












































































































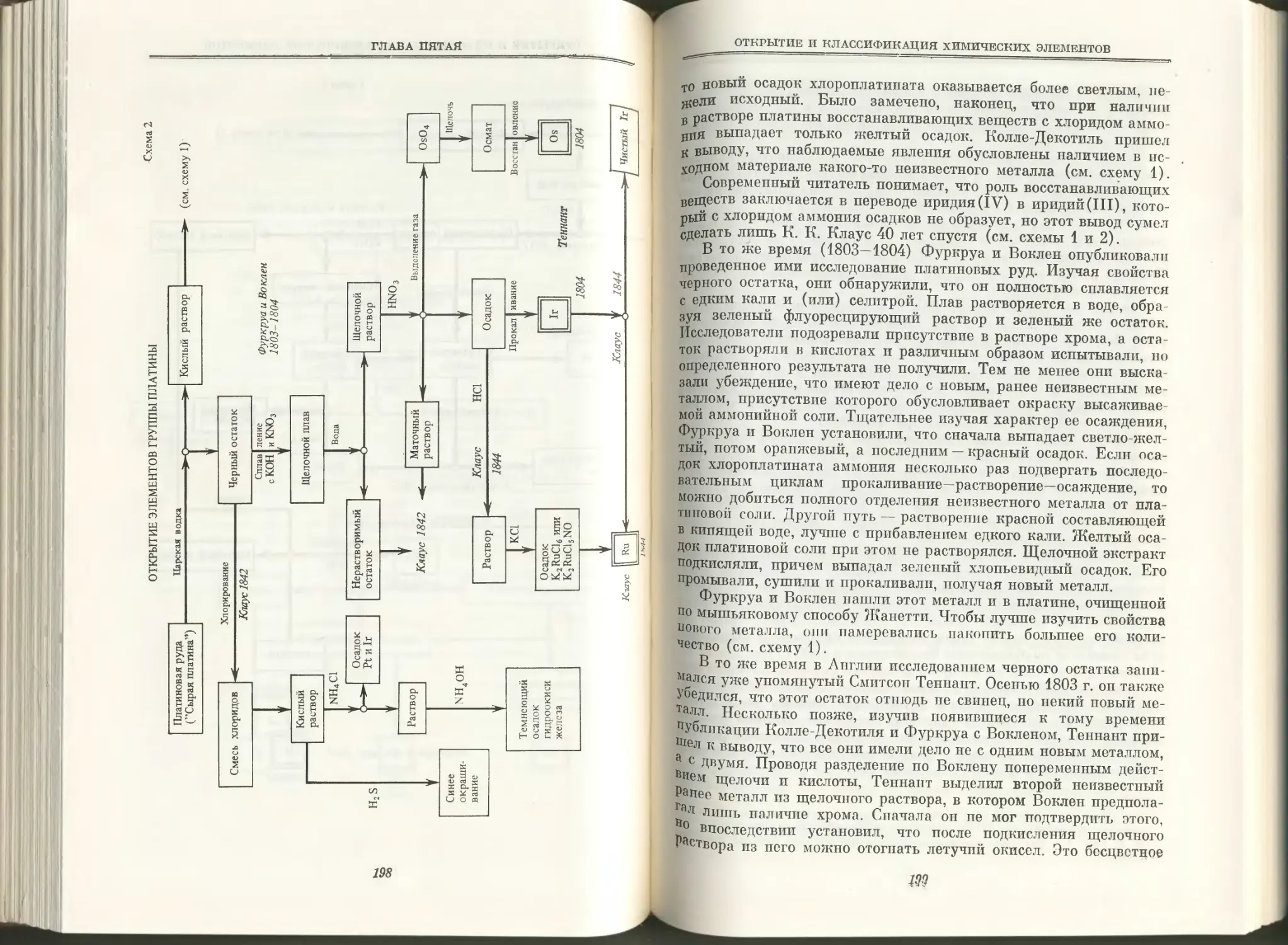










































































































































Text
СТАНОВЛЕНИЕ ХИМИИ КАК НАУКИ
(2-utyS' /у-' W (J ТИ./^.
