьеСглаживание можно осуществить также различными методами, исполь¬
зующими преобразование Фурье [4-71. Преобразование Фурье спектра, со¬
держащего шум, приводит к разложению по пространственным гармоникам
как полезного сигнала, так и шума. При этом можно уменьшить шумовую
компоненту путем умножения пространственного спектра на некоторую весо¬
вую функцию, после чего обратное преобразование приведет к сглаживанию
спектра.Сложность такого метода заключается в выборе подходящей весовой функ¬
ции. Ипю.'п. .окпись различны»* подходы 1 7 - 11], но выбор такой функции во
многом зависит от желаемой степени сглаживания. Увеличение отношения
П.М.А. Шервуд505Рис. П.3.2. Is-спектр кислорода, иллюстрирующий эффект сглаживания. Во всех трех
случаях спектр аппроксимируется тремя пиками, параметры которых при¬
ведены в табл. П.3.1.а - данные с хорошей статистикой, полученные сканированием спект¬
ра в течение большого промежутка времени; б — данные для того же ма¬
териала с плохой статистикой, полученные сканированием спектра в те¬
чение короткого промежутка времени; в - результат стократного сглажи¬
вания спектра "б" при сглаживающем интервале, равном 21.
Таблица П.3.1. Значения параметров из аппроксимации по нелинейному методу наименьших квадратов
спектра 01s, измеренного с хорошей статистикой, с плохой статистикой, с плохой статистикой и последую¬
щим сглаживаниемПараметрЭнергия связи, эВШирина на полувысоте, эВ
Отношение площадейПараметраПримечание, значения в скобках соответствуют доверительному интервалу 95%.Хорошая статистика
(рис. П.3.2, а)534,81(16)532,66(04)530,95(02)2,10(04)1/7, 77/16, 90
584,4534,79(76)532,74(18)530,99(08)2,04(15)1/7, 44/14, 94
15636,9Плохая статистика
(рис. П.3.2, б)Плохая статистика со сглажива¬
нием (рис. П.3.2, в)534,73(12)532,71(03)530,99(01)2,10(03)1/7, 08/14, 34
321,97
П.М.А. Шервуд507сигнал/шум приводит к уменьшению разрешения. Оптимальный S /W-фильтр
был предложен Тюриным [11].Любые методы сглаживания содержат многочисленные субъективные фак¬
торы и поэтому использовать их следует очень осторожно.П.3.5. Анализ сложных спектровЧасто информация, которую мы получаем, используя фотоэлектронную
спектроскопию, представляет собой сложный спектр, состоящий из несколь¬
ких перекрывающихся пиков различной формы и интенсивности. При спектро¬
скопии внутренних оболочек атомов пики будут связаны с химическими сдви¬
гами, сателлитами, характеристическими потерями энергии и переходами
Оже. В валентной области на спектр будет оказывать влияние вся сложная
структура валентной зоны основного состояния, а также в случае использо¬
вания фотонов малой энергии - и возбужденного состояния (т. е. объединен¬
ная плотность состояний). В обоих случаях однозначная интерпретация спект¬
ра невозможна и нельзя указать определенный способ проведения его анализа.
С точки зрения эксперимента лучше всего использовать источник монохрома¬
тических фотонов, особенно в рентгеновской области, однако такие источники
не всегда доступны, а существенное уменьшение интенсивности излучения
может привести к слишком большому времени регистрации спектра. Существу¬
ют два основных способа, с помощью которых можно пытаться расшифровать
такие спектры, а именно способ обратной свертки и эмпирическая подгонка.
Ни один из этих способов не может дать однозначного решения. До сих пор
основное внимание уделялось методам, основанным на эмпирической подгон¬
ке, хотя для некоторых спектров успешно применялся и метод обратной сверт¬
ки. Метод эмпирической подгонки в значительной степени зависит от началь¬
ной предположительной информации о спектре, т.е. о числе пиков и их пара¬
метрах, и при выборе этой начальной информации очень полезным может быть
метод обратной свертки. Для выбора этой начальной информации можно так¬
же использовать методы дифференцирования и синтеза кривых. Конечно, для
увеличения вероятности правильной интерпретации спектров необходимо ис¬
пользовать максимально возможное количество методов их анализа.П.3.5.1. Методы обратной сверткиВ наблюдаемую ширину линий в фотоэлектронных спектрах вносят вклад
факторы инструментального уширения. К ним относятся конечная разрешаю¬
щая сила спектрометра и ширина линии используемого источника фотонов
(см. гл. 3). Если вклад этих факторов известен и его можно описать некото¬
рой функцией (В), то наблюдаемый спектр будет сверткой "истинного" спект-
508Приложение 3. Обработка данных в РФЭСpa (ft) и этой аппаратурной функции. Истинный спектр можно было бы полу¬
чить путем применения операции обратной свертки к наблюдаемому спектру
(/0), конкретный вид которого определяется устройством экспериментальной
установки. Наблюдаемый спектр связан с "истинным” спектром уравнением
"свертки":(П.3.2)Восстановление функции /( по функции fQ и называется "операцией обратной
свертки". Статистический высокочастотный шум приводит к появлению шумовой
компоненты в уравнении (П.3.2), которая до некоторой степени учитывает ре«
альный шум, присутствующий в любых экспериментальных данных. Наличие
этой шумовой компоненты означает, что операция обратной свертки не явля¬
ется однозначной, при этом в спектре, получаемом в результате обратной
свертки, возможно появление шума и ложных осцилляций. Это служит объяс¬
нением того факта, что получаемое улучшение разрешения достигается за
счет уменьшения отношения сигнал/шум S /N точно так же, как увеличение
отношения S /N при сглаживании в общем случае сопровождается потерей раз¬
решения [ 12, 13].Использовались несколько методов выделения эффектов, приводящих к
искажению данных. Некоторые из них основаны на методах дифференцирова¬
ния. Однако чаще всего используются три метода:а. Прямое решение уравнения свертки.б. Фурье-анализ.в. Итерационные методики.Как оказалось, успешное решение этой задачи в меньшей степени зави¬
сит от используемого метода вычисления обратной свертки, чем от качества
экспериментальных данных. Поэтому важно, чтобы начальные данные имели
очень хорошую статистику.Читателям, желающим полнее ознакомиться с затронутыми здесь вопро¬
сами, мы рекомендуем прочитать работу Карли и Джойнера [14], в которой
содержится очень подробный их обзор.П.3.5.2. Дифференцирование спектровДифференцирование спектра является в некоторых случаях полезным ме¬
тодом локализации пиков. Ряд исследователей [15 - 21] неоднократно отме¬
чали полезность этого метода. Выли разработаны процедуры автоматическо¬
го нахождения пиков, основанные на свойствах идеальных производных, од¬
нако в рентгеновской фотоэлектронной спектроскопии они используются не¬
часто. Дважды дифференцированный спектр является простым методом быст¬
рого определения положения пиков [15, 16], а также сам по себе содержит
П.М.А. Шервуд509ценную информацию. В спектре вторых производных отрицательные выбросы
приблизительно соответствуют положению перекрывающихся пиков в первона¬
чальном спектре.Из спектра первых производных нельзя получить точное положение пи¬
ков, так как в случае перекрывающихся пиков наблюдаемый максимум каж¬
дого пика сдвинут из-за присутствия других пиков. Тем не менее спектр пер¬
вых производных содержит полезную информацию. Обычно наиболее точное
положение пика наблюдается при равенстве интервала свертки в дифференци¬
альном спектре полной ширине на полувысоте перекрывающихся пиков [15].На рис. П.3.3 показано, как можно использовать дважды дифференциро¬
ванный спектр для предсказания положения и относительных интенсивностейРис. П.З.Э. Вторая производная модельного спектра, состоящего из двух перекрыва¬
ющихся пиков различной интенсивности: а) 1 : 1; 6) 1 : 0,95, в) 1 : 0,90;
г) 1 : 0,5; д) 1 : 0,5. Интервал, на котором вычисляется свертка, равен 21,
полная ширина пика на полувысоте равна 60, расстояние между пиками
— 50 каналов.
510Приложение 3. Обработка данных в РФЗСпиков в простом двухкомпонентном модельном спектре, аналогичном спектру
О Is, состоящему из двух перекрывающихся пиков. Спектр вторых производ¬
ных имеет два пика, интенсивности которых соответствуют вкладу каждого
из перекрывающихся пиков в первоначальный спектр. Дополнительно к незна¬
чительным погрешностям в положении пиков, причины которых обсуждались
выше, относительные интенсивности пиков получаются также только прибли¬
женно. Это вызвано эффектами взаимной компенсации, которые возникают
при перекрытии положительного выброса от одного пика с отрицательным выб¬
росом от другого. В общем случае этот эффект будет зависеть от числа пе¬
рекрывающихся пиков, их ширины и интенсивности.На рис. П.3.4 приведено сравнение сглаженного дважды дифференцирован¬
ного спектра О Ls углеродной нити (см. рис. П.3.4, а), который состоит из
перекрывающихся пиков. Отношение Q = А/В указывает на асимметрию пика
С Is в той же степени, в которой может дать информацию об асимметрии пи¬
ка измерение его ширины. Однако точное измерение величины Q при помощи
дважды дифференцированного спектра значительно проще, чем любое измере¬
ние ширины.На рис. П.3.5 приведено сравнение дважды дифференцированного спект¬
ра О Is со спектром, полученным методом подбора кривых, которое показы-Рис. П.3.4. Вторая производная С Is-спектра углеродной нити, в - первоначальный
спектр; б- вторая производная спектра, демонстрирующая способ оцен¬
ки Q-фактора.
П.М.А. Шервуд511Рис. П.3.5. Сравнение предсказаний положения пиков в спектре 01s, полученных сиспользованием второй производной спектра (а) и подгонки к эксперимен¬
тальному спектру по нелинейному методу наименьших квадратов (б).вает полезность дважды дифференцированных спектров для получения пред¬
варительной информации о положении пиков с последующим ее использовани¬
ем в методе подбора кривых.Существуют два относительно простых метода вычисления дифференциро¬
ванных спектров.n.3.5.2.t. Фурье-методыЕсли фурье-образ у(х) есть F(s), то dy/dx имеет фурье-образ (i2irs)F(s)
[5, 22]. Обобщая это соотношение на случай п-й производной, получим(П.3.3)Таким образом, умножая фурье-образ на соответствующую весовую функ¬
цию и осуществляя обратное преобразование, мы получим га-ю производную
512Приложение 3. Обработка данных в РФЭСпервоначальных данных. Однако поскольку для более высоких спектральных
гармоник (большие 5) весовая функция имеет большее значение, то связан¬
ный с результирующей производной шум будет возрастать.П.3.5.2.2. Полиномиальные методыВычисление производной в некоторой точке спектра является просто вы¬
числением производной аппроксимирующего полинома п-й степени в этой точ¬
ке. Таким образом, весь дифференцированный спектр можно получить, сдви¬
гая процесс свертки аналогично тому, как это делается в методике сглажи¬
вания, основанном на обобщенном методе наименьших квадратов [ 3]. Этот ме¬
тод использовался во всех обсуждаемых в этом разделе примерах.Как и в случае преобразования Фурье, дифференцирование с применени¬
ем этого метода также изменяет характеристики шума [15], так что даже для
спектра с хорошим отношением сигнал/шум требуется предварительное сгла¬
живание.Повторение процесса дифференцирования позволяет получить производ¬
ные высокого порядка довольно удовлетворительного качества (т. е. повторе¬
ние процедуры однократного дифференцирования п раз дает аппроксимацию
производной порядка п) и, таким образом, знать начальные функции свертки
более сложные, чем для первой или второй производной, необязательно. Прак¬
тическое использование производных с п > 2 ограниченно.П.3.5.3. Синтез кривыхСпектр можно синтезировать цифровыми или аналоговыми методами пу¬
тем суммирования функций, соответствующих отдельным пикам, и получить
результирующую функцию, которая близка к экспериментальному спектру.
Функция, описывающая отдельный пик, конструируется таким образом, что¬
бы она зависела от таких параметров пика, как его положение, интенсивность,
ширина, тип функциональной зависимости и характеристики пика в хвостовой
части. Простой синтез кривой можно легко осуществить, используя микро¬
компьютерную систему или аналоговые устройства типа анализаторов кривых
Дюпона. Преимущество микрокомпьютерной системы заключается в возмож¬
ности использования более широкого класса функций, а также в возможности
быстрой оценки качества синтезированного спектра, поскольку его вместе с
соответствующей статистической информацией можно изобразить на дисплее.
Такой синтез кривых дает полезную начальную информацию о спектре для
дальнейшей его более строгой обработки по нелинейному методу наименьших
квадратов, который мы рассмотрим ниже.Для этой цели использовалось несколько видов функций, типичными из
которых являются функции Гаусса и Лоренца. В работе автора и др. [231 ис-
П.М.А. Шервуд513пользовалась смешанная функция Гаусса - Лоренца, которую, как оказалось,
с практической точки зрения удобнее всего представить в виде произведения(П.3.4)где xQ - положение центра пика, В - параметр, который приблизительно ра¬
вен 0,5 от полной ширины на полувысоте. Реальная ширина вычисляется по
параметру р методом итераций. Величина М определяет степень смешива¬
ния и равна 1 для чисто лоренцева пика и 0 для чисто гауссова пика.Описанный способ подгонки кривых предполагает, что профиль пика (га¬
уссов или лоренцев) полностью определен, если задана его ширина на полу¬
высоте и его нельзя разложить на составляющие. В 1968 г. Перрам [24] по¬
казал, что простой гауссов профиль (у = ехр(—х2/2.05)) можно выразить че¬
рез сумму двух отдельных гауссовых контуров (у = ± 0.515359 ехр [-(х±± 0,244622)2/2(0.988937)2]) при условии, что разложение в цифровом виде бес
структурного контура не является однозначным. Барья и Меддамс [25] пока¬
зали нетипичность этого результата, так как он основан на равенстве интен¬
сивностей и полуширин этих двух пиков и их симметричном расположении на
расстоянии, составляющем 10% полуширины по обе стороны от первоначаль¬
ного гауссова пика. Они сделали вывод, что в большинстве практических слу¬
чаев гауссов или лоренцев профили являются достаточными для аппроксима¬
ции кривых. Общие вопросы аппроксимации спектров и возникающие при этом
ограничения обсуждаются в работе Меддамса [13].П.3.5.3.1. Х2-методКачество аппроксимированных кривых можно оценить по взвешенному
параметру X2, который определяется следующем образом:(П.3.5)где уг - наблюдаемая скорость счета в точке х = х^, F(x/q) - выбранная
огибающая пика, k - полное число точек в спектре, wf - весовая функция,
которая в данном случае выбирается равной у~1, что приводит к равенству
параметрах2 статистическому значениюX2 [26-28].П.3.5.3.2. Качество аппроксимацииВеличина X2, или, более конкретно, разность ДХ2 между различными ап¬
проксимациями одинаковых данных, представляет собой статистическую ин¬
формацию о "качестве аппроксимации".
514Приложение 3. Обработка данных в РФЭССтрого говоря, непосредственно сравнивать можно только величины X2,
имеющие одинаковые степени свободы f(f=k есть количество свободных па¬
раметров). Хотя / может немного изменяться от одной аппроксимации к дру¬
гой, как правило, это несущественно. Также из самой природы параметра
X2 следует, что чем выше интенсивность в спектре, тем большее значение
будет иметь этот параметр для аппроксимаций одинакового качества. Таким
образом, хотя изменение параметрах2 при переходе от одной аппроксимации
к другой аппроксимации одних и тех же данных является информативным,
сравнение параметров X2 для различных спектров имеет смысл только в слу¬
чае равенства их интенсивностей.П.3.5.3.3. Параметры крыльев спектровВ работе автора для описания крыльев спектров использовались три па¬
раметра, а именно: постоянный коэффициент (СТ), экспоненциальный коэф¬
фициент (ЕТ) и коэффициент смешивания (ГМ). В результате функция кры¬
ла спектра принимает видТ=ТМ-СТ+(1- ГМ) exp (-D . ЕТ), (П.3.6)где Dx есть расстояние от центра пика, выраженное через количество кана¬
лов. Функция, описывающая пик с правой стороны от его центра, выбирает¬
ся таким образом, чтобы она не содержала хвостовой части и ее можно было
представить в виде Н • GL, где Н - высота пика, a GL - функция, описываю¬
щая форму пика. Эту функцию можно представить в видеY = H[GL + (I - GL)T]. (П.3.7)На рис. П.3.6 показано, как можно синтезировать форму пика путем из¬
менения параметров функции, представляющей собой произведение гауссовой
и лоренцевой функций вместе с изменением параметров крыльев.П.3.5.4. Аппроксимация кривых нелинейным методом
наименьших квадратовИспользование нелинейного метода наименьших квадратов представляет
собой попытку оптимизации синтеза кривых, в котором учитывается сложность
изучаемого процесса в том смысле, что параметры, описывающие этот про¬
цесс, входят в алгебраические выражения нелинейным образом. Такой слу¬
чай реализуется для фотоэлектронных спектров, которые можно представить
функцией F(x/q) [см. уравнение (П.3.5)], зависящей нелинейным образом от
упоминавшихся ранее параметров. Процесс минимизации X2 проводится опера¬
тором при помощи компьютера, а не вручную, путем синтеза кривых методом
проб. Существует несколько возможных нелинейных методов наименьших
п.М.А. Шервуд515Рис. П.3.6. Возможные формы пиков, полученные с использованием гаусолоренцева
произведения, для различных коэффициентов смешивания и параметров
крыльев.а- изменение коэффициента смешивания с шагом 10% от 10%-ного
до 100%-ного содержания лоренцева контура; б- аналогично "а” с тем
отличаем, что коэффициент смешивания изменяется с шагом 1% от 88% до
100%; в - изменение формы пика при варьировании постоянного парамет¬
ра крыла; г - изменение формы пика при варьировании экспоненциально¬
го параметра крыла.квадратов. Автор использует метод Гаусса — Ньютона. В этом методе тре¬
бование минимумах2 приводит к уравнениям(П.3.8)всего имеется k уравнений, где k - количество параметров q. Эта задача яв¬
ляется нелинейной и ее нелинейность связана с величинами q. Если положить(П.3.9)где q. — значение qy соответствующее минимуму у}, qЯ — его некоторое
начальное значение и д: — приращение, которое необходимо прибавить к qf,
чтобы получить <?.. Если(П.3.10)
516Приложение 3. Обработка данных в РФЭСто F(xr/q) можно разложить в ряд Тейлора и ограничиться линейными члена¬
ми, т. е.где j = 1, 2, ... , k. Тогда уравнения (П.3.8) можно представить в виде k ли¬
нейных уравненийЭти уравнения можно решить методом итераций, причем итерационный про¬
цесс останавливают, когда величины 5. [уравнение (П.3.9)] изменяются на
несущественную величину для всех 6f при переходе от одной итерации к дру¬
гой. Этот процесс не всегда обладает устойчивой сходимостью, и могут воз¬
никать проблемы типа операции с сингулярными матрицами или матрицами,
имеющими отрицательный детерминант. Преимущество такого процесса состо¬
ит в возможности расчета некоторых ценных статистических характеристик,
например стандартных отклонений всех вычисляемых параметров. Такая ин¬
формация придает некоторый количественный смысл вычисляемым парамет¬
рам. Дополнительно к конечному значению величины X2 можно оценить веро¬
ятность того, что величина X2 меньше, чем ее расчетное значение [24J.
Обычно значение меньше 95% считается статистически достоверным, хотя не¬
обходимо помнить, что точность этого теста увеличивается, когда скорость
счета велика. Обычно наиболее полезный подход заключается в сравнении из¬
менений х2 или / для различных аппроксимаций одного и того же спектра, а
не в использовании какой бы то ни было абсолютной информации. В целом
эта информация очень помогает при выборе "наилучшего приближения". Та¬
кой выбор должен основываться на статистической информации, непротиво¬
речивой в химическом и спектроскопическом смысле. Часто однозначное ре¬
шение отсутствует, однако спектр можно свести к нескольким несильно раз¬
личающимся вариантам, позволяющим получить существенную информацию,
содержащуюся в данном спектре. Пример такого случая был опубликован в
работе [29].Практическое осуществление этого процесса можно проиллюстрировать
с помощью спектра W4/, изображенного на рис. П.3.7. Этот спектр содержит
четыре ясно различимых пика, для последующей аппроксимации он разделен на
две группы, содержащие по два пика каждая: одна группа связана с оксидом
(W03), а другая - с металлом. Такое разбиение спектра на различные группы
необходимо, если известно, что пики имеют различные ширины и параметры
крыльев. Каждый пик имеет следующие связанные с ним параметры:(П.3.11)(П.ЗЛ2)
П.М.А. Шервуд517а. Положение центра пика.б. Высота пика.в. Ширина пика.г. Гаусс-лоренцево отношение, характеризующее форму пика.д. Постоянная, определяющая высоту крыла.е. Экспоненциальный коэффициент спада на крыле.ж. Коэффициент смешивания постоянного и экспоненциального коэффи¬
циентов на крыле.Таким образом, для п пиков мы имеем 1п параметров. Кроме того, име¬
ются также наклон линейного фона и его пересечение с осями координат. Та¬
ким образом, мы можем по своему желанию варьировать 1п + 2 параметров.
Однако на практике это не всегда возможно и некоторые параметры (как пра¬
вило, параметры от "г” до "ж") имеют фиксированное значение.Спектр аппроксимируется рассмотренной ранее функцией, представляю¬
щей гаусс-лоренцево произведение с коэффициентом смешивания М, равным
0,5 для всех пиков и фиксированным для каждого дублета отношением интен¬
сивностей, расчетное значение которого определяется спин-орбитальным рас¬
щеплением для 4/g/2 : 4/ , равным 1/1,333 (см. гл. 3). Параметры крыльев
пиков для оксида и чистого металла различны. Асимметрия в форме пика для
чистого металла обусловлена эффектами взаимодействия в зоне проводимо¬
сти [ 30 - 32], что в данном случае учитывается путем добавления к высоко-
энергетичной части пиков чистого металла дополнительного экспоненциаль-Рис. П.3.7. Спектр W 4/, полученный методом эмпирической подгонки. 1 — оксид, 2 -
металл.