$ »/ ¥ (/< X. * >, v
Ж/.
--
А -.иг.
Л» 7^7£
// <4--.6? /1‘
Т1ер1одическая система элементовъ по грулламъ и рядам ь
Ряды. 1 ГРУППЫ Э Л Е М Е Н Т 0 В Ъ:
0 I И ПТ IV V ! VI VII УШ
11 [ 2 1 3 1 1, ” Гемй. Не 40 Неон* Ne 19,9 Водород* н 1,008 Ли-Т1Й-Li 7,03 На-тр1й Na 28,05 Берил-Л)Н. Be 9.1 Маг-Н1Й. Мд 24,36 Бор*. в 11,0 Алю* МНН1Й. AI 27,1 Углерод*. С 12,0 Кремний. Si 28.2 Азот*. N 14,01 Фосфор* р 31,0 Кислород*. 0 13,00 Cipa S 32,06 Фтор*. F 19,0 Хлор* CI 35,45
4 5 Ар-гон*. Аг 38 Кд Л1Й к 39,15 М1дь Си 63,6 Каль Ц1Й. Са 40,1 Цимеъ, Zn 65,4 Скан Д1Й. Sc 44,1 Гал-Л1Й. ба 70,0 Тв-инь. Ti 48.1 Гер-л ан in Ge 72,5 Вана-Д1Й. V 51,2 Мышь-янь. As 75 Хрои*. Сг 52,1 Селен*. Se 79,2 Марганец*. Мп 55,0 Бром* Вг 79,95 1Ке- Ко- Ник-Л1зо. бальи, кель, Fe Со Ni (Си) 55,9 59 59
6 7 L—. Криптон*. Кг 81,8 Ру ЙИД1Й. нь 85,5 Сере бри Ад 107,93 Стров-ЩЙ. Sr b7.fi КаА Sift Cd 112,4 Ит- Тр1й. Y 89.0 Ин-Д1й. Jn 115,0 Цир К0»ПН. Zr 90,6 Ото-ьо. Sn 119,0 Hio-б)Й. Nb 94,0 Сурьма- Sb 120,2 Молибден*. Мо 96,0 Тел- лур* Те 127 1од* J 127 Ру- Ро- Пал-тен1й. Ain лаД1й. Ru Rh Pd (Ag) 101,7 103,0 106,5
8 9 К <4-НОВЬ. Хе 128 Це-31ft с$ 132,9 л» митв Ба-рш. Ва 137 4 JiilH Т^НЪ. La 12s,9 Це-pift. Се 1402 М'4М» 4WBMW1 Мт. » M.M. <МОМ
ho и —— Золото Аи. 197.2 Ртуть. H(j 2ц0 и Mlirp-61Й. Yb 173 Та lift Tl 1X14.1 Свн- Нгць РЬ 2о6,9 Тантал* Та 183 Внесут* Bi 208,5 Вольф рам*. W 184 тмт Ос- Ирм- Пла- М1й. д|й, тина. Os Jr Pt (Au) 191 193 194,8
1 12 — ач Рады) Rd 225 ***** Tvpift. Th 232,5 *— — Уранг, и 238,5
В ы с«ш i е солеобразные о к и с л и:
R R’O RO R=Oa Rfl2 Rifts RftS R2QT Rftl
Высш;» г а з « « f M 3 „ u , водсродныя соединен!»
RH* RH3 RIP R||
г
АКАДЕМИЯ НАУК СССР
ИНСТИТУТ
ИСТОРИИ ЕСТЕСТВОЗНАНИЯ И ТЕХНИКИ
ВСЕОБЩАЯ ИСТОРИЯ ХИМИИ
Редакционная коллегия
доктор химических наук Г. В. БЫКОВ | член-корреспондент АН СССР Ю. А. ЖДАНОВ академик Б. М. КЕДРОВ
доктор химических наук В. И. КУЗНЕЦОВ
доктор химических наук Ю. И. СОЛОВЬЕВ академик АН Латвийской ССР Я. П. СТРАДЫНЬ доктор химических наук II. А. ФИГУРОВСКИЙ
ИЗДАТЕЛЬСТВО «ПАУКА»
СТАНОВЛЕНИЕ ХИМИИ КАК НАУКИ
МОСКВА 1983
УДК 54 (091) «.../16»
Становление химии как науки. Всеобщая история химии.— М.: Наука. 1983.— 464 с.