518Приложение 3. Обработка данных в РФЭСТаблица П.3.2. Результаты эмпирической подгонки спектра W4f(x2 = 2855, f = 298)
(рис. П.3.7)ОксидМеталл5/27/25/27/2Энергия связиа), эВ37,66(0,01)35,75(0,01)33,5(0,01)31,50(0,01)Мультиплетное расщепление, эВ1,91(0,01)2,00(0,01)Ширина на полувысоте6), эВ1,43(0,01)1,01(0,01)Интенсивность х 10“7 (площадь
пика)31,720(1%)2,2931,127(1%)1,50321,323(1%)1,764Относительная интенсивность11,33311,333Параметр экспоненты хвостовой
части20,020,00,0320,032Относительно C1s = 284,6 эВ
' Без учета вклада экспоненциального слагаемого крыла пиканого крыла (его величина была определена из аппроксимации спектра чисто¬
го металла). Мы обнаружили, что для большинства случаев использование та¬
кого метода дает удовлетворительные результаты, хотя он и отличается от
метода, использованного Хафнером и Вертгеймом [31], которые вначале уст¬
раняли вклад инструментального уширения из наблюдаемого спектра методом
обратной свертки, а затем аппроксимировали результирующий спектр конту¬
ром Дониаха и Сунджика [30]. Данные, приведенные в табл. П.3.2, показыва¬
ют, что учет асимметрии крыла является весьма существенным при вычисле¬
нии интенсивности (площади) пика.Такая аппроксимация позволяет рассчитать наклон линейного фона и точ¬
ки его пересечения с осями координат, хотя можно использовать и процеду¬
ру вычитания нелинейного фона. Были учтены также вклады рентгеновских
сателлитов, причем в результирующей аппроксимации каждый основной пик
содержит в качестве слагаемых все сателлитные пики. Вклад сателлитов весь¬
ма заметен в низкоэнергетичной части спектра.Следует понимать, что аппроксимация кривых нелинейным методом на¬
именьших квадратов предпочтительнее метода аналогового синтеза кривых,
так как он обеспечивает количественный контроль качества аппроксимации
и в отличие от аналоговых методов идеальную воспроизводимость парамет¬
ров пиков. Но самым важным является существенное уменьшение влияния
субъективных факторов.
П.М.А. Шервуд519П.3.8. Вычитание фонаВычитание фоновой составляющей спектра является операцией, которую
необходимо проводить очень осторожно, так как, за исключением тривиаль¬
ного случая вычитания горизонтального фона, обсуждавшегося ранее, эта
операция при неправильном ее проведении может приводить к искажению дан¬
ных. Любое вычитание фона будет изменять абсолютные интенсивности пи¬
ков и применение любой количественной модели, которая строится с учетом
точных данных о фоне, будет сопряжено с трудностями. Строгого рецепта
вычитания фона не существует, и вся эта процедура все еще является спор¬
ной (см. гл. 5).Довольно грубо устранение фона можно осуществить путем вычитания
линейного фона, который представляет собой прямую линию, проведенную
через начальную и конечную точки спектра. Правильный выбор начальной и
конечной точек или соответствующего среднего значения по близлежащим
точкам влияет на результирующий фон. Такой выбор лучше всего проводить
не прямо, а включать его в процедуру аппроксимации кривой, как
это было описано ранее и проиллюстрировано на рис. П.3.7. При этом на¬
чальные данные не изменяются и если, как это будет описано выше, произ¬
водится вычитание нелинейного фона, то результирующий линейный фон обыч¬
но представляет собой горизонтальную прямую, как это показано ниже на
рис. П.3.9.Вычитание нелинейного фона требует глубокого понимания всех тех
процессов, которые приводят к появлению фоновых электронов. К таким про¬
цессам будут относиться процессы неупругой эмиссии электронов, од¬
нако связанный с ними фон необходимо отличать от любых широких
и трудноразличимых пиков типа различного вида сатбллитных пиков и элект¬
ронных оже-пиков, которые могут возникать из-за специфичных упругих про¬
цессов. Кроме топ-, любая угловая зависимость фона (которая может зави¬
сеть от прибора, поскольку параметры будут различаться для разных прибо¬
ров; см. гл. 5) и неоднородность материалов могут создавать дополнительные
проблемы. Смешивание плазмонных потерь и асимметричного уширения с не¬
упругим фоном могут создавать проблемы, что обсуждалось в работе [33]. В
наиболее часто используемом методе вычитания нелинейного фона, часто на¬
зываемом методом Ширли, фон в каждой точке связывается с неупругорас¬
сеянными электронами, которые, как предполагается, возникают исключитель¬
но из-за рассеяния электронов более высоких энергий, и, таким образом, фон
оказывается пропорциональным интегральной интенсивности фотоэлектронов
с большей кинетической энергией. Этот метод использовался рядом исследо¬
вателей [34-38], которые внесли в него различные усовершенствования.
520Приложение 3. Обработка данных в РФЭСРис. П.3.8. Пример неупругого фона. В(х) есть фон в точке х спектра, состоящего
из k эквидистантных точек.Бишоп [39] модифицировал эту модель, включив линейно уменьшающуюся составляю¬
щую фона неупругоэмитированных электронов в виде функции, уменьшающейся при
переходе от высокой к более низким энергиям. Как правило, вычитание нели¬
нейного фона становится все более чувствительным к возникновению ошибок по
мере увеличения интервала энергий, на котором он производится, поэтому,
если даже улучшенные модели типа модели Бишопа хорошо работают, при боль¬
ших интервалах энергии могут возникать проблемы типа появления отрица¬
тельных пиков. Важно гарантировать, чтобы начало и конец спектральной об¬
ласти, на которой нужно вычесть фон, являлись точками профиля фона (а не
частью некоторого характерного пика), иначе процедура вычитания фона при¬
ведет к значительному уменьшению интенсивности пика.Вычитание нелинейного фона можно проводить методом, который исполь¬
зовался автором [15] и который проиллюстрирован на рис. П.3.8. В спектре
такого типа величина фона в точке л; спектрального массива, состоящего из
k расположенных на одинаковом расстоянии h точек, равна(П.3.13)где а - усредненная начальная точка, b - усредненная конечная точка,
(Р+ Q) - суммарная площадь пика за вычетом фона (BS) и Q - площадь пика
(BS) от точки х до точки k за вычетом фона. Используя метод трапеций, по¬
лучим(П.3.14)Площади BS вычисляются вначале при постоянном линейном фоне, величина
которого равна b (линия В1 на рис. П.3.8). Подстановка полученных резуль-
П.М.А. Шервуд521татов в уравнение (П.3.13) приводит к фону В2, который затем используется
для вычисления новых площадей BS, приводящих к фону ВЗ. Этот процесс по¬
вторяется до тех пор, пока (P=Q) и последующие итерации не приводят к существен¬
ному нарушению равенства Р= Q. Обычно для уменьшения требуемого чис¬
ла итераций начальная точка выбирается вблизи пика. На рис. П.3.8 для ил¬
люстрации итерационного процесса начальная точка немного сдвинута от пи¬
ка, однако конечный результат при этом не изменяется.Член 0,5 (у^ + Ук) является поправкой к простой сумме точек по интер¬
валу от х до k в методе трапеций* При проверке условия Р = Q, когда х = 1,
а у и ук равны нулю, эта поправка несущественна. Однако по мере увеличе¬
ния х она становится все значительнее из-за увеличения ух.На рис. П.3.9, а показан вычисленный фон для области W 4/ спектра,
изображенного на рис. П.3.7, который сравнивается с линейным фоном, рас¬
считанным на компьютере методом эмпирической подгонки. Чаще всего ли¬
неаризация фона недооценивает ’’реальный фон” в высокоэнергетичной обла-Рис. П.3.9. Спектр \\4f. в — сравнение спектра, содержащего полный неупругий фон,
с линейным фоном; б- спектр и его разложение на составляющие после
вычитания полного фона.
522Приложение 3. Обработка данных в РФЭСсти пика, причем обратная ситуация имеет место в области, соответствующей
уменьшению энергии связи. Это может быть весьма существенным при вычис¬
лении площадей пиков и их отношений. В данном случае, как видно из эмпи¬
рической кривой на рис. П.3.9, б, этот эффект минимален. Следует, однако,
отметить, что в области пика, соответствующей малой энергии связи, все
еще присутствует составляющая, связанная с рентгеновским сателлитом. На¬
личие этого сателлита обнаруживается путем варьирования наклона эмпири¬
ческого фона и расчета интенсивности этого линейно падающего фона, кото¬
рый, однако, значительно меньше своего первоначального значения. Гори¬
зонтальный фон можно было бы оценить, приравнивая его к интенсивности
фактического фона на низкоэнергетичном крыле пика перед вычитанием.П.3.7. Разностный спектрВычисление разностного спектра может оказаться весьма полезным ме¬
тодом анализа данных для ряда одинаковых образцов после их различной хи¬
мической обработки. Простое вычитание одного спектра из другого является
тривиальной операцией, однако правильное использование разностного спект¬
ра требует, чтобы это вычитание проводилось аккуратно и в соответствии с
четкими критериями. Применение этого метода в РФЭС недавно рассматрива¬
лось в работе [15], где было указано на наличие ряда специфичных для РФЭС
моментов, которые необходимо учитывать.П.3.7.1. ВыстраиваниеПри любой процедуре вычисления разностного спектра прежде всего не¬
обходимо убедиться, что оба спектра правильно выстроены, т. е. что точки
этих спектров соответствуют конкретным значениям кинетической энергии.
Как правило, это условие требует, чтобы спектры содержали одно и то же
количество точек. Затем необходимо тщательно проверить калибровку спект¬
ров по энергии, чтобы убедиться в правильной их выстроенности. Было по¬
казано [15], что малые расхождения в выстроенности спектров могут приве¬
сти к большим изменениям результирующего разностного спектра, что под¬
черкивает необходимую тщательность и точность проведения этой операции.В работе [15] обсуждаются методы использования разностных спектров для
достижения хорошего спектрального выстраивания.П.3.7.2. НормировкаПосле выстраивания спектра необходимо прежде, чем вычислять разностный
спектр, произвести нормировку обоих спектров. При этом все точки каждого
спектра перед вычитанием умножаются на фиксированный коэффициент.
П.М.А. Шервуд523Процесс нормировки предполагает, что коэффициент пропорциональности меж¬
ду заданным спектром и спектром, из которого его необходимо вычесть, из¬
вестен. Процедура вычитания двух спектров, каждый из которых содержит
ясно различимые пики, обусловленные химическими сдвигами, является три¬
виальной, и в таких случаях мы едва ли выигрываем, вычисляя разностный
спектр. Обычно разностный спектр получают из спектра, который содержит
ряд перекрывающихся пиков, что делает нормировку нетривиальной и важной
задачей при вычислении разностного спектра. Таким образом, перед проведе¬
нием этой операции необходимо знать правильный ответ! Обычно оба спектра
предварительно обрабатываются такими методами, как сглаживание (с исполь¬
зованием одинаковых параметров сглаживания) и вычитание фона (с учетом
обсуждавшихся выше проблем). Затем в обоих спектрах находят точки, соот¬
ветствующие максимуму, и спектры располагают таким образом, что эти
точки имеют одинаковое значение (104 на приводимом здесь спектре). После
этой предварительной операции можно проводить нормировку. Существуют
три типа нормировок, которые проводятся по строго определенным правилам:
а) нормировка по высоте, б) оптимальная нормировка, в) нормировка по пло¬
щади.Нормировка по высоте и нормировка по площади являются легко осуще¬
ствимыми операциями. Оптимальная нормировка представляет собой попыт¬
ку получения "корректного" коэффициента нормировки методом последова-Рис. П.3.10. Модельный "окисленный” спектр ds. Спектр а состоит из "неокисленной"
компоненты в и смещенных из-за химического взаимодействия компо¬
нент в 1, В2 и в3.х (начальное значение) = h2/h,; х (истинное значение) =а3А, .
524Приложение 3. Обработка данных в РФЭСтельных приближений. Процесс нормировки можно проиллюстрировать
(рис. П.3.10) на примере С Is-спектров А и В, где В состоит из ряда перекры¬
вающихся смещенных из-за химического взаимодействия пиков В, Ву В2 и
Вз, а спектр А состоит из одного пика. Для выявления более слабых пиковб2 и В3 спектр В вычитается из спектра А.П.3.7.2.1. Нормировка по высотеПосле предварительного выстраивания спектров, процедура которого
была описана выше, можно приступить к вычислению коэффициента нормиров¬
ки х. Если высота наиболее интенсивного пика в спектре равна h3 (назовем
его спектром В) и этот спектр необходимо вычесть из другого спектра, со¬
стоящего из ряда пиков (назовем его спектром А) и наибольшая высота пика
А равна h^9 то, как это показано на рис. П.3.10,* = W (П.3.15)К сожалению, значение х, как правило, неизвестно, хотя оно должно быть
меньше единицы. Поэтому можно провести простую нормировку по высоте,
при которой х = 1. Такая нормировка по высоте может оказаться полезной
при выстраивании двух спектров [15].П.3.7.2.2. Оптимальная нормировкаОптимальная нормировка основана на предположении, что использова¬
ние итерационных методов, возможно, позволит получить для х более точ¬
ное, чем единица, значение. В методе, который использовался автором [15],
значение х выбирается меньше единицы, затем путем наблюдения за умень¬
шением некоторых характеристик результирующего разностного спектра вы¬
бирается то значение х, которое является оптимальным. Как было обнаруже¬
но для модельного спектра, наиболее удачным критерием отбора является ус¬
ловие отрицательности наклона разностного спектра в области, заключенной
между максимумом пика спектра В и правой границей спектра А (точки п* и
k на рис. П.3.10), т. е.(А - В)(п) > (А - В) (п - 1), (П.3.16)где п изменяется от п1 до (k - 1).Начиная с х = х (начальное) = h2/h1 (и гарантируя, таким образом, что
вычисленное значение х всегда будет меньше или равно единице), в рассмат¬
риваемом методе мы проверяем выполнимость критерия (П.3.16) [ 16]. Если
этот критерий не удовлетворяется для некоторой пары точек, то значение х
уменьшается на величину 0,01 от начального значения и вновь производится
проверка выполнимости критерия. Если условие (П.3.16) удовлетворяется
П.М.А. Шервуд525во всей области от п* до k, то значение х является оптимальным, а спектр -
оптимально нормированным (ON). Используя этот метод для модельного спект¬
ра, можно получить значение х, равное 0,854, которое очень близко к правиль¬
ному для рассматриваемого случая значению 0,855. В случае реальных спект¬
ров было обнаружено, что область проверки критерия должна быть уменьше¬
на в правой части спектра Л на С каналов (рис. П.3.10), иначе оптимальное
значение х может уменьшиться до нуля!П.3.7.2.3. Нормировка по площадиЗначение х выбирается равным отношению полной площади исследуемо¬
го спектра А к полной площади спектра сравнения В. В отличие от спектра,
нормированного по высоте и площади, такой коэффициент нормировки будет
больше единицы. Результирующий разностный спектр будет содержать отри¬
цательные пики, связанные с наличием у спектра А особенностей, не содер¬
жащихся в спектре В; чем меньше спектр В по сравнению со спектром А,
тем больше будут отрицательные пики. Если оптимальное значение х получе¬
но описанным выше способом, а также вычислено его значение из нормиров¬
ки по площади, то из этих двух коэффициентов нормировки можно получить
полезные характеристики. Так, доля спектра сравнения (В) в исследуемом
спектре (Л) будет равна х (нормировка по площади) /х (оптимальное), и, та¬
ким образом, разность между этим параметром и единицей равна доле пол¬
ной площади, связанной со спектральными особенностями, отличными от спект¬
ра сравнения. Таким образом, если бы спектр сравнения был спектром чисто¬
го металла, а исследуемый спектр содержал пики металла и оксида, то такой
расчет позволил бы оценить долю оксида в исследуемом спектре. Получен¬
ный результат с пользой можно было бы сравнить с данными, полученными
методом аппроксимации.П.3.7.3. Примеры разностных спектровНа рис. П.3.12 показан разностный спектр, полученный вычитанием двух
спектров, изображенных на рис. П.3.11. Визуально эти два спектра мало от¬
личаются друг от друга, однако разностный спектр выявляет это различие.Так, из спектра, нормированного по площади, видно, что в спектре С Is груп¬
па В1 смещена из-за химического взаимодействия в большей степени, чем
группа В2, которая соответствует "графитовому” углероду (химический сдвиг
приведен к началу координат). Вместе со спектром, нормированным по высоте,
представлены спектры, соответствующие меньшим значениям х. Критерий оп¬
тимальной нормировки удовлетворяется для х = 0,95 при С = 30. Как ожидалось,
оптимальное значение х изменяется при изменении С, как показано на
рис. П.3.13; значение 0,95 соответствует плато на графике, что позволяет
526Приложение 3. Обработка данных в РФЭСРис. П.3.11. Спектр С 1s необработанной углеродной нити типа II . а - В1, при тем¬
пературе окружающей среды; б- В2, после отжига при температуре
1400 °С. Дополнительно к попному экспериментальному спектру показа¬
на увеличенная и сглаженная плазмонная область (р= 21, 15),считать это значение оптимальным. В действительности спектр сх = 0,97,
по-видимому, более информативен и это может означать, что указанное зна¬
чение х является более подходящим в данном случае, чем оптимальное значе¬
ние. Все это может означать, что критерий подгонки является слишком жест¬
ким. Область I, как полагают, соответствует разности интенсивностей двух
спектров, а области II и III - различию в группах С/О на поверхности. Ясно,
что выбор оптимального значения х в реальном спектре является субъектив¬
ным и он определяется неоднозначно, как это имеет место и при нормировке
спектра по высоте и площади. Однако реальные данные в этом случае пред¬
ставлены, и при использовании других методов анализа данных они могут дать
полезную химическую информацию.П.3.8. ЗаключениеОчевидно, что анализ данных должен играть весьма важную роль при ин¬
терпретации рентгеновских фотоэлектронных спектров. В этом дополнении
Рис. П.3.12. Разностный спектр В1 — В2 (рис. П.3.11) при различных параметрах нор¬
мировки. а- нормировка по площади х = 1,03; б-нормировка по высо¬
те х= 1,0; в - х — 0,98; г - х = 0,97; д- оптимальная нормировка
X = 0,95; в - X = 0,93.Рис. П.3.13. Изменение оптимального значения х в зависимости от с для разнос¬
ти В 1(3) — В 2(1).
528Приложение 3. Обработка данных в РФЭСбыла сделана попытка представить основные используемые в настоящее вре¬
мя методы и дать характерные примеры их применения. Много времени и сил
можно затратить на анализ данных и поэтому следует соблюдать осторож¬
ность, чтобы не "переинтерпретировать" данные. Однако существует множе¬
ство систем, для которых спектральные изменения очень малы, но чрезвы¬
чайно существенны, и в таком случае тщательный анализ данных может ока¬
заться единственным средством выделения полезной информации. Доступ¬
ность дешевых систем сбора данных будет поощрять все большее число ис¬
следователей собирать данные в цифровом виде и использовать описанные
методы для повышения спектральной точности. Необходимо быть чрезвычай¬
но осторожным при использовании таких систем обработки, которые изме¬
няют первоначальные данные или автоматически выдают информацию о по¬
ложении пиков, их интенсивностях или даже об элементном составе. Подоб¬
ная обработка данных может основываться на приближениях и предположе¬
ниях об образце, делающих эти методы некорректными и приводящими к
неправильным результатам, если они применяются в данной ситуации.
Несмотря на необходимость соблюдения осторожности и благоразумия при
использовании методов анализа данных, их более широкое применение, бес¬
спорно, явится важным вкладом в развитие фотоэлектронной спектроскопии
в будущем.БлагодарностиЯ хотел бы поблагодарить Американское химическое общество за раз¬
решение воспроизвести многие приведенные в этом приложении рисунки. Я
также рад выразить свою благодарность д-ру А. Проктору за его вклад в
описанные здесь методы, который он внес за время своей студенческой и
аспирантской исследовательской работы.Литература1. Savitsky АGolay Anal. Chem., 36, 1627 (1964).2. Hildebrand F.B., Introduction to Numerical Analysis, Ch. 7, McGraw-Hill,
New York, 1956.3. Proctor A,, Sherwood Р.М.Л., Anal. Chem., 52, 2315 (1980).4. Maddams W,F., Appl. Spec., 34, 245 (1980).5. Lephardt J.O., Transform Techniques in Chemistry (ed. P.R. Griffits),Ch. 11, Plenum Press, New York, 1978, p. 285.6. H or lick G., Anal. Chem., 44., 943 (1972).7. Betty K*R; Horlick G., Appl. Spec., 30, 23 (1976).8. Maldacker У.Л., Davis J.E., Rogers L*F$., Anal. Chem., 46, 637 (1974).9. Kirmse D.W'., Westerberg A.W., Anal. Chem., 43, 1035 (1971).
П.М.А. Шервуд52910. Bush С.А., Anal. Chem., 46, 890 (1974).11. Turin G.L., IRE Trans. Inform. Theory, IT-6, 311 (1960).12. Ernst R.R., Advances in Magnetic Resonance (ed. J.S. Waugh), Vol. 2,
Academic Press, New York, 1966, p. 1.13. Maddams W.F., Appl. Spec., 34, 245 (1980).14. Carley A.F,.t Joyner R.W., J. Electron Spectrosc., 16, 1 (1979).15- Proctor A., Sherwood P.M.4*» Anal. Chem., 54, «13 (1982).16. Yule Я.Р., Anal. Chem., 38, <103 (1966).17. Inouye Т., Harper Т., Rasmussen N.C., Nucl. Instr. Methods, 67, 125 (1969).18. Westerberg A.W[; Anal. Chem., 41, 1770 (1969).19. Morrey J.R., Anal. Chem., 40, 905 (1968).20. Panlath A.E., Millard M.M., Appl. Spec., 33, 502 (1979).21. Pireaux J.J., Appl. Spec., 30, 219 (1976).22. Сhampery D.C., Fourier Transforms and Tbeir Physical Applications,
Academic Press, New York, 1973, p. 17.23. Ansell R.O.. Dickinson Т., Povey A., Sherwood P.M. A*» J. Electroanal.
Chem., 98, 79 (1979).24. Perram J.W., J. Chem. Phys., 49, 4245 (1968).25. Baruya J.W., Maddams W.F., Appl. Spec., 32, 563 (1978).26. Caulcott ESignificance Tests, Routledge and Kegan Paul, London, 1973.27. Bendat J.S., Piersol A.G., Random Data: Analysis and Measurement
Procedures, Wiley-Interscience, New York, 1971.28. Fisher F.E., Fundamental Statistical Concepts, Canfield Press, San
Francisco, Harper and Row, 1973.29. Proctor A., Sherwood P.M.A., Surf. Interface Anal., 2, 191 (1980).30. Doniach S., Sunjic М., J. Phys., C3, 285 (1970).31. Hufner S.K., Wertheim G.K., Phys. Rev., B11, 678 (1975).32. Wertheim G.K., Buchanan D.N.E., Phys. Rev., В16, 2613 (1975).33. Wertheim G.K., Hufner S., Phys. Rev. Lett., 35, 53 (1975).34. Shirley D.A., Phys. Rev., В 5, 4709 (1972).35. Krause M.O., Carlson T.A., Dismukes R.D., Phys. Rev., 170, 37 (1968).36. Fisher D.W., Advan. X-ray Anal., 13, 159 (1969).37. Barrie A., Street F.J., J. Electron Spectrosc., 7, 1 (1975).38. McIntyre N.S., Zetaruk D.G., Anal. Chem., 49, 1521 (1975).39. Bishop H. E., Surf. Interface Anal., 3, 272 (1981).
Приложение 4
СВОДКА ДАННЫХ
ПО ЭНЕРГИЯМ ОЖЕ-ЭЛЕКТРОНОВ,
ФОТОЭЛЕКТРОНОВ И ОЖЕ-ПАРАМЕТРАМК.Д. Вагнер 1}Это приложение является обзором экспериментальных данных по энерги¬
ям оже-линий типа N(E), взятых из литературных источников до 1982 г. В
нем содержатся данные по наиболее узким оже-линиям, если они имеются, и
по наиболее интенсивным фотоэлектронным линиям. Из этих данных рассчи¬
тываются модифицированные оже-параметры, которые представляют собой
разность энергий линий плюс энергия возбуждающего излучения, или, проще
говоря,а' = КЭ (Оже) - КЭ(фотоэлектрон) + hv = КЭ(Оже) + ЭС (фотоэлектрон)(КЭ - кинетическая энергия, ЭС - энергия связи).Здесь как для оже-электронов, так и для фотоэлектронов энергия отсчи¬
тывается от уровня Ферми. Эта величина весьма полезна, так как на ее зна¬
чение не влияют статические заряды, а ее химические сдвиги отражают из¬
менение в энергии экранирования [37]. Подборки данных, аналогичные
этой, представлялись ранее другими способами, включая графики зависимо¬
стей оже-энергий от энергии фотоэлектронов, графики зависимости от хими¬
ческого состояния [69, 80, 91, 92].Конечно, имеются некоторые сложности при отборе линий, которые сле¬
дует включить в таблицы. Мы ориентировались на выделение тех линий, ко¬
торые можно наблюдать в обычных ЭСХА- или РФЭС-спектрах. Было реше¬
но не включать линии валентного типа или CVV оже- линии, так как они обыч¬
но состоят из широких участков и форма этих линий очень чувствительна к
химическому состоянию. Мы не включили элементы до Z = 7 (азот), а также
не сочли необходимым повторять здесь имеющиеся в литературе сводки дан¬
ных для кислорода [119] и фтора [69]. Как исключение из принятого правила
не рассматривать LVV-лжж приведены некоторые данные по широким LVV-
линиям серы и хлора, LM23V-mmm Ti, V, Сг и Мп и LVV- линиям Fe, Со
и Ni. Приводятся также данные по М45РР-линиям Мо, Ru, Rh и Pd. Нако¬
нец, в WOO-серии приводятся данные по NVV-mmm Pt и Au. Данные по1 * C.D« Wagner, 29 Staryiew Drive, Oakland С A 94618, USA.
К.Д. Вагнер531оже-линиям переходов с энергией, превышающей энергию, достижимую при
использовании А1 £-рентгеновских фотонов, приведены для Al, Si, Р, S, С1,
Аг, Вг, Кг, отрывочные данные приведены по ^3М45М45-линиям Sr, Y, Zr,
Nb и Мо,и более высокоэнергетичные M5N6JN67-серии для тяжелых метал¬
лов Та, W, Os, Pt, Au, Hg, Tlf Pb и Bi. He приводятся данные для Rb и
для всех редкоземельных элементов. Для последних оже-линии слишком ши¬
роки, чтобы их использовать для аналитических целей. В литературе также
отсутствуют данные для актиноидов. Итак, в этом приложении приводятся
таблицы для всех стабильных элементов, за исключением элементов с Z < 10,
Rb, редкоземельных элементов,Hf, Re, Iг и актиноидов. Данные как для вы-
сокоэнергетичных, так и низкоэнергетичных оже-серий приведены для S, С1,
Аг, Pt, Au и РЬ. Для M45/V45^45 - и N6704S04s-серий приводятся данные
только для компонент M4N4SN45 и А!6045 0Л5, поскольку для непроводящих
соединений они оказались более резкими и точное измерение энергии было
значительно проще, чем для их М5- и ДО7-пар„Набор справочных фотоэлектронных линий выглядит проще. Он состоит
из 1 s-линий до Na включительно, 2рз/2-линий до Zn, Зс^-линий до Ва и
4/7/2-линий от Hf до Bi. В перекрывающихся областях этот набор в некотором
смысле произволен. Например, представлены также при наличии соответствую¬
щих данных энергии Is-линий для Mg и следующих за ним элементов до Аг
включительно, даже если для их получения необходимо было использовать
источники с большей энергией, чем в рентгеновских трубках с катодом из Mg
и Al. Данные для энергий 2^з/2-линий представлены для Ga, Ge, As, Se и
Вг. Включены некоторые данные для энергий М-линий сурьмы и теллура.
Представлены данные для 3^&/2-линиЙ соединений вольфрама.При составлении этих таблиц была сделана попытка представить данные
по энергиям линий в максимально возможном самосогласованном виде. В свя¬
зи с этим мы отметим следующее:а. Предполагается, что данные для газообразных веществ имеют хорошую
точность, и это обычно определяется использованием их в смеси с инертным
газом, выполняющим роль стандарта. Такие данные представлены без изме¬
нения. Исчерпывающий обзор по энергиям фотоэлектронных линий газовой фа¬
зы был опубликован ранее [120], также может быть весьма полезна информа¬
ция по газовой фазе, содержащаяся в ссылках этого обзора. Все данные для
газовой фазы приведены относительно уровня вакуума и отмечены в этих
таблицах символом v.б. Недавно в работах [102^ 129* 130*] были предприняты попытки получить
точные энергии линий относительно уровня Ферми для легкоприготавливае-
мых твердых образцов. В настоящих таблицах эти значения скорректированыи приведены относительно Au 4/? у2=84,00; абсолютные значения в вышеупомя¬
нутых работах приведены относительно 83,98; 84,1 и 84,00 соответственно.
532Приложение 4. Сводка данных по энергиямв. Для всех других данных по твердым образцам мы предположили, что
для них имеется общая энергетическая шкала с такой работой выхода спект¬
рометра, чтобы Au 4/7/2=84,0, Ag 3d5 /З^368,2 и Си 2ръ /2=932,6. Данные для
любого из этих элементов в соответствующей работе дают возможность при¬
вести шкалу напряжений к общему началу. Аналогично можно осуществить
коррекцию данных для диэлектриков, которые получены с привлечением стро¬
гих методов учета зарядки образцов типа декорирования золотом, использо¬
вания случайных примесей углеводородов или же использования "углеводо¬
родной” части образца, предполагая, что в углеводородах С 1s-284,8 эВ. Ког¬
да по указанным причинам производится изменение данных, то после соот¬
ветствующей ссылки в таблицах ставится буква г. Ссылки, в которых не ука¬
заны энергии естественных одиночных линий или нет связи ферми-уровней,
обозначены буквой п. Часто, хотя в статьях это может и не указываться,
предполагается, что в работах одной и той же лаборатории, разделенных ма¬
лым промежутком времени, шкала напряжений одним из своих концов "привя¬
зана" к одинаковому уровню отсчета (обычно Au 4/?/2). Данные для диэлект¬
риков, в которых не учтена зарядка или же правильность этого учета вызыва¬
ет сомнение, не включались в таблицы,, однако во многих случаях параметры
оже-линий были включены, так как они не зависят от поправки на зарядку об¬
разцов.Работы, содержащие данные для Is- и 2р-линий Na, Mg, Al и Si, а так¬
же для 2Р3/2- и З^-линий Zn, Ga, Ge, As и Se, представляют возможность
проверить масштаб шкалы напряжений, так как разности этих линий для ука¬
занных элементов должны быть равны энергии рентгеновских квантов. Сде¬
лать это оказалось возможным для некоторых работ, и с использованием данных
по энергиям рентгеновских квантов [3] была проведена незначительная кор¬
рекция линий, соответствующих большим энергиям связи, которые помечены
в таблицах буквой с. Возможность учесть нелинейности шкалы напряжений
отсутствует.Старые данные, вытесненные более поздними измерениями в той же ла¬
боратории, не учитывались. Не включены также некоторые результаты, нахо¬
дящиеся в явном противоречии с аналогичными данными большинства других
авторов. Обычно это относится к работам примерно десятилетней давности,
выполненным до появления современных измерительных средств. Некоторые
неопубликованные до настоящего времени данные включены под ссылкой [133].
Подавляющее большинство представленных данных получено с учетом работы
[ 92] и некоторые из них с учетом работы [91]; данные настоящих таблиц бы¬
ли получены тем же способом, который применялся в указанных работах. В
некоторых работах, в которых на одном и том же спектре представлены ли¬
нии, обусловленные изменением как химического, так и физического состояния об¬
разца, подчеркивается различие энергий этих линий. Такие работы были включены
К.Д. Вагнер533в таблицы с обозначением вида Mg -» Mg охи указанием величины изменения
энергии линии, обозначенной д. В том случае, когда имеется изменение фа¬
зового состояния, например Zn -> Zn(g), уровень отсчета энергии, конечно,
остается прежним.Порядок представления данных для соединений каждого элемента грубо
соответствует уменьшению значений оже-параметра, однако с учетом воз¬
можности образования кластеров однотипных соединений.НеонIsKL2^L23ol ЛитератураNe (импл. в Fe)863.4818.01681.4[3 7]rNe (импл. в алмаз)863.1818.71681.8[105]Ne (g)870.31 v804.56 v1674.87[128]Ne (g)870.37 v804.52 v1674.89[12]Ne (g)870.2 v804.8 v1675.0MNe (g)870.0 v804.15 v1674.15[1]НатрийNa1071.8 c944.32066.1 с[71]/'Na1071.5 c994.22065.7 с[13]Na994.5[78 ]nNal1071.6991.22062.8[37]rNal2062.2[27]Nal2063.7[86]NaBr1071.7990.62062.3[37]rNaBr2063.1[86]Na Na oxA +0.7-4.5-3.8[71]Na ox1072.7989.82062.5[71]Na ox2062.0[27]Na ox998.8[2]Na3Sb2064.8[101]Na2KSb2065.1[101]NaBi031071.3990.92062.2[91 ]rNa2Mo041070.9991.02061.9[91]rNa2Cr041071.2990.92062.1[91]/’Na2Cr2071071.6990.42062.0[91]rNa2PdCl41071.8990.22062.0[37]rNaCl1071.6990.32061.9[37]rNaCl2062.8[86]NaSCN1071.3990.52061.8[37]rNa2Se031070.8991.02061.8[3 7)rNa2S2041071.2990.62061.8[3 7]rNa2S2031071.6990.12061.7[3 7]r
Натрий —сот.Is^23^23<x ЛитератураNa?W041071.3990.42061.7[91]rNa2S031071.4990.22061.6[91]rNa2S032062.6 с[118]rNa тиоглюконат1071.2990.42061.6[91JrNa2Te041071.1990.52061.6[37)rNaAs021070.9990.72061.6[3 7]rNa2HP041071.6989.92061.5[9 2]r rNa2HP041071.5989.72061.2[131]Na2Sn03 • 3H201071.1990.32061.4[91]r rNaN021071.6989.82061.4[37]rNa2C031071.5989.82061.3[91]/' rNa2C2041070.8990.52061.3[91 ]r I rNa3P041071.1990.22061.3[131]Na соль EDTA*1070.8990.42061.2[91 ]rNaHC031071.3989.82061.1[91 ]rNa2IrCl6 • 6H201071.9989.22061.1[37jranb6MT(NaAlSi3Os)1072.2988.92061.1[135]rNaH2P041072.0989.12061.1Г131]NaP031071.7989.32061.0[371NaP031071.6989.42061.0[131]Na2S041071.2989.82061.0[37]Na2S042062.1c[118]Na2S042061.9[86]NaOAc1071.1989.92061.0[37]Naconb бензолсульфоновой кислотыЮ71.3989.72061.0[91]rNaH2P021071.1989.82060.9[131]NaOOCH1071.1989.82060.9[91 ]rHaTponnT(Na2Al2Si3O10 •2H20)1072.4988.52060.9[135]rNaN031071.4989.42060.8[91]rNaN032062.2[86]Na цеолитА (NaAlSiOJ1071.7988.92060.6[135]rгидроксисодалит1070.5989.82060.3[135]rNa2ZrF61071.5988.72060.2[3 7]rNa2TiF61071.6988.52060.1[3 7]rNa3AlF61071.9988.22060.1[91 ]rNaF1071.2988.62059.8[3 7]rMaF2061.0[86]NaBF41072.7987.12059.8[37]rNa2GeF61071.7988.12059.8[3 7]rNa2SiF61071.7987.72059.4[37]rNa (g)1078.6v976.7 v2055.3[25]* Этилендиаминтетрауксусная кислотаМагний2PKL 23^23aIs ЛитератураMg49.951185.51235.45Г921 rMg1185.9[791 nMg49.5*1185.51235.01303.2 c[781/7Mg49.61185.71235.3[59]nMg49.61185.61235.21303.2 c[56]* Из 2s = 88,6 в предположении 2s - 2р = 39,1
МагнийMgMgMgMgMg2Р49.449.4
50.0КЬгъЬгг1184.91186.5 с
1185.31185.6
1183.0 vа'1235.9 с1234.71235.6Is Литература
[55)г1303.0 с [33,41]1303.0 [35][37]/*[68]Mg2Cu49.51186.01235.51302.6[32]*Mg3Au49.41185.71235.11302.7[32]*Mg3Bi50.31184.91235.21303.6[32]Mg —► Mg oxА +1.3-6.1-4.8[91]Mg —> Mg oxЛ +1.4-5.9-4.5[59]Mg Mg oxА+0.8-5.1-4.3+ 1.3[56]Mg —> Mg oxА +1.2-5.2-4.0+ 1.5[55]/-Mg —► Mg oxА +1.5-6.2-4.7+ 0.6[33] ,[41]Mg Mg oxА-6.2[18]MgO50.41180.41230.81304.0[91]гMgO (монокрист.)1231.6[59]Mg ацетилацвтонат50.11180.51230.61304.0[91]гMg циклогексаноутират50.71180.01230.71304.2[91]гMg соль эруковой кислоты 50.71180.21230.91304.4[91]гMgSe0451.11180.71231.81304.7[91]гMgS04*7H2051.61178.81230.41305.2[91]/-тал ьк, Mg3Si4011 • H2050.461180.31230.76[135]лMgF250.951178.151229.11305.0[113]гMgF250.91178.01228.91304.8[91]гMg(g)1167.0 v[68]Mg(g)1167.1 v[22]Mg (s) -» Mg (g)А-16.0[68]* Энергии линий из работы [32] на основании более поздней работы из той же лаборатории [56]были скорректированы по кинетической энергии на +0,3 эВ.Алюминий2РKL2i /-2заIsol ЛитератураА172.921393.211466.13[128]г[135]гА172.851393.291466.14[132]А11393.2[79] лА11393.2[55]А11393.0[40]гА11466.2[95]А11558.22953.2 [97]AlAsAIN73.6 1391.2 1464.8
74.0 1388.9 1462.9(132][125]
АлюминийKL23L23 *\s а ЛитератураAl ox74.15*1387.481461.63[128]гAl -+ Al oxА+2.7-7.4-4.7[132]Al -* Al oxА +2.8-7.4-4.6+3.0155)Al -> Al ox-6.5+3.0-3.5[48]A1203 сапфир74.101387.871461.97[128] гА12Оэ сапфир,при 450 °C74.321387.681462.00[128] гor-Al20373.851388.241462.09[128)гA1203 корунд2948.5[1341ai2o31461.9[95]y-Al20373.721387.831461.55[128] гAIO(OH)бёмит74.221387.601461.82[128]гAl(OH)3байерит73.901387.621461.52[135]гА1(ОН)3байерит74.31387.71462.0[132]А1(ОН)3 гиббсит74.001387.431461.43[128]>A1F31460.7[95]Каолинит74.681386.731461.41[135]гКаолинит2948.6[97]Пирофиллит74.711386.751461.46[1351тПирофиллит2948.9[134]Мусковит74.251387.061461.31[1351тМусковит2948.8[134]Альбит74.341386.471460.81[135]тАльбит2947.7[134]Натролит74.251386.531460.78[135]/-2947.8[134]Сподумен74.321387.131461.45[135]тСиллиманит74.581386.861461.44[135]тАндалузит2948.5[134]Альмандин2948.9[134]Анортит2948.5[134]Берилл2949.2[134]Кордиерит2948.7[1341Эпидот2948.6[134]Кианит2948.85[134]Микроклин2946.9[134]Плагиоклаз2948.0[134]Ставролит2949.1[1341Стильбит2946.9[134]Содалит2947.9[134]• Зарядка учтена по С1 s
Алюминий 2р KL2iL2i ql Is а ЛитератураМолекулярное сито
типа АМолекулярное сито
типа XМолекулярное сито
типа Y
Н-Цеолон
Гидроксисодалит73.6674.1374.4574.8273.951386.901386.251385.851385.521486.351460.561460.381460.301460.341460.3[128]г[128)г[128]г
[135]г
[135]гКремнийSiSiSi99.4499.699.61616.681616.31616.41716.121715.91716.0[128]г[124][104]лSi,осажденный из
газовой фазыSiSi99.71616.21616.41715.91839.3 с3455.5 с
3456.3[26]г[55][97]PdSiUi6MoSi2MoSix99.899.5699.41617.4
1617.201617.41717.21716.761716.8[114]г
[128]г
[128]гSiCSiCSi3N4Si,N4101.9*1612.21714.11714.1
1713.73453.73454.15[104][134][124][104][134]ФенилсиликоноваясмолаМетилсиликоноваясмолаПолидиметил-силикон102.74102.92102.401609.961608.801609.381712.701711.721711.78[135]г[135]г[135]гSi oxSi ox (наSi 100)
Si02 (на Si 100)
Si02,осажденный
из газовой фазы103.43103.4103.41608.271608.61608.81711.701712.01711.91712.21842.7 с3451.5 с[128]г[124][124][26]гSi02 а-крис-
тобалит
Si02 а'-кварц
Si02 кварц103.25103.651608.641608.61711.891712.251712.23452.4[128 }г
[135]г
[Ю4]Si02 Vycor103.51608.51712.0[80]г* Получено N2 -бомбардировкой пластины Si (100)
Кремний2р KL2iL23 *Is а ЛитератураSi02 гель
Si02 гель
Si02 гель103.59104.11607.871607.41711.461711.51711.3[135]г[80]г[104]ZnSi031711.8[104]Каламин101.961610.521712.48[135]г3452.7[134],Волласточит102.361609.991712.35[135]гПсевдовол-ластонит102.161610.271712.43[135]гТальк103.131608.931712.06[135]гТальк1712.33453.6[104],[134]Каолинит102.981609.031712.01[135]гКаолинит1711.93451.5[104],[97]ПиоосЬиллит102.881609.201712.08[135]гПирофиллит1712.13453.1[104],[134]Калиевая слюда102.361609.641712.00[135]гМусковит1712.03452.5[104],[97]Силлиманит102.641609.481712.12[135]гСподумен102.461609.591712.05[135]/Альмандин1712.43453.0[104],Анортит1712.33452.4[134]Биотит1712.15[104]Бентонит1712.1[104]Лепидолит1712.0[104]Микроклин1711.953452.0[104],[134]Берилл1711.73452.1[104],[134]Стильбит1711.73451.9[104],[134]Андалузит3452.6[134]Ставролит3453.6[134]Эпидот3452.7[134]Уваровит3452.2[134]КианитКордиерит3452.6 [134]3452.7 [134]
Кремний2р КЬ2ЪЬ2Ъ a Is а ЛитератураОливин3452.2[134]Энстатит3452.6[134]Асбест3452.5[134]Циркон3452.65[134]Серпентинит3453.1[134]Натриевое стекло102.951608.721711.67[135]rАльбит102.631609.261711.89[135]rАльбит3452.3[134]Натролит102.221609.621711.84[135]rНатролит3452.4[134]Гидроксисодалит101.651610.71712.35[135]rСодалит3452.35[134]Плагиоклаз3452.4[134]Молекулярное ситотипа А101.431610.091711.52[128]rМолекулярное ситотипа X102.161609.401711.56[128]rМолекулярное chtqтипа Y102.841608.631711.47[128]rН-Цеолон103.281608.401711.68[127]rSiCI4 (g)110.17v1600.16 v1710.33[Ю8]Si(OMe)4 (g)107.70 v1601.81 v1709.51[Ю8]Si(OEt)4 (g)107.56 v1602.27 v1709.83[Ю8]SiMe4 (g)105.94v1603.74v1709.68[108]SiEt4 (g)106.03 v1604.3v1710.33[108]SiCl3Ph (g)108.81 v1601.95v1710.76[108]SiCl3C3H5 (g)108.92 v1601.51 v1710.43[Ю8]SiCl3Et (g)108.97 v1601.34v1710.31[108]SiCl3C2H3 (g)109.05 v1601.29v1710.34[Ю8]SiCl3Me (g)109.15v1600.96 v1710.11[108]SiCl3H (g)109.44 v1600.3 v1709.74[108]SiCl2MeC2H3 (g)108.07 v1602.08 v1710.15[108]SiCl2Et2 (g)107.85 v1602.52 v1710.37[108]SiCl2Me2 (g)108.10v1601.82v1709.92[108]SiCl2MeH (g)108.53 v1601.17v1709.70[108]SiClMe2C3H5 (g)106.98 v1603.18 v1710.16[Ю8]SiClEt3 (g)106.6v1603.8v1710.4[Ю8]SiClMe3 (g)107.06 v1602.80 v1709.86[108]SiMe3CH2Cl (g)106.23 v1603.62 v1709.85[Ю8]SiMe(OEt)3 (g)107.09v1602.70v1709.79[108]SiMe2(OEt)2 (g)106.69 v1603.00 v1709.69[108]SiMe3OEt (g)106.29v1603.29 v1709.58[108]SiF4 (g)111.70v1595.34v1707.04[108]SiH4 (g)107.1 v1601.2v1708.3 1847.03448.2[30],[84]
Фосфор2pKL2}L23aIs ЛитератураGaP128.71858.91987.6[114]rР (красный)130.21857.01987.2[123] rNaP03134.71848.31983.0[80 ]rNa2HP04133.11850.81983.9[114]/'SPC13 (g)141.15v1842.6v1983.75[84]OPCl3 (g)141.35 v1841.3v1982.65[84]PC13 (g)140.15 v1842.4v1982.55[84]SPF3 (g)142.85 v1839.1 v1981.95[84]C5H5P, фосфазен (g)136.1 v1845.3 v1981.4[81]PF5 (g)144.65 v1836.2 v1980.85[84]OPF3 (g)143.25 v1836.9 v1980.15[84]PF3 (g)142.05 v1837.4v1979.45[84]PH3 (g)137.35 v1842.0v1979.352150.5 v[84]PH3 (g)137.3v1841.4v1978.7[81]СераЛитератураws2162.82115.62278.4[80]rNiWS2162.62115.92278.5[80]rNiS162.82116.12778.9[114]rNa2S203 (S в центре)168.62107.82276.4[80]rNa2S203 (периферийная S) 162.52112.52275.0[80] гNa2S03166.62108.52275.1[136]rCuS04169.12108.02277.1[114]/’SF6174.42100.452274.85[136]rSF6 (g)180.4v2092.6 v2273.02490.1 v[43]SF6(g)180.28 v2092.52 v2272.80[50]CS2163.62111.652275.25[136]rCS2 (g)170.03 v2101.40v2271.43[38]so2167.42106.22273.6[136]rso2 (g)174.8v2095.5 v2270.32483.7 v[43]S02 (g)174.84 v2095.40 v2270.24[50]cos (g)170.8v2099.2 v2270.0[38]H2S (g)170.2 v2098.7 v2268.92478.5 v[43]H2S (g)170.44 v2098.42 v2268.86[50]H2S (g)2099.1 v2477.7 v[84]LLVS (s)164.25152316[133]Хлор2Рэ/2LVVIs ЛитератураПоли(винилхлорид)200.1182.3382.4[92] гKC1199.3181.0380.3[66] rКСЮ3206.5181.0387.5[66] гксю4208.7180.7389.4[66] г
Хлор2р3/2 KL23L23 o' Is ЛитератураСС14 (g)207.04 v2375.72 v2582.76[99]CHC13 (g)206.86 v2375.52 v2582.38[99]CC13F (g)207.20 v2374.93 v2582.13[99]t-C4H9Cl (g)205.38 v2376.64 v2582.02[99]i-C3H7Cl (g)205.62 v2376.17 v2581.7980-1n-C3H7Cl (g)205.81 v2375.77 v2581.58[99]CH2C12 (g)206.62 v2375.15 v2581.77[99]cci2f2 (g)207.47 v2374.18 v2581.66[99]C2H5C1 (g)205.92 v2375.46 v2581.38[99]Cl2 (g)207.82 v2373.72 v2581.54[99]CC1F3 (g)207.83 v2373.30 v2581.13[99]CH3C1 (g)206.26 v2374.51 v2580.77[99]C1F (g)209.18 v2370.73 v2579.91[99]HC1 (g)207.38 v2371.98 v2579.36[99]HC1 (g)2372.2 v2829.2 v [84]Аргон2^3/2l3m2m*3 l2m2m\aIsЛитера*
KL23L23 тураАг(импл. в Fe)241.7216.9*458.6[37]rАг (импл. в Be)211.0v 212.8v[93]/?Аг (ад. на Ад)210.6v 212.7v[47]Аг (полислой на Ад)207.6 v[47]Аг (g)248.62 v203.49 v 205.61 v453.17**[12]Аг (g)3205.9v2660.51 v [4,51]* Центр неразрешенных пиков.•• Рассчитано для среднего значения приведенных оже-пиковКалийк547.9**[101]K3Sb546.8**[101]Na2KSb547.0**[101]KI292.8250.8*543.6[3 7]rКВг293.1248.3 250.7542.6[92]rКС1292.5250.4*542.9[133]KNO3292.9249.3*542.2[3 7]rk2so4542.5[118]K2so3542.3[118]KF292.5249.6*542.1[3 7]rKSbF6293.7248.6*542.3[37]/K(g)300.7 v236.67 v 239.51 v540.2**[127]• Пики состоят в основном из ^3^23^23^2 и L2M23M23D2' Значения* расположенные между этими
колонками, соответствуют неразрешенному дублету.•* Оже-параметр получен из ЦА^А^з- Остальные оже-параметры основаны на оже-энергиях
неразрешенных дублетов
Кальций2^3/2l3m2m; l2m2m3*сг'ЛитератураСа345.9298.2644.1[110] гСаС12348.3291.9640.2[37] гСаО346.5291.9638.4[133]СаО347.3292.5639.8[110]гСа —*• СаОА+1.4-5.7-4.3[110]CaS04347.6291.2638.8[133]гСаС03Волластонит347.0291.8638.8[92]г(силикат Са)347.0291.5638.5[135]гCaF2347.9288.9636.8[133]Скандий8с20з401.9334.9736.8[92 ]гSc оксалат403.3333.5736.8[133]Sc ацетилацетонат402.2333.4735.6[133]ScF3405.0329.8734.8[133]* Пики состоят в основном из ^р^з^з1 ^2 и ^2^23^23 ^2‘ Значения, расположенные
между этими колонками, соответствуют неразрешенным дублетам.Титан2^3/2l3m23 м2Ъakl23l23а Литератур!Ti836.677-6ТЮ2840.3[54]ЦМ2ЪУTi454.0419.1873.1[92 )rTiN455.7420.0875.7[91 }rTiC454.6418.2872.8[91]rТЮ2458.74; 4.9873.6[133]ТЮ2458.5414.7873.2[91]/'ТЮ ацетилацетонат458.4414.8873.2[91]rТ итаноцендихлорид457.2414.9872.1[91]rNa2TiF6462.6409.8872.4[91]rK2TiF6462.1409.4871.5[91]rTi TiCA +1.3-1.5-0.2 [5]Ti -> TiNA+1.5-1.9-0.4 [5]Ti -> TiOA+1.0-1.6-0.6 [5]Ti Ti02A+4.9-5.8-0.9 [5]ВанадийV512.15472.0984.15[92]rV-» VCA + 1.8-2.4-0.6 [5]ХромCr574.3527.21101.5[92]rМарганец2^3/2l3m22vaЛитератураMn639.0586.41225.4[92]rMn582.8 v[126]Mn02642.4584.91227.3[133]Mn02642.3583.91226.2[91]r
МпС12642.8580.91223.7[91]rMnSi03642.3582.41224.7[91 )rK3Mn(CN)6641.7582.01223.7[91]rMn(C24H27N7)(PF 6)2*640.8582.21223.0[91 IfMn (g)561.6v[126]Mn (s) Mn (g)A-21.2[126]• Азотный лиганд содержит три пиридиновых кольца.Железо^Рз/ 2L3VV*а ЛитератураFe706.95702.41409.35*[92]rFe203711.0703.11414.1[91]rFe203710.9702.01412.9[133]FeW04711.5703.11414.6[91 ]rFe2(W04)3711.1702.51413.6[91]rFeS710.4703.21413.6[91 ]rFeS2707.4702.71410.1[91 IfFe11 ацетилацетонат711.5700.81412.3[91]rFe111 ацетилацетонат711.8700.31412.1[91VFe циклогексанбутират712.0700.81412.8[91 ]rFe дитиодибутилкарбамат711.3701.21412.5[91 ]rFeS04-7H20711.0700.41411.4[91 ]rFe(C10H8N2),(PF6)2t708.2699.61407.8[91 ]rK3Fe(CN)6709.9698.41408.3[91]rK4Fe(CN)6708.5698.91407.4[91]rK3FeF6713.8698.61412.4[91]r* Оже-линии для всех соединений, кроме Fe и FeS«, очень широкие, их ширина приблизительноравна 8 эВ, и поэтому точность измерения энергии линии ограниченна.•* Лигандом является CgHgNCH =nch3.Кобальт2Рз/гL3VV а ЛитератураСо778.1773.2 1551.3[92 )rСо778.2773.0 1551.2[611Со304780.0773.6 1553.6[133]Со304780.3773.9 1554.2[91]rСо304779.3773.6 1552.9[61]СоО780.2773.6 1553.8[133]CoSi03781.5770.4 1551.9[91 ]rCo2Si04781.3770.6 1551.9[91 ]rСо циклогексанбутират781.6770.Q 1551.6[91 ]rСо дибутилдитиокарбамат779.5770.2 1549.7[91]rCo(NH3)6Cl3781.7768.6 1550.3[91]rCo(N4-тетраметилэтилендиамин) (N03)2780.0770.1 1550.1[91]rK3Co(CN)6781.9766.8 1548.7[91]rCo(C24H27N7)(PF6)2*780.5773.4 1553.9[91]rCoSiF6783.6768.2 1551.8[91]г* Азотный лиганд содержит три пиридиновых кольца.
Никель?Рз/2L3VVaЛитератураNi852.7*846.2*1698.9*[129]VNi852.5846.21698.7[92]rNi852.6845.91698.5[42]Ni852.7845.81698.5[23]rNi852.9846.11699.0[106]Ni (s) -* Ni (импл. в С)A+0.9-0.7+0.2[106]NiO853.5846.0*1699.5[133]NiO853.5846.4*1699.9[91]rNiO854.1845.81699.9[23]Ni циклогексанбутират856.3842.51698.8[92]rNi ацетилацетонат855.7842.91698.6[92]rKNi биурет856.8841.91698.7[92]rNi трифторацетат856.9841.81698.7[92]rNiSi03856.9841.41698.3[92] гNi диметилглиоксим854.8842.41697.2[92] гNKCmHjtNtKPFJj**855.4842.11697.5[92] гK2Ni(CN)4855.4840.31695.7[921 гNiF2857.4842.41699.8[92] гNiSiF6858.7840.41699.1[92]* Соответствует центру широкой линии.** Азотный лиганд содержит три пиридиновых кольца.Медь?Рз/2L3Ma 5 M45aЛитератураСи932.67*918.65*1851.32*[130]*Си932.68*918.62*1851.30*[102]*Си932.6*918.8*1851.4*[129]*Си932.6918.41851.0[93]Си932.8918.51851.3[57] гСи932.S918.31851.1[12]Си932.6918.61851.2[37] гСи932.6918.81851.4[58]Си932.7 с918.61851.3[42]Си932.2919.01851.2[17]Си932.4919.01851.4[44]Си933.0918.41851.4[771Си933.1918.21851.3[74]Си932.8918.01850.8[15]Си933.0918.11851.1[12]Си932.6918.91851.5[122] гСи918.9[8]А12Си933.9918.01851.9[57] гCuAgSe932.3917.61849.9[77]Cu2Se932.3917.51849.8[771CuSe932.4918.31850.7[77]Cu2S932.5917.41849.9[37] г
Медьу 2OLЛитератураСи Cu2SА + 0.07-1.371.30[133]Си —> Cu,SА + 0.1-1.81.7[29]CuS932.6917.81850.4[77]Cu20932.6916.61849.2[133]Си20932.4917.21849.6[58]Си20932.2917.61849.8[44] гСи20933.1916.21849.3[74] гСи Си20А-0.11-2.00-2.11[133]Си —> Си20Л + 0.1-2.3-2.2[29]Си —* Си20А-2.3[122]Си —> Си20А 0.0-2.2-2.2[15]СиО933.8917.91851.7[133]СиО933.6918.11851.7[58]СиО933.8917.81851.6[74] гСиО933.5917.91851.4[58] гСиО933.0917.91850.9[17]Си СиОА +0.96-0.88+ 0.08[133]Си —> СиОА +1.2-1.0+ 0.2[29]Си СиОА+1.3-0.8+ 0.5[122]Си -> Си(ОН)2А+2.5-2.7-0.2[122]СиС1932.4915.61848.0[58]СиС1932.6915.01847.6[37] гСиС12934.4915.51849.9[58]СиС12935.2915.11850.3[91] гСиС12934.8915.31850.1[121] гСиВг2933.3916.91850.2[121 ]гCuCN933.1914.51847.6[37] гСи2Мо3О10932.0916.51848.5[74] гСиЮ4935.5915.91851.4[133]CuS04935.5915.61851.1[66] гCu(N03)2935.5915.31850.8[66] гСиОТ3935.0916.31851.3[91] гCuMo04934.5916.61851.1[74] гCuSi03934.9915.21850.1[91] гCuC(CN)3933.2914.51847.7[66]гCu(C24H27N7)(PF6)2*934.0915.91849.9[91] гCuF,937.0914.81851.8[91]/'Cub2936.1916.01852.1[58]CuF2936.8914.41851.2[121] гCu -* атомы Си в Si02А 0.7-4.1-3.4[98]Си -* Си (g)А 2.5-13.2-10.7[139]* Азотный лиганд содержит три пиридиновых кольца
Цинк?Рз/2L3M45 M45а ЛитератураZn1021.65992.22013.85[92уZn1021.6992.32013.9[63уZn1021.7992.52014.2[161Zn1021.4 c992.02013.4 c[42]Zn1022.0991.82013.8[27]Zn1021.7 c992.02013.7 c[58]Zn1021.9992.42014.3[23]гZn1021.4 c992.32013.7 c[60]лZn988.4 с[65]Zn988.2 v[68]Zn991.7[8]ZnTe1021.6 c991.32012.9 c[60]лZnTe2012.2[6]ZnSe1022.0 c989.52011.5 c[60]пZnSe2010.2[6]ZnS1021.6 c989.72011.3 c[58]ZnS1021.9 c988.22010.1 с[60 ]пZnS2011.9[6]Znl21022.5 с988.72011.2c[58]Znl22011.1 с[60]ZnBr22010.3 c[60]ZnBr21023.4987.32010.7[37] гZnO1021.7988.22009.9[16]ZnO1022.1 с987.62009.7 с[60]пZnO1022.1 с987.72009.8 с[58]ZnO2010.3[6]ZnO1021.9988.62010.5[133]ZnO1022.2987.42009.6[27]Zn —> Zn oxA + 0.3-4.2-3.9[91]Zn Zn oxA-4.2[65]Zn —► Zn oxA + 0.2-3.5-3.3[23]ZnCl22009.2 с[60]Zn ацетилацетонат1021.4987.72009.1[37УZn(C24H27N7)(BF4)2*1021.3988.32009.6[91]гГемиморфит (Znсиликат)1021.96987.302009.26[13]гZnF21021.8c986.22008.0 с[58]ZnF22008.2 с[58]лZnF21022.8986.72009.5[37]гZnF22007.4[27]Zn (g)973.3 v[21]Zn (g)974.5 v[68]Zn -> Zn (g)A 3.15-13.1-9.9[87]• Азотный лиганд содержит три пиридиновых кольца
Галлий3dL3M45M45 a 2p 3/2ЛитератураGa18.71068.11086.8[92]Ga18.71068.11086.8 1116.6 c[133,37]rGa18.61068.31086.9 1116.5 c[16]Ga18.41068.21086.6[70]Ga18.51069.01087.5 1116.4 c[23]rGa18.51068.11086.6[76]rGaAs (скол)19.21066.41085.6[83]nGaAs (скол)19.41066.21085.6[76]rGaAs (катодн.распыл.) 19.01067.11086.1[76 ]rGaAs (хим. травл.)19.31066.21085.5[76уGaP19.31066.21085.5[133]GaN19.541064.51084.04[107JrGa -* Ga oxA +2.0-6.3-4.3[133]Ga -* Ga oxA+2.6-6.4-3.8[23]Ga20321.01061.91082.9[76]/’Ga20320.31062.81083.1 1117.9[ЩгGaAs Ga oxA+1.0-3.5-2.5[83]Германий3dl3m45m45<*' 2^3/2сг'** ЛитератураGe29.01145.41174.4[6 туGe29.151145.21174.35 1217.2 c2362.4 с [92 ]rGe29.5*1145.0*1174.5 1217.4*2362.4 [64]Ge29.01145.01174.0 1217.0 c2362.0 с [23]rGe29.21144.91174.1[70]GeTe230.01144.81174.8[6 туGeSe230.91143.81174.7[6 туGeS230.51143.71174.2[67]гGe -► Ge ox A +4.1-8.0-3.9 +3.84.2 [52]Ge Ge ox A +3.8-7.9-4.1 +3.74.2 8 [133, 37][3 7]гGe0232.71137.71170.4 1220.62358.3 [133][3 туGe022358.9 [45]Na2GeF633.31135.71169.0 1221.32357.0 [133,3 туK2GeF62357.15 [45]GeBr4 (g)38.95 v1130.32 v1169.27[14]GeCl4 (g)39.6v1129.01 v1168.61[14]GeMe4 (g)35.63 v1132.64v1168.27[14]GeH3Br (g)37.65 v1129.81 v1167.46[14]GeH3Cl (g)37.77 v1129.4v1167.17[14]GeH3Me (g)36.44 v1130.55 v1166.99[14]GeH4 (g)36.9 v1129.5v1166.4[14]GeF4 (g)41.55 v1124.28v1165.83[14]* Значение относительно вакуумного уровня, скорректированное на работу выхода 4,3 эВ
** Основано на 2р3/2 и L3MA5MA5'
Мышьяк3dL3M45M45a2p3/ 2ЛитератураAs41.81225.01266.81323.4 c[39]As41.51226.11267.6[36уAs41.51225.21266.71323.1[133, 91 ]rAs42.11225.01267.11323.7 c[23)rAs41.61225.41267.01323.3[132]GaAs (скол)41.31224.51265.8[83]лGaAs (хим. травл.)41.21225.01266.21323.0[132]GaAs (катодн. распыл.)41.01225.51266.51322.7[139]GaAs (катодн. распыл.)40.91225.41266.31322.8[92].NbAs40.81226.01266.8[39]As2Te31225.0[39]As2Se343.01223.31266.31324.7[39]As2S343.51222.01265.51325.6[39]rAs4S443.11222.71265.81325.1[39]rPh3AsS44.11220.01264.11325.9[39]rMe3AsS44.01219.31263.31325.8[39]rAsl343.51222.91266.41325.6[39]rMeAsI243.51222.31265.81325.1[39]rAsBr345.31218.11263.41327.4[39]rPh3As42.41221.11263.51324.3[39]/As20344.41218.91263.-31326.7[39]As20345.01218.81263.81326.4[91]rAs20344.91218.71263.61326.6[132]rАф546.21217.41263.61328.1[39]rAS20544.91218.61263.51328.8[23)rNaAs0244.21219.41263.61325.6[91VNaAs0244.31219.61263.9[132 ]rNa2HAs0445.51217.11262.61326.8[91]rPh3AsO44.31219.51263.81325.5[39]PhAsO(OH)245.21218.41263.61326.8[39]Ph2AsOOH44.41219.01263.41326.8[39]Me2AsOOH44.61218.41263.01326.3[39](C10H21)2AsOOH44.01219.01263.01325.5[39]BuAsO(OH)245.11218.31263.41327.0[39]KAsF647.81213.81261.61330.0[91УGaAs —> As203A 3.1-5.9-2.8[83]3sL2M45M45C5H5As (арсабензол) (g)211.21248.2[81]AsMe3 (g)211.11247.9[81]AsH3 (g)212.41245.1[81]Селен з d5/2 l,3Л/45 M45a2p 3/2«'** ЛитератураSe 55.51307.01362.5[103]/’Se 55.71306.71362.4[91 ]rSe 55.51306.6*1362.1*1434.2 с 2740.8 с [92)rSe 56.31305.81362.11435.0 c 2740.8 c [23)r
Селен 3dSf2 1/3^45^45 Зрз/2 L3Af23Af45 ЛитератураSeUS® 1.88161.9 1178.2 [66]f
161.1 1180.0 [66]rPh2Se56.01303.81359.8[103]rPh2Se256.01304.11360.1[103]rSe=C(NH2)255.01305.21360.2[103]rPh2SeI258.31301.91360.2[103]/'Ph2SeCl257.91302.71360.6[103]rC7H8SeCl358.11302.61360.7[103]rSe0259.01301.41360.4[103]rH2Se0359.21300.81360.0[103JrH2Se0461.21297.91358.1[103]rPh2SeO57.81301.71359.5[103]rNa2Se0358.51301.21359.7[91]rNa2Se03164.9 1173.1 [66]rNa2Se0460.61298.91359.5mrMgSe0459.51299.21358.7[91]r(NH4)2Se0459.21300.11359.3[91]rK2Se04165.9 1173.1 [66]r* ±0,5 эВ*• Основано на 2py2 и LzMA5MA5Бром3^5/2L3M45 m4Sa2py2 ЛитератураKBr68.71388.01456.7[114]rLiBr68.91389.21458.11548.» [80]rNaBr68.91388.31457.21549.0 [80]rC16H33NMe3Br67.51390.11457.61547.5 [80]rKBr0374.81384.41459.21553.9 [80]rТетрабром фенол-сульфофталеин*70.51387.91458.41550.5 [80]rСВг4 (g)76.7v1378.9v1455.6[7]СНВгз (g)76.8 v1378.6v1455.4mСН2Вг2 (g)76.6 v1378.1 v1454.7[7]CH,Br(g)76.3 v1377.6v1453.9[7]КриптонKr(g)93.8v1460.4 v1554.2[12, 10]СтронцийSrFj1640.6[80]rИттрийy2o31736.5[80]rЦирконийZr OX1831.0[Q0]rНиобийNb ох1919.7[80]r
Молибден 3ds/2 L3Mi5Mib a'Мо
Мо
Мо охМо Мо ох
MoSi2227.9228.0232.7
A+4.7227.72038.82032.2-6.62039.02266.82264.9-1.92266.7M45VV a
222.8 450.7 [92]r
[114 ]r
[114 )r
[114]
[135] г* Индикатор, бромфеноловый голубой.Рутений 3d5/2M45VVaЛитератураRu280.2274.2554.4[92]rRu3(CO)12280.9271.8552.7[133]РодийRh307.2301.3608.5Литература[92]rRh ацетилацетонат310.0298.8608.8[91]rRh(N03)3-2H20310.7297.7608.4[91]rRhCl3-3H20310.0298.2608.2[91]rNa3RhCl6310.0297.7607.7[91]rRh6(co)16308.6298.7607.3[91]/'Rh(NH3)5Cl3310.0297.1607.1[91 J/'(PH3P)3RhBr309.5297.5607.0[91]r(PH3P)3RhHCO309.5297.4606.9[91]r(PH3P)3RhCl307.5297.3604.8[91]rПалладий3^5/2A/4 N45 N4§OLЛитератураPd335.1327.8662.9[92]rPd334.6328.5663.1[94]nPd335.1327.8662.9[115]Mg75Pd25336.2326.4662.6[115]AlgoPd2o337.4325.5662.9[115]Ag3oPdso334.6328.8663.4[115]AggoP^20334.9329.8*664.7[115]Ag90Pd10334.9329.7*664.6[115]Р150^50334.6327.5662.1[94]pAu40Pd60334.5327.5662.0[94]nPd ox336.4326.3662.7[94]/?PdS04338.7324.8663.5[91]rPd ацетилацетонат338.1324.9663.0[91]rPd(N03)2338.2324.7662.9[91]rPdBr2337.7324.9662.6[91]rPd(CN)2338.7323.7662.4[91]rPdCl2338.0325.2662.2[91 ]r(cyC8H12)2PdCl2338.5323.8662.3[91]/Pd(NH3)4Cl2338.4323.8662.2[91]/'(Ph3P)2PdCl2338.0323.6661.6[91]rNa2PdCl4338.0323.4661.4[91]/'K2PdCl4337.9323.1661.0[91]r(Ph3P)4Pd335.1324.4659.5[91 J/’* Энергетическое положение линии необычно
Серебро3*5/2m4n45n4SaЛитератураAg368.28 *357.84*726.12*[130]*Ag368.1357.9726.0[92]rAg368.2358.1726.3[57 ]rAg368.3358.0726.3[58]Ag368.3358.0726.3[11]Ag368.4357.5*725.9[77]Ag368.2357.6*725.8[58]Ag368.2357.9726.1[89]Ag357.8[75]rAg359.3[9]Ag353.7 v[47]Mg21Ag79368.3358.1*726.4[116]Mg3oAg50368.7357.9*726.6[116]Mg97Ag3368.8358.2*727.0[116]AlAg2368.7357.7726.4[57}rAl40Ag60368.8357.7*726.5[116]Al95Ag5369.0357.5*726.5[116]Ag2S368.2356.8*725.0[77]Ag2S724.7[118]Ag2Se367.9357.0*724.9[77]AgCuSe367.9356.9*724.8[77]Ag20368.5356.0*724.5[77]Ag20367.9356.6724.5[11]Ag20367.8356.7*724.5[58]AgO367.6357.2724.8[11]AgO367.4356.6*724.0[58]Agl368.0356.1*724.1[58]AgOOCCF3368.8355.1723.9[37]rAg2S04368.3354.2722.5[37]rAg2S04367.9355.1*723.0[58]Ag2S04722.5[118]AgF367.7355.3*723.0[58]AgF2367.3355.6*722.9[58]Ag (g)341.8*[112]Кадмий3*5/2m4n45n45a'ЛитератураCd405.0383.6788.6[92]rCd404.9383.7788.6[37]гCd405.0384.0*789.0[58]Cd405.3383.5*788.8[57]* Для получения энергии А^Л^Л^-линии к энергии ^5^45^45-линии было добавлено 6 эВ.
Кадмий3*5/2M 4N 45N 45OLЛитератураCd385.6[9]Cd380.5 v*[68]Cd380.4 v[72]Cd380.1 v[46]CdSe0 65Te0 35404.9382.2787.1[138]rCdTe405.2382.4787.6[138]rCdTe405.0382.6*787.6[58]CdSe405.3381.5*786.8[58]CdSe405.0381.7786.7[138]rCdS405.3381.3*786.6[58]Cdl2405.4381.2*786.6[58]CdO404.2382.4*786.6[58]Cd(OH)2405.1380.0785.1[91]/'CdF2405.8378.8784.6[37]rCdF2405.9379.0784.9[58]Cd —► Cd (g)A +2.95-11.8-8.85[87]Cd(g)368.2 v[72]Cd(g)368.3 v*[68]Cd(g)367.9 v[20]• Для получения энергии M4^4^N^линии к энергии M5N45/V45-линии было добавлено 6 эВИндийЛитератураIn443.84410.41854.25[88]In443.8410.4854.2[92]rIn444.0410.5854.5[62]rIn444.3410.1854.4[37 ]rIn444.0410.2854.2[89]In443.6410.5854.1[109]лIn443.4411.5854.9[96]/?In411.1[9]InTe444.3409.2853.5P1VIn2Te3444.5408.9853.4[91 JrInSe445.0408.0853.0[91]rIn2Se3444.8408.3853.1[91]rInS444.5408.3852.8[91]rIn2S3444.7407.3852.0[91]rIn2S3444.3408.0852.3[96]nIn2S3444.4408.9853.3[109J/7InP444.0410.0854.0[109]/?In In oxA+1.2-3.6-2.4[37]ln20444.3406.8851.1[69]rln20444.0406.6850.6[109]/7
Индий3*5/2M4N45N45aЛитература1п203444.3406.4850.7[69]r1п2Оэ444.9406.3851.2[62 Jr1п2Оэ444.9407.2852.1[1091л1п2Оэ443.7407.2850.9[96]n1п(ОН)3445.0405.0850.0[96 ]n1п(ОН)3445.8404.8850.6[109]/?1п(ОН)3444.6405.4850.0[96]nInl3445.8405.8851.6[69]rInl3 (красный)445.3406.5851.8[96]лInl3 (желтый)445.7405.8851.5[96}n1пВг3446.0404.8850.8[69J г1пВг3445.7405.2850.9[96]nInCl444.8405.7850.5[69]r1пС13446.0404.6850:6[69]r1пС13445.8403.8849.6[109]/?InCl3445.7404.8850.5[96]nlnF3446.2403.7849.9[3 7]rInF3445.9403.7849.6[96 ]n(NH4)3InF6445.6404.1849.7[691r(NH4)3InF6445.3404.0849.3[96]nInCl3 (g)394.1 v *[100]InCl (g)393.95 v *[100]In (g)393.25 v *[100]• Для получения энергии М4Л/45Л/45-линии к энергии МдА/^/У^-линии было добавлено 7,6 эВОлово3*5/2m4n45n45aЛитератураSn484.87437.27922.14[88]Sn484.85437.6922.45[92]rSn485.0437.4922.4[3 7]rSn484.9437.5922.4[62)rSn484.8437.4*922.2Г891Sn434.0 v[85]rSnS485.6435.7921.3[3 7]rSn -* Sn oxA+1.5-4.7-3.2[37]Sn Sn oxA-4.1[85]SnO486.8432.15918.95[133]SnO486.7432.0918.7[91VSn02487.3431.8919.1[91]rSn02486.6432.4919.0[133]Sn02486.6432.6919.2[62]r
Олово3*5/2QLЛитератураNa2Sn03486.7431.7918.4[3 7]г(R3Sn)20485.9432.4918.3[91VNaSnFj487.4430.8918.2[3 7]гSnCI2 (g)420.24 v[88]* Для получения энергий Л^ЛМУ^-линии к энергии M^N^N|5>линии было добавлено 8,5 эВ.Сурьма3*5/2A/45 A^45а4dЛитератураSb528.02464.29992.3131.94[88]гSb528.25463.9992.15[92]гSb528.2464.2992.4[37]гSb465.7[9]Cs3Sb991.5[101]K3Sb990.7[101]Na2KSb990.6[101]Na3Sb990.6[101]Sb2S3529.5462.1991.6[37]гSb2S5529.3462.2991.5[3 7]гSb2Os530.0459.7989.7[37]гKSbF6532.9454.4987.3[3 7]гSb4 (g)452.95 v[81, 82]ТеллурЛитератураТе572.85492.131064.9840.26[88]Те572.9492.21065.1[92]гТе573.1491.81064.940.5[52]Те492.2[9]Те —► Те охЛ+3.7-5.2-1.5[37]CdTe572.7490.81063.5[138]гCdSe0 65Te0 35572.6491.11063.7[138]/*Те02576.1487.11063.243.4[53]ТеОэ577.3485.51062.844.6[53]Те(ОН)6577.1485.11062.245.0[53]гPh2Te2573.9488.51062.442.8[53]* гPhTeI3575.8488.21064.044.8[53]гPh2TeI2575.4487.81063.244.4[53]гEt2TeI2575.3487.61062.944.2[53]гMe2Tel2575.6487.61063.242.8[53]гTeBr4576.7487.31064.044.0[53]PhTeBr3576.6486.81063.443.9[53]гPh2TeBr2576.2486.71062.943.5[53]гFC6H4TeBr3576.3487.01063.343.7[53]г
Теллур 3dm M4Ni}N4S a Ad ЛитератураBuTeBr3576.6486.51063.143.9[53]rMePhTeBr2576.0486.61062.643.2[53]rTeCl4576.9486.11063.044.3[53]p-MeOC6H4TeCl3576.7485.91062.644.4[53]fPh2TeCl2576.2486.31062.543.8[53]r(NH^TeCI*576.9486.41063.345.3[53]rТе (тиомочевина)2С12574.7488.31063.042.0[53]rp-MeC6H4TeOOH576.1486.61062.743.7[53]rNa2Te04576.8485.51062.3[3 7]rTe2 (g)479.20 v[82, 88]* Предполагается, что все данные работы [53] с пометкой г приведены относительно линийпримесного углерода С 1s, нуждающихся в коррекцииИод3*5/2M4N4S ^V45or'ЛитератураLil619.7517.01136.7[92]rNal618.9517.21136.1[91]rК1618.7517.01135.7[91]rNil2619.0518.8*1137.8[58]Cul619.0518.6*1137.6[58]Znl2619.8517.5*1137.3[58]Agl619.4518.3*1137.7[58]Cdl2619.2518.5*1137.7[58]Inl3619.9517.21137.1[9]rkio4624.1513.31137.4[9VI2 (g)^I(g)A-3.25[90]* Для получения энергий A^/V^/V^-линий к энергии ^4^45^45’линии было добавлено 11,5 эВ.КсенонЛитератураХе (импл. в графит)669.65545.21214.85[92 }rХе (импл. в алмаз)668.9545.4*1214.3[105]лХе (импл. в Fe)670.2544.81215.0[37]rХе (аде. на Pt)669.5544.21213.7[24]nХе (g) Хе (аде. на Мо)A -1.2+6.3+5.1[13 7]Хе (g) Хе (аде. на МоСХ>)A-3.5+7.1+3.6[13 7]Na4Xe06674.1541.41215.5[69]Na4Xe061216.3*[34]Xe(g)676.4 v532.7v1209.1[4], [Ю]Xe(g)532.8v[19]• Для получения энергий М4Л/45Л/45-линии к энергии М5Л/45Л/45-линии было добавлено 12,7 эВЦезий3*5/2 M4NA5NA5aЛитератураCs1296.6[101]Cs1297.1[117]Cs3Sb1296.1[101]Csl1294.0[101]Cs2041293.1[117]CsCl724.9568.71293.6[133]CsOH724.15568.71292.85[92]rCs2S04723.9568.41292.3[69]r
БарийВаВа oxВа —► Ва ох
ВаО779.3*
779.1
Л-0.2
779.85602.0*598.4
-3.6597.5Литература
1381.3 [110]г
1377.5 [110]r
-3.8 [110]
1377.35 [92]rBaS04780.8596.11376.9[60]гВасоль эруковой кислоты 780.2596.21376.4[133]Ва циклогексанбутират780.1596.11376.2[133]Ва хлоранилат779.6596.71376.3[133]BaF2779.7597.21376.9[133]* Предполагается, что эти данные точнее данных работы [111]Тантал4/7/2М5 NblNbla MANblNbl 3d5/2 ЛитератураТа21.91674.651696.55[114]гВольфрамWC32.51729.11761.61791.51807.7[80]/'ws233.01728.51761.51790.81807.7[80]rNiWS232.81728.71761.51791.11807.7[80]гK4W(CN)832.81725.61758.41787.91807.5[80]r(C5H5)2WC121725.91787.91808.5[80]fwo336.11723.81759.91786.31810.6[80]rh2wo436.11723.91760.01786:31810.5[80]rW(OPh)637.31723.81761.11786.21811.3№CoW0436.01725.01761.01787.21810.7[80 ]rCuW0436.11725.31761.41786.91810.7[80]rNiW0436.01724.31760.31786.61811.2[80]rFe2(W04)336.31723.81760.11786.31810.8[80]rNa2W04-2H2036.41722.81759.21785.31810.4[80]rLi2W0435.71722.81758.51785.11810.6[80]rОсмийЛитератураK2OsCl656.21907.7[80]rK4Os(CN)657.21909.8[60]rПлатинаN61WЛитератураPt71.163.4[133]Pt71.31960.72032.02041.1[80]rK2PtCl473.42035.2[37]rЗолотоЛитератураAu84.0*2015.9*2099.9*[129]VAu84.02015.72099.7[114]rAu84.069.4*[133]Au84.02015.82099.82101.2[80]rAu2015.92101.3[123]r* Вероятно, основная компонента /У7УУ-линии.Ртуть4/7/2 MANblNbl or'^6^45^4 5 *N7045045ЛитератураHg 99.9Hg(g)80.55 180.45 77.75 [133]
63.5 v [49]
ТаллийTl117.8Литература
85.1 [133]СвинецPb137.02180.5 2317.5Литература[114]/'Pb136.896.25233.0592.95 [133]Pb136.896.25233.05[140]PbTe137.2595.45232.7[140]PbSe137.694.75232.35[140]PbS137.594.55232.05[140]PbO137.2592.85230.1[140]Pb02137.493.05230.45[140]Pbl2138.3593.35231.7[140]PbBr2138.892.6231.4[140]PbCl2138.992.1231.0[140]PbF2138.590.6299.1[140]Pb(OH)2137.9591.95229.9[140]Pb(N03)2138.591.7230.2[140]PbSi03138.6591.1229.75[140]PbS04140.090.1230.1[140]РЬТЮ3138.092.6230.6[140]РЬСЮ4138.392.75231.05[140]PbZr03138.591.7230.2[140]РЬ(Ю4)2138.292.7230.9[140]PbW04138.791.8230.5[140]PbNCN137.594.0231.5[140]Pb(OAc)2138.591.45229.95[140]* Полагают, что эту компоненту, которая дает основной вклад в линию N$^45^45 ’ легче всего
измерить для большинства соединений (ср. с [49])Висмут 4Л/2 N6°4S°*S* *' W7045O45 Литература
Bi 15Г0 103Л 260.7 100Л [133]* Полагают, что эту компоненту, которая дает основной вклад в линию легче всегоизмерить для большинства соединений (ср с [49]).Литература1. КЪгЬег ft., Mehlhom W„ Z. Physik, 191, 217 (1966).2. Fahlman A., Nordberg R., Nordling C., Siegbahn K., Z. Physik, 192, 476
(1966).3. Bearden J.4., Rev. Mod. Phys., 39, 78 (1967).4. Siegbahn Nordling C., Johansson et al., ESCA Applied to Free
Molecules, North Holland Publishing Company, Amsterdam, 1969.5. Ramqvist L., Hamrin K., Johansson G. et alJ. Phys. Chem. Solids., 30,
1835 (1969).6. Longer D.W., Vesely C.J., Phys. Rev., B2. 4885 (1970).7. Spohr R., Bergnark TV, Magnusson N. et al., Phys. Scr., 2, 31 (1970).8. Aksela S., Pessa M„ Karras М., Z. Physik, 237, 381 (1970).
558Приложение 4. Сводка данных по энергиям9. Aksela S., Z. Physik, 244, 268 (1971).10. Werme I.Q., Bergmark Т., Siegbahn K., Phys. Scr., 6, 141 (1972).11. Schon G., Acta Chem. Scand., 27, 2623 (1973).12. Johansson G., Hedman J., Bemdtsson A. et al., J. Electron Spectrosc.Relat. Phenom., 2, 295 (1973).13. Kowalczyk S. P., Ley L., McFeely F.R. et al., Phys. Rev., В 8, 3583(1973).14. Perry W.8., Jolly W.L., Chem. Phys. Lett., 23, 529 (1973).15. Kowalczyk S.P., Poliak R.4; McFeely F.R. et al., Phys. Rev., B8, 2387
(1973).16. Schon G., J. Electron Spectrosc., 2, 75 (1973).17. Schon G., Surf. Sci., 35, 96 (1973).18. Wagner C.D-, Biloen P., Surf. Sci., 35, 82 (1973).19. Werme L.O., Bergmark Т., Siegbahn K., Phys. Scr., 8, 149 (1973).20. Aksela H., Aksela S., J. Phys. B. Atom. Mol. Phys., 7, 1262 (1974).21. Aksela S., Aksela H., Phys. Lett., 48A, 19 (1974).22. Breukmarm B., Schmidt V., Z. Physik, 268, 235 (1974).23. Castle J.E., Epler D., Proc. Roy. Soc. London, A339, 49 (1974).24. Erickson N.E>> J» Vac. Sci. Technol., 11, 226 (1974).25. Hillig H., Cleff В.Ц., Mehlbom IV., Schmitz W., Z. Physik, 268, 225 (1974).26.Klasson М., Bemdtsson A., J. Hedman J. et al., J. Electron Spectrosc., 3,
427 (1974).27. Kowalczyk S. P., Ley L., McFeely F.R. et al., Phys. Rev., В 9, 381 (1974).28. Thomas T.D., Shaw R.W., J. Electron Spectrosc., 5, 1081 (1974).29. Larson P., Electron Spectrosc., 4, 213 (1974).30. Perry W.B., Jolly W.L., Inorg. Chem., 13, 12111 (1974).31. Fiermans L., Hoogewijs R., Vetmik J., Surf. Sci., 63, 390 (1977).32. Fuggle J.C., Watson L.M., Fabian D.J., Affrossman S., J. Phys. F. Metal
Phys., 5, 375 (1975).33. Haider H.C., Alonso J., Swartz W.E., Z. Naturforsch, 30a, 1485 (1975).34. Jorgenson C.K., Berthou H., Chem. Phys. Lett., 36. 432 (1975).35. Ley L., McFeely F.R., Kowalczyk S.P. et al., Phys. Rev., В11, 600 (1975).36. Roberts E.D., Weightman P., Johnson C.E., J. Phys. C. Sol. State Phys., 8,
1301 (1975).37. Wagner C.D., Faraday. Disc. Chem. Soc., 60, 291 (1975).38. Asplund L., Kelfve P., Siegbahn H. et al., Chem. Phys. Lett., 40, 353
(1976).39. BahlM.K., Woodall R.O., Watson R.L., Irgolic J., J„ Chem. Phys., 64, 1210
(1976).40. Dufour G., Mariot J.-M., Nilsson-Jatko P.-E., Kamatak R.C., Phys. Scr., 13,
370 (1976).41. Haider H.C., Alonso L., Swartz W.E., Phys. Rev., В13, 2418 (1976).
К.Д. Вагнер55942. Kim K.S., Gaarenstroom S.W., Winograd N*, Chem. Phys. Lett., 41, 503
(1976).43. Keski-Rahkonen 0., Krause M.O., J. Electron Spectrosc., 9, 371 (1976).44. McIntyre N.S., Rummery T.E., Cook M.G., Owen D., J. Electrochem., Soc.,
2123, 1165 (1976).45. Reisfeld R., Jorgenson C.D., Bomstein A., Berthou H., Chimia, 30, 451
(1976).46. Weightman P., J. Phys. C. Sol. State Phys., 9, 1117 (1976).47. Nutall J.D., Gallon T.E., J. Phys. C. Sol. State Phys., 9, 4063 (1976).48. Castle Hazel L.B., Whitehead R.D., J. Electron Spectrosc., 9, 247(1976).49. Aksela H., Aksela S., Jen /.5., Thomas T.D., Phys. Rev., A15, 985 (1977).50. Asplund L., Kelfve P., Blomster В. ?t at., Phys. Scr., 16, 273 (1977).51. Asplund L., Keldve P., Blomster В., Siegbahn H. et al., Phys. Scr., 16, 268(1977).52. Bahl MWatson R.L., J, Electron Spectrosc., 10, 111 (1977).53. Bahl M.K., Watson R.L., Irgolic K.J., J. Chem. Phys., 66, 5526 (1977).54. Armstrong N.R., Quinn R.H., Surf. Sci., 67, 451 (1977).55. Carlson Т.Л., Dress W.B., Nyberg G.b, Phys. Scr., 16, 211 (1979).56. Fuggle /.C., Surf. Sci., 69, 581 (1977).57. Fuggle /.C., Kallne £., Watson L.MFabian D./., Phys. Rev., В16,750 (1977).58. Gaarenstroom Winograd NJ. Chem. Phys., 67, 3500 (1977).59. Hoogewijs RFiemans LVennik /., J. Electron Spectrosc., 11, 171 (1977)60. Hoogewijs R., Fiermans LVennik J. Microsc. Spec. Electron 1, 109
(1977).61. Haber Ungier LJ. Electron Spectrosc., 12, 305 (1977).62. Lin A.W.C., Armstrong N.R*, Kuwana Г., Anal. Chem., 49, 1228 (1977).63. Mariot J.M., Dufour G.. Chem. Phys. Lett., 50, 219 (1977).64. McGilp J.F., Weightman P., J. Phys. C. Sol. State Phys., 9, 3541 (1977).65. Fox J.H., Nuttall J.D., Gallon T.E., Surf. Sci., 63, 390 (1977).66. Nefedov V.L, Zhumadilov A.K., Konitova T.If., J. Struct. Chem., 18, 692
(1977).67. Shalvoy R.B., Fisher G.B., Stiles P.J., Phys. Rev., В 15, 1680 (1977).68. Vayrynen J., Aksela S., Aksela H., Phys. Scr., 16, 452 (1977).69. Wagner C.D., in: Handbook of X-pay and Ultra-violet Photoelectron Spectro¬
scopy (ed.D. Briggs), Chap. 7 and Appendix 3, Heyden and Sons, 1977, pp.387 ~ 392.70. Antonides E., Janse E.C., Sawatzky G.A., Phys. Rev., В15, 1669 (1977).71. Barrie A., Street F.J., J. Electron Spectrosc., 7, 1 (1977).
560Приложение 4. Сводка данных по энергиям72. Aksela S., Aksela НVuontisjarvi М. et al., J. Electron Spectrosc., 11,137 (1977).73. Bahl M.K., Watson R.L., Irgolic K.J., J. Chem. Phys., 68, 3272 (1978).74. Haber J., Machej TUngier L., Ziolkowski Sol., State Chem., 25,207 (1978).75. Mariot ].Ц., Dufour GJ. Electron. Spectrosc., 13, 403 (1978).76. Mizokawa Y., Iwasaki H., Nishitani R., Nakamura S., J. Electron Spectrosc.,
14, 129 (1978).77. Romand R., Roubin М., Deloume J.P., J. Electron Spectrosc., 13, 229 (1978).78. Steiner P., Reiter F.J., Hochst H. et al., Phys. Lett., 66A, 229 (1978);Phys. Stat. Solidi, 90, 45 (1978).79. Van Attekum P.M.Jh. М., Trooster J.ty., J. Phys. F. Metal Phys., 8, L165(1978).80. Wagner C.Z)., J. Vac. Sci. Technol., 15, 518 (1978).81. Ashe A.J., Bahl M.K., Bomben K.O• et.aU, J. Am. Chem. Soc., 101, 1764(1979).82. Aksela H., Vayrynen J., Aksela S., J. Electron Spectrosc., 16, 339 (1979).83. Brundle C.R., Seybold D., J. Vac. Sci. Technol., 16, 1186 (1979).84. Cavell R.G., Sodhi R., J. Electron Spectrosc., 15, 145 (1979).85. Barlow S.Af., Bayat-Mokhtari P., Gallon T.%., J. Phys. C. Sol. State Phys.,
12, 5577 (1979).86. Kuroda H., Ohta Т., Sato Y., J. Electron Spectrosc., 15, 21 (1979).87. Kumpula R., Vayrynen J., Rantala Т., Aksela S., J. Phys. C. Sol. State
Phys., 12, L809 (1979).88. Pessa М., Vuoristo A., Villi M. et al., Phys. Rev., В20, 3115 (1979).89. Parry-Jones A.C., Weightman P., Andrews P.T., J. Phys. C. Sol. State
Phys., 12, 1587 (1979).90. Rantala T„ Vayrynen J., Kumpula R., Aksela S.f Chem. Phys., Lett., 66,384 (1979).91. Wagner C.D., Gale L.H., Raymond R.H., Anal. Chem., 51, 466 (1979).92. Wagner C.D., Riggs W.M., Davis L.E. et al., Handbook о X-Ray Photo-
electron Spectroscopy, Perkin-Elmer Corporation, Physical Electronics
Division, Eden Prairie, .Minnesota, 1979.93. Zehner D.M., Madden H.H., J. Vac. Sci. Technol., 16, 562 (1979).94. Hilaire ]., Legate P., Holl Y., Maire G., Solid State Comm., 32, 157 (1979).95. Castle J.E., West R.H., J. Electron Spectrosc., 16, 195 (1979).96. Nichols G.D., Zatko D.A., Inorg. Nucl. Chem. Lett., 15, 401 (1979).97. Castle J.E., Hazell L.B., West R.H., J. Electron Spectrosc., 16, 97 (1979).98. Young V.Y.y Gibbs R.A., Winograd N., J. Chem. Phys., 70, 5714 (1979).99. Aitken E.J., Bahl M.K., Bomben K.D. et al., J. Am. Chem. Soc., 102, 4874(1980).
К.Д. Вагнер561100. Aksela S., Vayrynen ]., Aksela H., Pennanen S., J. Phys. B. Atom. Molec.
Phys., 13, 3745 (1980).101. Bates C.W., Galan L*E., Proc. Ninth IMEKO Symp. on Photon Detectors,
Budapest, Hungary9 9—12 September, 1980, pp. 100 — 129.102* Bird R.J., Swift P., J. Electron Spectrosc., 21, 227 (1980).103. Bahl M.K., Watson R.1^., Irgolic K.J., J. Chem. Phys., 72, 4069 (1980).104. Castle J.E., West R.H., J. Electron Spectrosc., 18, 355 (1980).105. Evans S., Proc. Roy. Soc. London, A370, 107 (1980).106. Gibbs R.AWinograd N., Young V., J. Chem. Phys., 72, 4799 (1980).107. Redman J., Martensson N., Phys. Scr., 22, 176 (1980).108. Kelfve P., Blomster B.9 Siegbahn H. et al., Phys. Csr., 21, 75 (1980).109. Kazmerski L.L., Ireland P.]., Sheldon P. et al., J. Vac. Sci. Technol.,17, 1061 (1981).110. VanDoveren H., Verhoeven J.A. Th., J. Electron Spectrosc., 21, 265
(1980).111. Verhoeven J.A.Th., Van Doveren H., Appl. Sur. Sci., 5, 361 (1980).112. Vayrynen ]., Aksela S., Kellokumpu М., Aksela H., Phys. Rev., A22,1610 (1980).113. Wagner C.D., J. Electron Spectrosc., 18, 345 (1980).114. Wagner C.D., Taylor J.A., J. Electron Spectrosc., 20, 83 (1980).115. Weightman P., Andrews P.Т., J. Phys. C. Sol. State Phys., 13, L815,L821 (1980).116. Weightman P., Andrews P.Т., J. Phys. C. Sol. State Phys., 13, 3529 (1980).117. Yang S.J., Bates C.W., Appl. Phys. Lett., 36, 675, (1980).118. Turner N.H. Murday J.S., Ramaker D.E., Anal. Chem. 52, 84 (1980).119. Wagner C.D., Zatko D.A., Raymond R.H., Anal. Chem., 52, 1445 (1980).120. Bakke A.A., Chen H.-W., Jolly W.L., J. Electron Spectrosc., 20, 333 (1980).121. Van der Laan G., Westra C., Haas С., Sawatzky G.A., Phys. Rev., В23,
4369 (1981).122. McIntyre N.S., Sunder S., Shoesmith D.W., Stanchell F.W., J. Vac. Sci.
Technol., 18, 714 (1981).123. Scharli М., Brunner J., Z. Physik, В42, 285 (1981).124. Taylor J.A., Appl. Surf. Sci., 7, 168 (1981).125. Taylor J.A., Rabalais J.W., J. Chem. Phys., 75, 1735 (198D.126. Vayrynen ]., J. Electron Spectrosc., 22, 27 (1981).127. Aksela S., Kellokumpu М., Aksela H., Vayrynen J., Phys. Rev., A23,2374 (1981).128. Wagner C.D., Six H.A., Hansen W.J*f Taylor J.A., Appl. Surf. Sci., 9,203 (1981).129? Powell C.J., Erickson N.E., Jach Т., J. Vac. Sci. Technol., 20, 625
(1982).
562Приложение 4. Сводка данных по энергиям130?Seah М.Р., Anthony М.Т., см. приложение 1.131. Swift P., Surf. Inter. Anal., 4, 47 (1982).132. Taylor J.A., J. Vac. Sci. Technol., 20, 751 (1982).133. Wagner C.D., неопубликованные данные.134. West R.H., Castle J.E., Surf. Inter. Anal., 4, 68 (1982).135. Wagner C.D., Passoja D.E., Hillery H.F., et. al., J. Vac. Sci., Technol.,
21., 933 (1982).136. Wagner C.D., Taylor J.A., J. Electron. Spectrosc., 28, 211 (1982).137. Henry R.M., Fryberger T.A., Stair P.C., J. Vac. Sci. Technol., 20,818 (1982).138. Polak М., J. Electron Spectrosc., (в печати).139. Aksela S., Sivonen S., Phys. Rev., A23, 1243 (1982).140. Pederson L., J. Electron Spectrosc., 28, 203 (1982).
Приложение 5
ЭМПИРИЧЕСКИЕ ФАКТОРЫ
ЭЛЕМЕНТНОЙ ЧУВСТВИТЕЛЬНОСТИ
ДЛЯ РФЭСПриведенный ниже набор эмпирических факторов элементной чувствитель¬
ности по отношению к F Is =1,00 выведен из совокупности данных, получен¬
ных на спектрометрах типа Вариан IEE и Перкин - Эльмер 550. В этих спект¬
рометрах сканирование осуществляется путем варьирования тормозящего на¬
пряжения, прикладываемого к эмитируемым электронам; анализатор при
этом работает при постоянной энергии пропускания. Это приводит к измене¬
нию функции пропускания спектрометра, обратно пропорциональному кинети¬
ческой энергии электронов. Поэтому приводимые здесь факторы следует ис¬
пользовать для спектрометров с аналогичными характеристиками пропускания.
[ ср. Seah М.Р., Surf. Interface Anal., 2, 222 (1980) ] и они будут непригодны
для спектрометров, работающих в других режимах. Эти данные взяты из рабо¬
ты Wagner C.D., Davis L.E., Zeller M.V. et al., Surf. Interface Anal., 3, 211
(1981).Интенсивные линии Побочные линии6ПлощадьIsВысота3IsПлощадьЬВысотаЪLi0.0200.020Be0.0590 059В0.130.13С0.250.25N0.420.42О0.660.660 0250 025F1.001 000 040 04Ne1.51.50 070 07Na2.32 30 130 12Mg3.5*в3 30,200 15
Интенсивные линииПобочные линии6ПлощадьВысота3ПлощадьВысота32^3,22P2^/22s2sMg0.120.120.200.15Л10 1850.180.230.17Si0 270.250.260.19Р0.390.360.290.21S0.540 490.330.24CI0.730.610.370.25Аг0.960.750.400.26К0.831.240.830.430.26С а1.051 581.050.470.26Sc(1.1)(1.65J(1.1)0.500.26Ti(1.2)(1.8)(1.2)0.540.26Ър3РTi(1.2/(1.8)(1.2)0.210.15V(1.3)(1.95)(1.3)0.210.16Сг(1.5)(2.3)(1.5)(0.21)(0.17)Mn(1.7)(2.6)(1.7)(0.22)(0.19)I'C(2.0)(3.0)(2 0)(0.26)(0.21)Со(2.5)(3.8)(2.5)(0.35)(0.25)Ni(3.0)(4.5)(3.0)(0.5)(0.3)Cu(4 2)(6.3)(4 2)(0.65)(0.4)Zn4.84.80.750.40Cja5.45 40.840.40Ge6 1.6 0*0.920.40As6.8.6 8*1.000.43ПлощадьПлощадьВысотаВысота3^23 dЗР1/23 р3^3/2Ga0 310.310.840.40Ge0.380.370.910.40As0 530.510.970.42Se0 670.641.050.48Br0 830.771.140.54Kr1 020.910.821.23я0.60Kb1 231.070.871.300.67Sr1 481.240.921.380.69V1 761.370.981.470.71Zr2.11.51.041.560.72
Интенсивные линииПобочные линии6ПлощадьВысота3^5/2ПлощадьВысота3^5/23^5/23 dЗРз/z3 pNb1.442.41.571.100.72Мо1.662.751.741.170.73Тс1.893.151.921.240.73Ru2.153.62.151 300.73Rh2.44.12.41.380.74Pd2.74.62.71.430.74Ag3.15.23.11.520.75Cd3.53.51.600.75In3.93.91 680.75Sn4.34.31.770.75ПлощадьВысота8ПлощадьВысота4d4dSb4.84.81.000.86Те5.45.41.230.97I6.06.01.441.08Xe6.66.61.721.16Cs7.27.02.01.25Ba7.97.52.351.35La(10)r(2)Ce(10)(2)Pr(9)(2)Nd(7)(2)Pm(6)(2)Sm(5)(2)Eu(5)(2)Gd(3)*(2)Tb(3),(2)4 dif 2Dy(2)r(0 6)ГHo(2)(0 6)Er(2)(0.6)Tm(2)(0.6)Yb(2)(0 6)Lu(2)(0.6)
Интенсивные линииПобочные линии6ПлощадьПлощадьВысота4^5/24Л/24/4/7/24^5/24dHf2.051.701.422.350.90Та2.41.891.502.500.90W2.752.01.572.60.90Re3.12.11.662.750.90Os3.52.21.752.90.90Ir2.253.952.41.840.90Pt2.554.42.551.920.90Au2.84.952.82.050.90Hg3.155.53.152.150.95ПлощадьПлощадьЙиллто ПплшО ПLВысотаВысотаDblUU I d 1 lilиЩа/ц b4/7/24/4/7/2 4d5/24^5/2*«5/25 d5^5/2TI3.56.153.5 2.250.950.90.55Pb3.856.73.82 2.351.001.00.6Bi4.257.44.25 2.51.001.10.65Th7.87.8 3.51.20.91.50.9U9.09.0 3.851.31.01.61.0а Факторы чувствительности по высоте основаны на ширинах линий, которые для интенсивных линий
равны примерно 3,1 эВ, характерные линии получены из обзорных спектров непроводящих образцов
В случае неразрешенных спиновых дублетов приведенные данные соответствуют высоте огибающего
пикаФакторы для интенсивных линий не зависят от источника излучения (Mg или AI). Факторы для
побочных линий (2s, 3р, 4d и 5d) до некоторой степени зависят от энергии фотонов
Представленные значения усреднены для AI и Mg Для более точных расчетов эти факторы следует
умножить на 0,9 при использовании излучения Mg и на 1,1 при использовании излучения AI
в Данные, отмеченные звездочкой, соответствуют пикам, полученным с использованием только Al-
рентгеновского излучения
г Данные в скобках указывают на сильную зависимость от химического состояния, так как в этом
случае доминируют многоэлектронные процессы Данные, представленные для ряда Ti—Си,
соответствуют диамагнитным формам, данные для парамагнитных форм будут, как правило, меньше.
Данные для редкоземельных элементов основаны на нескольких экспериментальных точках и
поэтому их следует рассматривать только как грубую оценку.
д Многие данные, основанные на измерении площади пиков, приводятся в случае 3р- и 4(У-переходов
для спиновых дублетов вследствие значительной ширины этих линий Приводятся данные для
суммарного спинового дублета 2р-серий переходных металлов и ЗсУ-серий редкоземельных металлов,
это связано с преобладанием линий «встряски», вследствие чего желательно рассматривать эти
дублеты как одно целое
Приложение 6а. Положение линий3 при использовании Mg-рентгеновского излучения
(шкала ЭС).
Приложение 6.б. Положение линий* при использовании Al-рентгеновского излучения
(шкала ЭС).%Ь9
Приложение в.571а Линии в прямоугольниках являются наиболее интенсивными и лучше всего подходят для
идентификации химических состояний.Для сокращения обозначений 2р^2 заменяется на 2р3, Зс/^ — 3dg и т. д.
в Включает KVV, если 12 з не является остовным уровнем.
г Обозначение упрощено. ’А Включает LVV, если M-уровни не являются остовными и MVV, если ^-уровни не являются остовными.
9 4с/-дублеты этих элементов имеют сложную структуру и вследствие мультиплетного расщепления и
многоэлектронных процессов зависят от химического состояния.
ж Часто наблюдаются, возбуждаются тормозным излучением.•* Простые 4р^/2*линии отсутствуют для этой группы элементов(Wagner С. U., Riggs W. М., Davis L £., Moulder V. F., Handbook of X-ray Photoelectron Spectroscopy (ed
G. E. Muilenberg), Perkin — Elmer Corporation, Eden Kaire, 1979)
Приложение 7а. Энергий линий в порядке их возрастания при использовании
магниевого рентгеновского источника.*17Hf4f7102Si 2p?206Nb Ъdz359Lu 4p3575Те 3db863Ne Is23О ъ105Ga 3p420SKr 3p3359Hg Ad,577Cr 2py872Cd (A)25Та 4/7108Ce4 d5213HF4d5362Gd (A)594Ce (A)875N(A)30F2s110Rb 3db229S 2s364Nb 3p3599F (A)882Ce 3dk31Ge Ъdь113Be \s229Та Adb368Ag 3d,618Cd 3p3897Ag (A)34W4/7113Ge (A)230Mo ldb378К 2s6191 3dj920Sc (A)40V 3p114Pr 4d2'заRb Ърз380U4fi632La (A)928Pd (A)41Ne Is118Tl4/7241Ar 2/73385TI Adb641Mn 2/?3930Pr 3db43Re 4/7119Al 2s245W Ad5396Mo Зр,657Ba (A)934Cu 2p}44As 3d5120Nd Ad263Re Ad,402N Is666In 3p3954Rh (A)45Cr 3p3124Ge Ър з264Na (A)402Eu (A)670Mn (A)961Ca (A)48Mn 3p3132Sm 4d265Zn (A)402Sc 2pя672Xe 3d,970U (A)501 Ad5133P2p3269Sr Зря405Cd 3db677Th Adb980Nd Ъd^51Mg 2p133Sr Ъd5270Cl 2s'410N1 (A)684Cs (A)981Ru (A)52Os4/7136Eu 4d279Os Adb413Pb Ad5686F Is993С (A)55Fe 3p3138Pb 4/7282Ru Ъdb435Ne (A)710Fe 2p31003К (A)56Li Is143As Ър j284Tb 4p3439Ca 2s711Xe (A)1005Th (A)57Se 3d5150Tb Ad287С Is440Sm (A)715Sn Зр 31022Zn 2p,61Со Зр я153Si 2s293Dy 4/7 3443Bi Ads724Cs 3ds1035Ar (A)62Ir 4/7 *154Dv Ad293К 2p3445In Ъd,729Cr (A)1071Cl (A)63Xe Ads158Y Ъd5297lr Ads458Ti 2p37371(A)1072Na Is64Na 2s159B»4/7301V3/73463Ru Зря739U Adb1082В (A)67Ni 3ря161Ho Ad306Но 4p3483Co (A)743О (A)1083Sm Ъd,69Br Ъd'5163Se 3p3309Rh Ъdъ486Sn Ъd5765Те (A)1088Nb (A)73Pt4/7165S 2p 3316Pt Adb498Rh 3p3768Sb3p31103S (A)74Al 2p169Er 4Л319Ar 2s501Sc 2s780Ba Ъd51117Ga 2p375Cs Adb180Tm 4d320Er 4p3515V 2/? 3781Co 2p.1136Eu ъа577Cu 3ря181Zr 3J5331Zr 3p3519Nd (A)784V (A)1155Bi (A)85Au4/7182Br 3p3333Tm 4p3530Sb Ъd5794Sb (A)1162Pb (A)87Zn 3p3185Yb Ad^335Th 4/7531О Is819Sn (A)1169TI (A)88Kr Ъd5189Ga (A)336Au Ads534W3p3822Te3p31176Hg (A)90Ba Ads191В Is337Pd Ъd5553Fe (A)834La 3d51184Au (A)90Mg 2s191P 2s337Cu (A)555Pr(A)839Ti (A)1186Gd 3ds100Hg 4/?197Lu 4*/s342Yb 4/7 3565Ti 2s846In (A)1192Pi (A)101La 4^f5199Cl 2/7 3347Ca 2pi573Ag 3p3855N1 2p3
б. Энергии линий в порядке их возрастания при ирпо/]ьзовании
алюминиевого рентгеновского источника.17Hf 4/7110Rb 3d5229Та Ads385TI Ad,667Th 4d>IQ72Na h23О 2s113Be Is230Mo 3ds396Mo 3p3686F \91072Ti (A)25Та 4/7114Pr Ad238Rb 3p3402N Is710Fe 2p}1079In (A)30F 2s118TM/7241Ar 2p3402Sc 2p3715Sn 3p31083Sm34w 4/7119Al 2s245W Adb405Cd 3ds716Co (A)1105Cd (A)40V 3p120Nd Ad263Re Ad,413Pb Ad5724Cs3db1108N (A)41Ne 2s124Ge 3jp3265Tb (A)422Ga (A)739U Adb1117Ga 2рз43Re 4/7132Sm Ad266As (A)439Ca 2s752Nd (A)1130Ag (A)44As 3d5133P 2p3269Sr 3p3443Bi 4db768Sbapj1136Em 3ds45Cr Ърг133Sr 3ds270Cl 2s445In 3d}780Ba3ds1153Sc (A)48Mn 3p*136Eu Ad279Os Ad<458Ti 2p3781C° 2p31161Pd (A)50144,138Pb 4/7282Ru 3d,463Ru 3Рз786Fe (A)1186Gd 3ds52Os 4/7141Gd Ad287С Is486Sn 3d788Pr(A)1187Rh (A)55Fe Ъръ142Ho (A)293К 2p.497Na (A)822Те Зл31194Ca (A)56Li Is150Tb Ad297lr Adb498Zn (A)827Ce (A)1205U(A)57Se 3d5153Si 2s301Y3p3498Rh 3p3832F (A)1214Ru (A)61Со Зрг154Dy Ad305Mg (A)501Sc 2s834La 3ds1219Ge Ъ362lr 4/7158Y 3d5306Ho 4p,515V?p3855N. 2p31226O(A)63Xe Ad5159Bi 4/7309Rh 3ds530Sb 3ds863Ne Is1230Th (A)64Na 2s161Ho Ad316Pt Ad5531О Is865La (A)1236К (A)67Ni 3p3163Se 3p3319At 2s534М3Рз882Ce 3ds1244Tb34s69Br 3d5165S 2p3320Er 4p3565Ti 2s890Ba (A)1268Ar (A)73Pt4/7169ErAd331Zr 3p3570Cu (A)903Mn (A)1295Dy 3rfj74Al 2p180Tm Ad333Tm 4p3573Ag 3p3917Cs (A)1301Mo (A)75Cs Ads181Zr 3ds335Th 4/7575Те 3ds930Pr id51304C| (A)77Cu 3p3182ВгЗрз336Au Ads577Cr 2p3934Cu 2p}1305Mg U85Au4/7184Se (A)337Pd 3d5595Gd (A)944Xe (A)1315В (A)87Zn 3p3185Yb Ad5342Yb 4p3618Cd 3p3962Cr (A)1321Nt> (A)88Kr 3ds191В Is346Ge (A)6191 3^59701(A)1326As 2p390Ba 4ds191P 2s347Ca 2p3635Eu (A)976О (A)1336S (A)90Mg 2s195Dy (A)359Lu 4p3641Mn 2ръ980Nd 3ds1388Bi (A)99Er (A)197Lu Ad5359Hg Ads643N1 (A)998Те (A)1395Pb (A)100Hg 4/7199c\ 2p3364Nb 3p3.666In 3p31017V (A)1402"П (A)101La Ads206Nb 3d5368Ag 3^5668Ne (A)1022Zn 2рз1409Hg (A)102Si 2p3208КгЗрз378К 2s672Xe 3db1027Sb (A)1417Au (A)105Ga 3p3213Hf Adb380U4/7673Sm (A)1052Sn (A)1425Pt(A)108Ce Ad5229S 2s* Оже-линии обозначены буквой А в скобках В таблицу включены самая интенсивная оже-линия и
две наиболее интенсивные фотоэлектронные линии для каждого элемента. Для упрощения состояния
2рз/2 обозначаются 2р3,3d5/2 — 3с/5 и т.д Для всех линий приведена энергия связи (Wagner С. D, Riggs W. М., Da¬
vis L Е., Moulder J. Е, Handbook of X-ray Photoelectron Spectroscopy (ed.G. E. Muilenberg), Perkin-Elmer Corporation,
Eden Praire, 1979)
Приложение 8
КИНЕТИЧЕСКИЕ ЭНЕРГИИ
ОЖЕ-ЭЛЕКТРОНОВ:
ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ
ИЗ СПЕКТРОВПРИ РЕНТГЕНОВСКОМ ВОЗБУЖДЕНИИ
К.Д. Вагнер ”Приведенная ниже таблица содержит, по мнению автора, самые луч¬
шие из имеющихся данных по наиболее заметным оже-линиям в спектрах,
возбуждаемых рентгеновским излучением. Представленные линии являются
в своем большинстве линиями остовного типа и автор не пытался включить
линии валентного типа, такие, как KVV для Li - О, LVV для Mg - Cl, MW
для Сг - Y и NVV для 1г - Hg. Йе были включены низкоэнергетичные N00-
линии для Tl-Bi и довольно заметные линии переходов Костера - Кронига
для тяжелых металле®. Не были включены также некоторые линии очень
малой интенсивности типа KLV-, LMV- и MNV-линий и сателлиты на крыльях
интенсивных линий.Данные представлены в основном для элементов в проводящем состоя¬
нии. Данные для газовой фазы используются в том случае, если они значи¬
тельно более детальны, чем данные для твердой фазы. Для некоторых эле¬
ментов также включены данные для оксидов в тех случаях, когда спектры
для элементов и оксидов значительно различаются. Для других данных сле¬
дует учитывать химические сдвиги.Подавляющее большинство данных получено из информации, содержа¬
щейся в работе [23]. Упоминаются также другие работы, имеющие важное
значение для распределения энергий линий. Данные приводятся, если это
не оговорено особо, относительно уровня Ферми. Для многих линий эти энер¬
гии близки к энергиям линий, наблюдаемым при обычном электронном воз¬
буждении, т. е. данным с/ДО/</£-типа, отсчитываемым от уровня вакуума и
часто используемым в электронной оже-спектроскопии, так как работа вы¬
хода, равная приблизительно 4-5 эВ, смещает разность между положени¬
ем пика и точкой перегиба в высокоэнергетичной части линии. Используют¬
ся следующие сокращения:(g) газовая фаза,v по отношению к уровню вакуума,
i интерполированное или экстраполиро¬
ванное значение,Ь широкая линия,1 низкая интенсивность.* ^ Wagner C.D,1, 29 Starview Drive, Oakland, СА 94618, USA.
К.Д. Вагнер579БлагодарностиПри составлении этих таблиц использовались неопубликованные дан*
ные, полученные в отделении физической электроники фирмы Перкин-Эль-
мер, а также данные, представленные в работах [23] и [261Литература1. Korber Н., Mehlbom WZ. Phys., 191, 217 (1966).2. Albridge R.G., Hamrin КJohansson G., Fahlman A., Z. Phys., 209, 419
(1968).3. Gleff £., Mehlhom W., 219, 311 (1969).4. Spohr R; Bergmark Т., Magnus son N. et al., Phys. Scr., 2. 31 (1970).5. Aksela S., Z. Phys., 244, 268 (1971).6. Coad J.P., Phys. Lett., 37A, 437 (1971).7. Johansson C., Hedman JBemdtsson AJ. Electron Spectroscopy, 2, 295
(1973).8. Schon G„ J. Electron Spectr., 2, 75 (1973).9. Aksela Aksela S., J. Phys., fe7, t262 (1974).10. Barrie A., Street J. Electron Spectrosc., 7, 1 (1977).11. Fuggle J.C., Watson L.M., Fabian D.J., Affrossman S., J. Phys., F5 375
(1976).12. Wagner C.D., Faraday Disc. Chem. Soc., 60, 291 (1975).13. Dufour G., Mariot /. М., Nilsson*]atko P.-E., Kamatak R. C., Phys. Scr.,
13, 870 (1976).14. Antonides Eu Janse E.C., Sawatzhy G.4.# Phys* Rev. В15, 1669 (1977).15. Asplund L., Kelfve P., Blomster В. et ah, Phys. Scr., 16, 268 (1977).16. Asplund L., Kelfve P., Blomster В. et aL, Phys. Scr., 16, 273 (1977).17. Bahl M.K., Watson R.I+., J. Electron Spectrosc., 10, 111 (1977).18. Carlson T.A., Dress W.B., Vyberg G.L., Phys. Scr., 16, 211 (1977).19. Mariot J.-M., Dufour G., Chem. Phys., Lett., 50, 219 (1977).20. Roberts E.D., Weightman P., Johnson C.£., J. Phys. C. Sol. State Phys.,
8, 1301 (1975).21. Van Attekum PM.ThMTrooster ].Ц., J. Phys., F8, L169 (1978).22. Wagner C.D., J. Vac. Sci. Technol., 15, 518 (1978).23. Wagner C.D., Riggs W.M., Davis. L.E. et al., Handbook of X-ray Photo¬
electron Spectroscopy, Physical Electronics Division, Perkin-Elmer —
Corporation, Eden Prairie, Minnesota, 1979.
580Приложение 8. Кинетические энергиии оже-электронов24. Barlow S*M; Bayat-Mokhtari P., Gallon T.E., J. Phys. C. Sol. State Phys.,
12, 5577 (1979).25. Van Doveren HVerhoeven J.A.Th., J.Electron Spectrosc., 21# 265 (1980).26. Wagner C.D., Taylor J.A., J. Electron Spectrosc., 20, 83 (1980).27. Aksela S., Kellokumpu Aksela //., Vayrynen ]., Phys. Rev., A23, 2374
(1981).
ИМЕННОЙ УКАЗАТЕЛЬАбико (АЫко) 291
Адамс (Adams) 157
Андерсон (Anderson) 364
Анжевен (Angevine) 341, 357, 392, 393
Ансель (Ansell) 471
Аптекарь Е. J1. 385
Армстронг (Armstrong)) 384
Армур (Armour) 323
Асами (Asami) 448, 449, 435, 458, 466,
467, 469Бадринараджанан (Badrinarajanan) 374Барбулеско (Barbulesco) 463Барр (Barr) 340, 369, 393Барри (Barrie) 67Барья (Baruya) 512Батиста-Лкаль (Batista-Lcal) 354Башенко О. А. 224, 225Берд (Bird) 480, 481Берешейм (Berresheim) 231, 235Беркстренд (Burkstrand) 269Бернардини (Bemardini) 306, 307Бернс (Bums) 334Берч (Burch) 353Бетцольд (Baetzold) 360, 361, 393Билон (Biloen) 144, 355, 392, 393Бимсон (Beameon) 26Биндер (Binder) 420Бишоп (Bishop) 96, 205, 230—232, 519Бозон-Вердурас (Bozon-Verduraz) 353Бом (Bohm) 17Бонель (Bonnelle) 379Борисов В. Т. 306Боумэн (Bouwman) 392Браво (Bravo) 372Брайант (Brtant) 310, 311Брайнен (Brinen) 322, 335, 341, 358, 361,
364, 380, 384
Брандл (Brundle) 20, 333, 377
Бреди (Brady) 267, 269
Брехт (Brecht) 420
Бриггс (Briggs) 335
Брунауэр (Brunauer) 296
Бударт (Boudart) 334
Бхасин (Bhasin) 322, 362
Бэр (Baer) 465Вагнер (Wagner) 139, 144, 145,
232—236, 324, 330, 480, 492
Вайнберг (Weinberg) 332
Вальтере (Walters) 358
Вандет (Wandett) 378
Ван Сантен (Van Santen) 321
Ван Циттерт (Van Cittert) 196, 194, 197
Вартули (Vartuli) 341, 392
Ватанабе (Watanabe) 291
Вебер (Weber) 16
Ведрен (Vedrine) 362, 388, 392
Вейбулл (Weibull) 291
Венник (Vennik) 378
Вернер (Werner) 186
Вертгейм (Wertheim) 194, 517
Виндэви (Windawi) 337, 340, 341
Виноград (Winograd) 117, 351, 352, 360
Вихтерлова (Wichterlova) 372
Вудраф (Woodruff) 304
Вулси (Woolsey) 378Гаренструм (Gaarenstroom) 117
Гаярдо (Gajardo) 384, 393
582Именной указательГейл (Gale) 480Герц (Hertz) 18Геттинс (Gettins) 226Гиббс (Gibbs) 306Гильдебранд (Hildebrand) 502Глэнд (Gland) 330—332Голей (Golay) 502Голиков В. М. 306Гомер (Gomer) 333Грабке (Grabke) 310Гранж (Grange) 323, 383, 384, 393Грехем (Graham) 486, 469Грижинский (Gryzinski) 206, 207Гримбло (Grimblot) 379Гриффитс (Griffiths) 354Густафсон (Gustafson) 373Гутман (Guttmann) 302, 303, 305, 310Даутценберг (Dautzenberg) 358
Де Ангелис (De Angelis) 377
Де Бройль (de Broglie) 15, 18
Де Врие (De Vries) 240
Деклерк-Гримс (Declerck-Grimce) 379
Делане (Deiannay) 384
Делафос (Delafosse) 322, 365
Дельво (Delvaux) 383, 384
Дельгас (Delgass) 322, 341, 357, 392, 393
Дельмон (Delmon) 383, 384, 388, 393
Деминг (Deming) 296
Демпси (Dempsey) 372, 377
Денч (Dench) 210, 212, 283
Дефоссе (Defossfc) 393
Дженкинс (Jenkins) 116, 463
Джермер (Germer) 15
Джеффрис (Jeffries) 89
Джойнер (Joyner) 507
Джонсон (Johnson) 288, 293, 457
Джордано (Giordano) 358, 380
Джоши (Joshi) 324, 446
Дилкс (Dilks) 154, 406, 409, 420, 421,
433Долби (Dolby) 69
Дониах (Doniach) 517
Дуайер (Dwyer) 372
Дуайт (Dwight) 420Дьюк (Duke) 75
Дэвис (Davis) 119, 212
Дэвиссон (Davisson) 14, 15
Дюмулен (Dumoulin) 302Заира (Zaera) 343, 345, 352, 393
Замбаулис (Zambaulis) 372
Захтлер (Sachtler) 321, 345—355
Зенер (Zehner) 463
Зетарук (Zetaruk) 468, 469
Зигбан (Siegbahn) 19, 20, 101, 318, 326,
489Зигмунд (Sigmund) 181Иейтс (Yates) 319, 323, 392
Иманака (Imanaka) 380
Иннес (Innes) 18Итимура (Ichimura) 207, 238, 283Йохансон (Johanson) 480Казмерский (Kazmerski) 247
Каннесон (Cannesson) 323
Карасима (Karashima) 291
Карберри (Carberry) 321
Каретт (Carette) 102, 104
Карли (Carley) 507Кастл (Castle) 146, 340, 446, 466, 470Келли (Kelley) 73, 241, 335, 337, 338, 340Кемпф (Kempf) 167Керкхоф (Kerkhof) 341, 364, 392Ким (Kim) 117, 380, 378Кимура (Kimura) 291Киршнер (Kirschner) 182Китамура (Kitamura) 291Киши (Kishi) 331Кларк (Clark) 353, 403, 406, 409, 420,
433Клаузинг (Clausing) 116
Клейн (Klein) 385, 386
Клейтон (Clayton) 466
Коад (Coad) 96, 296
Именной указатель583Кобурн (Coburn) 23, 241
Коглан (Coghlan) 116
Коел (Кое!) 148
Коллинс (Collins) 420
Коннер (Conner) 464
Коннор (Connor) 491, 492, 498
Контур (Contour) 322, 359, 365
Крамер (Kramer) 293
Кринер (Сгеапег) 353
Кузницки (Kuznicki) 374
Кук (Cook) 378
Кумар (Kumar) 296
Кучинский (Kuczynski) 321
Кэй (Кау) 23Ландер (Lander) 15Ландольт (Landolt) 178Лансфорд (Lunsford) 373, 393Ларкине (Larkins) 117, 374Лейграф (Leygraf) 460Лемонс (Lemons) 269Лехериси (Le Hericy) 450, 452Ли0-еа)179, 180, 192, 299, 305, 307, 311Либагл (Lebugle) 480Ликоргиотис (Lycourghiotis) 384, 393Линдсей (Lindsay) 322Липски (Lipski) 362Литмарк (Littmark) 181Лонжерон (Longeron) 450, 452Лоу (Lowe) 420Льюис (Lewis) 194, 337, 338, 340Мадден (Madden) 194
Макгайр (McGuire) 213
Макдональд (McDonald) 269
Мак-Доуэлл (McDowell) 378
Мак-Доугалл (McDougall) 469
Макинтайр (McIntyre) 378, 468, 469
Мак-Кейб (McCabe) 333
Мак-Ки (McKee) 333
Мак-Кэррол (McCarrol) 321—323
Маклин (McLean) 280, 295, 2%, 303
Макмагон (McMahon) 310Мартинес (Martinez) 321
Маскет (Musket) 54
Массиньон (Massignon) 131
Массос (Massoth) 379, 384
Матьенцо (Matienzo) 322
Мацумото (Matsumoto) 362
Меддамс (Maddams) 512
Мейди (Madey) 319, 323, 324, 351, 392,
480Мейдикс (Madix) 319, 334, 352Мейер (Mayer) 420Мейсон (Mason) 360, 361, 393Мелера (Melera) 322Менсон (Menson) 224Мерфи (Murphy) 468Метью (Mathieu) 178Миллард (Millard) 421Миллер (Miller) 333Миначев X. М. 368Митчел (Mitchell) 381, 452, 469Мозли (Moseley) 18Морабито (Morabito) 169, 193, 211Мосс (Moss) 358Мортенсон (Mortensson) 480Моулдер (Moulder) 269Мсезан (Msezane) 224Мулин (Moulijn) 341, 364, 392Мюллер (Muller) 88, 468Hr (Ng) 326Нефедов В. И. 224, 225, 234, 235, 341
Николас (Nicholas) 420
Новицки (Nowicki) 269
Нортон (Norton) 458Оверайндер (Overeijnder) 240
Оже (Auger) 15, 19Окамото (Okamoto) 364, 374, 379, 380,
382, 383, 385
Оку (Oku) 233
Очкур В. И. 206
Оуэн (Owen) 469
Оуэнс (Owens) 429, 433
584Именной указательПалмберг (Palmberg) 16, 17, 100
Паул (Paul) 269
Педерсон (Pederson) 373, 393
Пенн (Penn) 211, 212, 233, 238
Пеплински (Peplinski) 480
Пеплоу (Peplow) 470
Периа (Peria) 16
Перрам (Perram) 512
Пиро (Pireaux) 409
Пичард (Pichard) 301
Понс (Pons) 450, 452
Портер (Porter) 26
Поуп (Pope) 358
Поуэлл (Powell) 351, 450
Праттон (Prutton) 82
Преветт (Prewett) 89
Проктор (Proctor) 502Ратнер (Ratner) 416Резерфорд (Rutherford) 18Рейли (Reilley) 410Рейлман (Reilman) 224Реймонд (Raymond) 430Рейтер (Reuter) 207, 211Рёлль (Roll) 185Ривьер (Riviere) 96, 205Риггс (Riggs) ЩРихтер (Richter) 430Робертс (Roberts) 20, 331, 333, 334Робинсон (Robinson) 18, 19Родин (Rhodin) 16Рой (Roy) 102, 104Роуланде (Rowlands) 304Роулинсон (Rawlinson) 18Савитский (Savitsky) 502Сайкс (Sykes) 192Сандер (Sunder) 471Саниман (Saniman) 467Свифт (Swift) 338, 480, 481, 489, 490Ceo (Seo) 460Серве (Servais) 471Секстон (Sexton) 330, 331, 332Серфас (Serfass) 19Сикафус (Sickafus) 99, 215, 218
Симицу (Shimizu) 207, 238, 283
Симодаира (Shimodaira) 448, 466, 467
Сих (Seah) 20, 50, 179, 180, 192, 210,
212, 238, 293, 2%, 298, 299, 302, 305,
307Скаррел (Scurrell) 364
Скелтон (Skelton) 312
Скотт (Scott) 156Скофилд (Scofield) 134, 233, 235, 337
Скохарт (Scohart) 393
Смарт (Smart) 288
Смит (Smith) 468Сомораи (Somorjai) 319, 321, 333, 342,
344, 345, 392, 393
Спенсер (Spencer) 311
Стаблер P. 206
Стейнхард (Steinhardt) 19
Стивенс (Stevens) 322
Стодарт (Stoddart) 358
Сузуки (Suzuki) 291
Сунджик (Sunjic) 517Тайлер (Tyler) 73
Тайсон (Tyson) 305
Такасу (Takasu) 360
Таусик (Tauszik) 36p
Тейлор (Taylor) 139
Теллер (Teller) 2%Темпре (Tempere) 322, 368
Тераниси (Teranishi) 380
Тернер (Turner) 26
Тодд (Todd) 333Томас (Thomas) 157, 245, 248, 267, 332Томлин (Tomlin) 205, 206Томпкинс (Tompkins) 321Тонкс (Tonks) 88Трейси (Tracy) 17Тюрин (Turin) 506Уайт (White) 148, 319, 334
Уолтон (Walton) 392
Уортингтон (Worthington) 205, 206
Уотсон (Watson) 345, 392
Именной указатель585Уэлс (Walls) 180, 192 Цинг (Zingg) 382, 383, 392, 393Уэст (West) 146, 340Фагл (Fuggle) 480Файула (Fajula) 374Фанг (Fung) 341, 361, 392Фарж (Farge) 75Фарнсворт (Farnsworth) 14, 15Федли (Fadley) 151Филлипс (Phillips) 228Финстер (Finster) 353Фирмане (Fiermans) 378Фишер (Fischer) 319, 330, 331, 392Френкель (Frenkel) 88Фрост (Frost) 378Фэдли (Fadley) 322, 326Хабер (Haber) 378
Халтгрен (Hultgren) 307
Хаммонд (Hammond) 472
Харрис (Harris) 15, 16
Хасимото (Hashimoto) 448, 455, 466,
467, 469
Хафнер (Hufner) 517
Хензель (Haensel) 349
Хирокава (Hirokawa) 233
Хо (Но) 194
Ховланд (Hovland) 267
Хоге (Hoge) 267
Холл (Hall) 192, 211
Холлоуэй (Holloway) 99, 215, 245
Хониг (Honig) 23, 293
Хондрос (Hondros) 296, 307, 311
Хоуалла (Haualla) 388
Хоустон (Houston) 194
Хофер (Hofer) 182
Хуанг (Khuang) 378
Хугевийс (Hoogewijs) 378
Хьюз (Hughes) 228, 322Чанг (Chung) 116
Чаран (Czaran) 353
Чимино (Cimino) 351, 352, 377
Чин (Chin) 386—388, 390, 391Шарпен (Scharpen) 341
Шербединский Г. В. 306
Шервуд (Sherwood) 502
Шён (Schoen) 462
Шимерска (Szymerska) 362
Шимонски (Szymonski) 240
Шипиро (Shipiro) 354, 392
Ширли (Shirley) 219, 229, 231, 232
Шмидт (Schmidt) 333, 468
Шмит (Schmitt) 358, 361
Шнабель (Schnabel) 353
Шон (Schon) 480
Шусмит (Shoesmith) 470Эбель (Ebel) 233Эванс (Evans) 156, 157, 234, 235, 354
Эверхарт (Everhart) 410
Эдмондс (Edmonds) 322, 381, 382, 385
Эйре (Eyre) 296
Эйринг (Eyring) 374
Эклунд (Ekelund) 460
Энтони (Anthony) 374
Эплер (Epler) 470
Эренберг (Ehrenberg) 15
Эриксон (Erickson) 351, 480
Эркюль (Hercules) 326, 381, 385—388,
390, 391, 393
Эртль (Ertle) 334, 393
Эрхарт (Erhart) 310
Эскар (Escard) 353
УКАЗАТЕЛЬ ХИМИЧЕСКИХ
СОЕДИНЕНИЙAg 50, 122, 180, 207
Al 50, 68, 69, 75, 91, 148, 224, 254,
413, 530
AlGaAs 267А1203 184, 232, 328, 338, 341, 347, 353,
463
A12Cu 259
А12(Мо04) 380, 385
Al/Zr 70
Ar 530As 50, 145, 530Au 50, 152, 180, 239, 250—252, 528,
530
Au203 471
В 50
Ba 236
BaO 232
Be 50, 207, 239
Bi 530Br 407, 413, 530С 50, 69, 125, 207, 239, 250—252Ca 50, 144CaC03 232, 413Cd 50, 122, 207, 239Ce 50Ce02 151C2H 344C2H3 344Cl 407, 530Cl/Rh 364CO 345C02 378Co 50, 121, 150, 162, 528
CoA1204 378, 384, 388, 392
CoMo04 378, 386, 463
[Co (NH^H^H^H^NO^JNO-, 141
CoO 378
Co304 384, 391Co(OH)2 378
CoOOH 378
Co9S8 383Cr 50, 68, 120, 3 239, 250—252, 413,
528
Cr3C2 125
CrF2 232Cr203 456, 458, 465
Cr204 456
Cr03—NiO 468
CsOH 232Cu 50, 68, 121, 180, 220, 236, 239
CuO 229, 308, 457, 462
Cu20 457, 462
Cu (OH)2 462, 470
CuS 157Cu/Si02—A1203 322
Dy 239
Er 50, 239
F 236, 407Fe 50, 120, 230, 239, 348, 460, 466, 528Fe203 230Fe (COOH)2 460a-Fe203 460, 465Fe304 460a-FeOOH 460FeMo04 460, 467Fe—0,22%Sn 300GaAs 157, 267, 268, 530Gd 50Ge 50, 236, 413, 530
H2 334, 348, 360
HC1 364
H20 162
H2PtCl6 353
H2S 380
H2S/H2 383
H2S04 286
Указатель химических соединений587H2S04-CuS04 468
Не 160Hf 50, 236, 239
Hg 50, 530
In 50, 122, 145, 327
ln203 327
InSb 327
I2 50, 239
К 413
KBr2 232K2Cr207/H20/H2S04 424Kr 530La 50LaB6 83LaRh03 345Li 50, 236Lil 232Lu 50Mg 50, 68, 91, 138, 144, 148, 224, 530,
531
MgO 184
Mg(OH)2 470
Mg/Zr 70
Mn 50, 120, 528
MnO 459
Mn02 459Mo 50, 68, 308, 462, 528
Mo2C 288, 289
Mo02 378
Mo03 378, 380
Mo03—A1203 382
Mo03—ТЮ4 388
MoS2 383
N 50N2 334, 348
Na 413
NaC104 157
Na2Mo04 378
Na2S04 469
NaxW03 144
Nb 50, 68
Nb205 189
Nd 50
Ne 532
NH3 334, 348Ni 50, 68, 121, 239, 413, 461, 528Ni3C 125
NiCI—NiAl 43
NiCr 167, 191
NiFe204 461, 468
NiO 461, 469, 470
NiOH 461
Ni(OH)2 461, 469
NH2 141
NO 331, 332, 345
N02 141
N03 141О 50, 254, 333, 352
Os 50, 239, 530
P 50, 413, 530
P205 261, 264
Pb 530
Ph 50(Ph3P)2Pt(C2Cl4) 157
(Ph3P)PtCl(C2Cl3) 157
Pd 122, 180, 528
Pd—A1203 362
PdCl4“ 354
PdCl—A1203 360, 374
Pr 50Pt 50, 152, 333, 343, 345, 347, 356, 528,
530Pt(NH3)4Cl2 353
Pu 50
Re 50, 239
Re03 144Rh 50, 122, 345, 347, 364, 528RhCl 364RhH 364Rh203 345Ru 50, 332, 528Ru—A1203 357, 374Ru—Si02 357Ru02 373S 50, 308, 413Sb 50, 413, 236Sc 50, 119Sc03 232Se 50, 301, 530Si 50, 68, 207, 217, 239, 250—254, 414,
530Si02 184, 253, 255, 263, 341, 354, 355
588Указатель химических соединенийSm 239Sn 50, 122, 236, 361, 413
Та 50, 530
ТаО 184
Та02 184Та2Ос 167, 183, 184
ТЬ 50
Те 301
Th 50, 239Ti 50, 68, 119, 239, 413, 458, 528
TiC 125
Т Ю 458
ТЮ2 353, 458TI 50, 530
U 50, 236, 239V 50, 119, 239, 528
VC 125VO 143, 458
v205 458
W 50, 530Y 50, 68
Y203 232
Zn 50, 413
ZnSe 157, 531
Zr 50, 68, 224
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬАвтоионный микроскоп 24
Анализ послойный 57, 160, 246, 405,
453 загрязнения 178 калибровка шкалы концентраций168 количественная интерпретация165, 239 на краю кратера 190 по изменению угла регистрации187 относительной интенсивностипиков 190
сегрегация 289 с использованием шаровых шли¬
фов 191 скорость распыления 166 калибровка 166Анализатор с задерживающим полем
(АЗП) 91, 93
Асимметрия угловая в РФЭС 221
Атомное перемешивание 177Валентная зона 135, 143 структура 118, 143Волокна углеродные 437, 525
Встряска 150, 233, 326, 408, 435
Вторичная ионная масс-спектрометрия
(ВИМС) 21, 89, 246, 412, 448— эмиссия электронов (ВЭ) 14, 113Демпси поле 372, 374
Десорбция 23Дифракция быстрых электронов (ДБЭ)
17— медленных электронов (ДМЭ) 15,
23, 91, 93, ИЗ, 156, 321, 342, 357
Духи 147EXAFS 20
S-EXAFS 21, 23Загрязнение 30, 43, 51, 178, 189, 287,
403, 488Излучение синхротронное 20, 74, 75,
330, 333— тормозное 139Интенсивность сигнала РФЭС 228, 235 ЭОС 118, 213, 235Ионизация 113, 114— пики потерь 113
Ионные источники 83, 163Катализ 318, 321, 323, 351
Катализатор биметаллический 322,
326, 335, 350, 375 кобальт-молибденовый 322, 326,335, 375— бифункциональный 322, 326, 335,
361, 375— никелевый 322— платиновый 357, 361— промышленный 360
Квантовое число спиновое 107
Конфигурационное взаимодействие 149
Коронный разряд 428, 433
Коррозия 444
590Предметный указатель— ингибирование 471
Костера — Кронига переходы 134,
137, 207
Краевой угол 424, 425, 434Легирование диффузионное 248— ионной имплантацией 248
Локальная плотность состояний (ЛПС)
124, 135Ыаделунга энергия 140
Масс-спектральный анализ рассеянных
нейтральных частиц (МСАРН) 22, 23
Масс-спектроскопия с бомбардировкой
быстрыми атомами (МСББА) 24,
412Метод анализа тонких пленок (АТП)
246Металлизация 249
Многослойная структура 191
Момент орбитальный 107, 108, 133— спиновый 107
Монте-Карло метод 207Нарушенное полное внутреннее отра¬
жение (НПВО) 411
Насос диффузионный 36, 49— ионный 36, 49— титановый сублимационный 36— турбомолекулярный 36, 49
Неупругого рассеяния длина 168, 208Образование производных соединений
409, 433Обратная свертка 143, 193, 506
Оже-линий тонкая структура 123
Оже-параметр 145, 330, 340, 528
Оже-переходов интенсивность 118, 214,
452 ширина линии 458 энергии 117, 118, 479, 528Оже-пиков серии 118, 136
Окисление, металлы легкие 463 переходные и их сплавы 463 тяжелые 471— полимеры 424, 433— полупроводники 247
Омический контакт 250
Осаждение плазменное 261— химическое из газовой фазы 261
Остовные уровни 114, 133 время жизни 134 ионизация 114Отжиг СВВ-систем 31
Относительной чувствительности фак¬
торы, РФЭС 235, 562
ЭОС 235, 562Плазменное осаждение 261— травление 248
Площадей измерение 500
Полиацетилен 442
Полимеризация в плазме 422
Полимеры 402, 413, 418, 472— анализ поверхности 413— взаимодействие с металлами 440— модификация поверхности 418, 440— проводимость 437— фторированные 418
Полиолефины 422Политетрафторэтилен (ПТФЭ) 33, 403
Постоянная функция пропускания
(ПФП) 104, 134
Постоянное относительное разрешение
(ПОР) 104, 134
Потери ионизационные 113, 131— плазмонные 113, 128, 153, 525
Протонное возбуждение рентгеновско¬
го излучения (ПРИ) 23, 24Пушка электронная 78, 81, 82, 104 компенсационная 340, 371, 404,493Работа выхода 66. 77, 132, 136
Разностный спектр 521
Разрушение образца 84 электронным пучком 362, 368,404, 411, 412, 456, 464
Предметный указатель591Распыление ионное 48 загрязнения 178 зарядка диэлектриков 177 избирательное 177 изменения топографические 50,51, 177 химические 50, 51 количественная интерпретация 165 коэффициент 50, 51, 239 зависимость от угла падения180 скорость 51 калибровка 166 усиление диффузии 177 сегрегации 177 эффект «вбивания» 51, 181Растрескивание коррозионное 311— при отжиге 312Расщепление мультиплетное 148, 326,
458— спин-орбитальное 107, 133, 150
Резерфордовское обратное рассеяние(СОРР) 22, 160
Ричардсона уравнение 76Сверхбольшие интегральные схемы
(СБИС) 244
Сверхвысокий вакуум (СВВ) 30, 33,
343 , 403
Связь у—у 108, 115, 133— Россель—Саундерса (LS) 108, 127
Сканирующая оже-микроскопия (СОМ)245, 247, 446, 453, 463— электронная микроскопия (СЭМ) 17,
359, 412Спектрометр электронный 90, 209
анализатор 90 концентрический полусфериче¬
ский (ПСА) 90, 98, 101, 104 коммерческий 224, 225 полусферический 90, 98, 104 пропускание 104 разрешающая способность 91,98, 212 разрешение 91, 99, 104, 134, 457 с задерживающим полем (АЗП)91, 93, 94 светимость 104 типа цилиндрическое зеркало(АЦЗ) 90, 98, 213, 215, 226, 482 торможение 92, 99 функция пропускания 134, 212,224Спектроскопия инфракрасная отража¬
тельная 411, 414, 425— ионно-нейтрализационная (ИНС) 23— потенциала появления (СПП) 17
—чпотерь (СП) 17— рассеянных ионов (СИР) 21, 25— рентгеновская 19— ультрафиолетовая фотоэлектронная
(УФЭС) 20, 25, 332, 357, 388— характеристических потерь энергии
электронов (СХПЭЭ) 17, 23, 25, 76,
323, 333, 342, 347, 362, 393Стряхивание 153Угловые эффекты 153Фаулера—Нордгейма уравнение 77
Ферми уровень 113, 136, 340, 479, 481
Фоновой составляющей вычитание в
РФЭС 229, 501, 518 ЭОС 218, 518Фотоэлектронов дифракция 155, 156— энергии 405, 479, 528Хемосорбция 319, 333, 342
Химический сдвиг в металлах 458 полимерах 406 оже-линий 124, 244, 473, 528 РФЭС-спектра 139, 330, 456, 528Цеолиты 322, 332, 362, 365Чувствительность к поверхностному
слою 153
592Предметный указательШаровой шлиф 191, 192Электронно-зондовый рентгеновский
микроанализатор (ЭЗРМА) 22
Электронный парамагнитный резонанс
(ЭПР) 364, 388Электронов источники 76
Электрохимия 444Эмпирической подгонки метод 506, 511
Ядерных реакций метод анализа 448
СОДЕРЖАНИЕПредисловие редакторов перевода 5Предисловие 9Глава 1. Перспективы анализа поверхности и границ раздела(М.П. Сих, Д. Бриггс) 111.1. Введение 111.2. Основные сведения о методах применения электронных
пучков для исследования поверхности твердых тел 141.2.1. Электронная оже-спектроскопия 141.2.2. Другие методы 171.3. Основные сведения о методах применения пучков фотонов
для исследования поверхности твердых тел 181.3.1. Рентгеновская фотоэлектронная спектроскопия .... 181.3.2. Другие методы 201.4. Основные сведения о методах применения ионных пучковдля исследования поверхности твердых тел 211.5. Заключение 23Литература 26Глава 2. Оборудование (Дж. К. Ривьвр) 292.1. Введение 292.2. Вакуумные условия 292.2.1. Системы СВВ 302.3. Подготовка и установка образца 392.3.1. Подготовка образца 392.3.2. Введение образца в спектрометр 442.3.3. Подготовка образца- в вакууме 472.3.4. Управление положением образца 632.4. Источники 662.4.1. Источники рентгеновского излучения 662.4.2. Источники электронов 762.4.3. Ионные источники 832.5. Электронные энергоанализаторы 902.5.1. Требования к энергетическому разрешению 912.5.2. Анализатор с задерживающим полем (АЗП) 932.5.3. Анализатор типа цилиндрическое зеркало (АЦЗ) .... 952.5.4. Полусферический концентрический анализатор (ПСА) 101
Литература 105Г лава 3. Интерпретация спектров (Д. Бриггс, Дж.К. Ривьер) ... 1073.1. Введение 1073.2. Терминология 1073.2.1. //-связь 1083.2.2. LS-связь 109
594Содержание3.3. Спектр вторичных электронов, возбуждаемый электронным
ударом 1123.3.1. Спектры оже-электронов 1133.4. Рентгеновские фотоэлектронные спектры 1313.4.1. Первичная структура 1313.4.2. Информация, получаемая из первичной структуры
спектров 1393.4.3. Вторичная структура спектров 1473.4.4. Угловые эффекты 1533.4.5. Спектры, зависящие от времени 157Литература 157Глава 4. Послойный анализ (С. Хофман) 1604.1. Введение 1604.1.1. Неразрушающие методы анализа 1604.1.2. Разрушающие методы 1614.2. Практическое использование послойного анализа с ЭОС и
РФЭС 1624.2.1. Требования к вакууму 1624.2.2. Ионная пушка 1634.2.3. Способы проведения анализа 1644.3. Количественная интерпретация данных послойного анализа 1654.3.1. Калибровка скорости распыления [ z = /(f)] 1664.3.2. Калибровка шкалыконцентраций,[с= /(/)] 1684.4. Сравнение профиля концентраций, возникающего при распы¬
лении, с первоначальным профилем концентраций: понятие
разрешения по глубине 1724.4.1. Разрешение по глубине 1724.4.2. Факторы, ограничивающие разрешение по глубине .. 1784.4.3. Суперпозиция различных эффектов, влияющих на раз¬
решение по глубине 1854.5. Специальные методы послойного анализа . . . . 1864.5.1. Изменение угла регистрации эмитированных электро¬
нов и использование зависимости глубины выхода
электронов от энергии 1874.5.2. Проведение послойного анализа на краю кратера
травления с помощью СОМ с использованием границ
кратеров 1904.5.3. Послойный анализ с использованием шаровых шлифови СОМ 191
Содержание5954.6. Построение профилей концентрации с помощью алгоритма
обратной свертки при послойном анализе с использованием
ионного травления .. о о 1934.7. Заключение 198Литература 199Глава 5. Количественная оже-электронная и рентгеновская фото¬
электронная спектроскопия (М.П. Сих) 2035.1. Введение 2035.2. Количественный анализ с помощью ЭОС в случае однород¬
ных бинарных твердых тел 2045.2.1. Основные соображения 2045.2.2. Измерение интенсивности 2135.3. Количественный анализ с помощью РФЭС в случае однород¬
ных бинарных твердых тел 2215.3.1. Основные соображения 2215.3.2. Измерение интенсивности и банки справочных данных 2285.4. Количественный анализ состава образцов с тонкослойными
покрытиями 2365.5. Количественная оценка результатов послойного анализа . . 239
Литература 241Глава 6. Применение ЭОС в микроэлектронике (P.P. Олсон,П.В. Палмберг, С.Г. Ховланд, Г.Е. Бреди) 2446.1..Введение 2446.2. Технология изготовления полупроводниковых ИС 2456.2.1. Оценка качества исходного материала 2456.2.2. Окисление 2476.2.3. Фотолитография, нанесение рисунка схемы и удаление
фоторезиста 2486.2.4. Химическое или плазменное травление 2486.2.5. Легирование диффузией или ионной имплантацией . . . 2486.2.6. Системы металлизации 2496.2.7. Химическое осаждение из газовой фазы и плазменное
осаждение 2616.2.8. Герметизация и прикрепление кристалла 2676.3. Другие области микроэлектронной технологии 2676.4. Заключение 268Литература 269Г лава 7. Оже-Слектросколия в металлургии (М.П. Сих) 2777.1. Введение 2777.2. Характеристика сегрегаций 2837.2.1. Измерения на границах зерен 284
596Содержание7.2.2. Измерения на свободной поверхности 2927.3. Теория сегрегации 2947.3.1. Теория поверхностных и зернограничных сегрегацийв двойных системах Ленгмюра - Маклина 2957.3.2. Изменение свободной энергии зернограничных сегре¬
гаций &Ggb в двойных системах 2967.3.3. Изменение свободной энергии сегрегации на поверхно¬
сти в двойных системах 2987.3.4. Конкуренция мест в кристаллической решетке в про¬
стых тройных системах 2997.3.5. Более сложные двойные системы 3007.3.6. Сегрегация в тройных системах 3027.3.7. Кинетика сегрегации 3037.4. Влияние материала на сегрегацию 3067.4.1. Сегрегации и основные параметры материала 3067.4.2. Отпускная хрупкость 3097.4.3. Коррозионное растрескивание под напряжением ... 3117.4.4. Растрескивание при отжиге для снятия напряжений и
охрупчивание при ползучести 3127.5. Заключение 313Литература 313Глава 8. Применение электронной спектроскопии в гетерогенномкатализе (T.J1. Барр) 3188.1. Введение 3188.1.1. Краткий обзор исследований катализа методами
электронной спектроскопии, проведенных до 1975 г. 8218.2. Основы электронной спектроскопии применительно к изуче¬
нию катализа 3238.2.1. Введение 3238.2.2. Обзоры по энергиям связи и интенсивностям линий 3248.2.3. Новые методы и катализ 3268.2.4. Хемосорбция и фундаментальные исследования
катализа 3338.3. Электронная спектроскопия и прикладной катализ 3348.3.1. Современные обзоры 3348.3.2. Проблемы и методы использования электронной
спектроскопии в катализе 3368.3.3. Катализ на платиновых металлах 3428.3.4. Цеолиты: каталитический крекинг и другие процессы 3658.3.5. Кобальт-молибденовые катализаторы: обессеривание 375
Содержание5978.4. Заключение 392Литература 394Глава 9. Применение РФЭС в технологии полимеров ( Д. Бриггс) 402
9J. Введение 4029.2. Работа с образцами 4029.3. Аппаратура 4049.4. Спектральная информация . . . 4059.4.1. Эффективная толщина анализа образца (информаци¬
онная глубина) 4059.4.2. Энергии внутренних уровней 4069.4.3. Сателлиты электронной "встряски” (shake-up) 4089.4.4. Фотоэлектронные спектры валентных оболочек .... 4099.4.5. Образование производных соединений для пометки
функциональных групп 4099.5. Сравнение РФЭС с другими методами 4119.6. Применение РФЭС в задачах анализа поверхности полимеров 4139.6.1. Некоторые общие положения 4139.6.2. Модификация поверхности полимеров 4189.7. Заключение 437Литература 438Глава 10.Применение оже-электронной и фотоэлектронной спектро¬
скопии в исследованиях коррозии (Н.С. Макинтайр) .... 44410.1. Введение 44410.2. Особенности применения РФЭС и ЭОС для исследования
коррозии 44610.2.1. Чувствительность анализа поверхности 44610.2.2. Чувствительность к элементному составу 44710.2.3. Количественный анализ 44810.2.4. Пространственное разрешение 45210.2.5. Профилирование по глубине 45410.2.6. Экспериментальные методы 45610.2.7. Химические эффекты 45710.3. Обзор работ по применению РФЭС и ЭОС в исследованиях
коррозии 46410.3.1. Легкие металлы 46410.3.2. Металлы первого ряда и их сплавы 46410.3.3. Тяжелые металлы 47210.3.4. Органические покрытия 47210.4. Заключение 473Литература 475
598СодержаниеПриложение 1. Калибровка спектрометров (М.Т. Энтони) 480П.1Л. Калибровка РФЭС -установок 480П01о2. Калибровка ЭОС-установок 483Литература 487Приложение 2. Методы учета статической зарядки {П. Свифт,Д. Шатпуорс, М.П. Сих) 488П.2.1. Введение 488П.2.2. Примесные поверхностные слои 489П.2.3. Напыленные поверхностные слои 491П.2.4о Смеси 492П.2.5. Внутренние стандарты 493П02.6. Низковольтная компенсирующая электроннаяпушка 494Литература 495Приложение 3. Обработка данных в рентгеновской фотоэлектрон¬
ной спектроскопии (П.М.А. Шервуд) 497П.ЗоЬ Введение 497П.3.2. Системы сбора данных 497П.3.3. Простые операции с данными 500П.3.4. Сглаживание 502П.3.5(. Анализ сложных спектров 507П.Зобо Вычитание фона 519П.3.7. Разностный спектр 522П.3„8. Заключение 526Литература 528Приложение 4. Сводка данных по энергиям оже-электронов, фото¬
электронов и оже-параметрам (К.Д. Вагнер) 530Литература 557Приложение 5. Эмпирические факторы элементной чувствительностидля РФЭС 563Приложение 6< 567Приложение 7 572Приложение 8. Кинетические энергии оже-Эпектронов: эксперимен¬
тальные данные из спектров при рентгеновском воз¬
буждении (К.Д. Вагнер) 574Литература 579colИменной указатель Указатель химических соединений 586Предметный указатель . 589
Научное изданиеМ.П. Сих, Д. Бриггс, Дж. К. Ривьер и др.АНАЛИЗ ПОВЕРХНОСТИ
МЕТОДАМИ ОЖЕ- И РЕНТГЕНОВСКОЙ ФОТОЭЛЕКТРОННОЙ
СПЕКТРОСКОПИИ
Под ред. Д. Бриггса и М.П. СихаСт. научный редактор В.И. Самсонова
Мл. редакторы И.А. Зиновьева, В.И. Аксенова
Художник В.Б. Прищепа
Художественный редактор K.B. Радченко
Технические редакторы M.A. Анциферова, B.H. Ефросимова
Корректоры Н.А. Вавилова, И.С. ГолубеваИБ № 5761Подписано к печати 23.03.87. Формат 60 х 90/16
Бумага офсетная № 1. Гарнитура тайме. Печать офсетная.
Объем 18,75 бум. л. Уел. печ. л. 37,5
Уел. кр.-отт. 37,5. Уч.-изд. л. 40,45
Изд. N® 2/4342. Тираж 3800 экз. Зак.249. Цена 6 р. 40 к.Набрано в издательстве ’’Мир” на участке оперативной полиграфии
129820, ГСП, Москва, 1-й Рижский пер., 2Тульская типография Союзполиграфпрома
при Государственном комитете СССР
по делам издательств, полиграфии и книжной торговли.
300600, Тула, проспект им. В.И. Ленина, 109.