Предлагаемая книга «Всеобщей истории химии» посвящена развитию химии в XVII—XIX вв. В ней подробно рассмотрен процесс становления химии как науки. Дан анализ атомно-корпускулярных представлений ученых XVII в., рассмотрены история флогистонной теории, развитие аналитической химии, становление и развитие атомно-молекулярного учения, открытие периодического закона, формирование важнейших ветвей химической науки первой половины XIX в.
В Приложении впервые на русском языке публикуются фрагменты из двух трактатов Р. Бойля: трактата по философии науки и трактата «Происхождение форм и качеств согласно корпускулярной философии», три фрагмента из работ Г. Шталя, раздел из «Учебника химии» Я. Берцелиуса, а также статья А. Лавуазье «О горении вообще».
Книга предназначена для химиков, преподавателей и учащихся высших учебных заведений и всех читателей, интересующихся историей химии. Ил. 68. Табл. 14. Библиогр. 490 назв.
Ответственный редактор доктор химических наук Ю. И. СОЛОВЬЕВ
1801000000-086 055(02)-83
121-82, кн. 2.
© Издательство «Наука», 1983 г.
ПРЕДИСЛОВИЕ
Предлагаемая вниманию читателей монография многотомного издания «Всеобщая история химии» посвящена истории становления химии как науки.
Книга открывается главой «Особенности развития науки в XVI—XVII вв. Место химии в естествознании Нового времени». Эта глава продолжает описание истории развития химии, начатое в монографии «Возникновение и развитие химии с древнейших времен до XVII века» \ где мы закончили изложение алхимического периода развития химии и подошли к XVII в., когда появился принципиально новый стиль научного мышления, когда химическое ремесло и натуральная философия, на фоне заката алхимии, подготовили необходимые предпосылки для становления химии как науки.
Всматриваясь в даль XVII и XVIII веков, мы видим не туманные очертания спокойно текущей научной мысли, а нарастающий поток новых знаний, новых открытий и изобретений. То была эпоха острой борьбы между сторонниками отживающих алхимических доктрин и новых научных представлений. Ньютоновское фундаментальное положение (масса вещества определяется по весу тела, ибо она пропорциональна весу) имело историческое значение для последующего развития химии. Определяющая роль массы как коренного свойства частиц материи и основанные па этом положении количественные весовые измерения революционизировали теоретические представления химии XVIII века.
Созданная Ньютоном новая концепция о взаимодействии тел, обусловленном проявлением сил различной природы, послужила руководящим принципом для химиков XVIII в. и последующих поколений.
XVIII век, впитав в себя знания предыдущих столетий, оставил весьма солидное химическое наследство. Именно это наследство на почве новых практических запросов создало основные предпосылки для становления химии как самостоятельной науки — науки, имеюще!! свои задачи: изучение состава и свойств химических элементов и их соединений, анализ и синтез природных и искусственных продуктов, исследование химических реакций и явлений, их сопровождающих.
Процесс становления химии как пауки, основанной па кор-пУокулярных представлениях, начался во второй половине XVII в.
всеобщая история химии. Возникновение и развитие химии с древнейших времен до XVII века. М.: Наука, 1980. 399 с.
5
ПРЕДИСЛОВИЕ
и связан главным образом с именем английского ученого Роберта Бойля. Точку зрения Бойля, что «многие химические эксперименты могут быть удачно объяснены с помощью понятий о корпускулах», принял и далее развил М. В. Ломоносов. В основе химии как науки об изменениях, происходящих в веществе в процессе химического взаимодействия, по мнению М. В. Ломоносова, должны лежать фундаментальные законы атомно-корпускулярного учения.
Но для того чтобы атомно-корпускулярные представления приобрели в химии реальный смысл, необходимо было детально изучить качественный и количественный состав различных веществ. Вот почему с конца XVII в. главной задачей химии было изучение состава и свойств веществ минерального происхождения с целью выяснения, из каких веществ состоят тела, какие общие составные части они содержат и каковы их свойства. Для этого предстояло значительно усовершенствовать качественный и количественный анализ, убедиться в воспроизводимости экспериментальных результатов.
Почвой, взрастившей учение о химическом элементе, был опыт, с очевидностью доказывающий, что элементы обладают определенными, для них характерными, химическими и физическими, свойствами.
К концу XVII в. стала отчетливо проявляться потребность свести многообразные изменения вещества, которые происходят в процессе горения, к одному общему принципу. Решение этой проблемы мы находим в трудах немецкого ученого Георга Шталя.
Основной вклад Г. Шталя в развитие химии заключается в том, что разработанная им теория флогистона установила глубокую связь между процессами горения и окисления. Флогистонная теория впервые сблизила и объединила процессы горения с явлениями обжига и кальцинации (окисления) металлов, не имеющие, казалось бы, ничего общего.
С помощью учения о флогистоне удалось свести явления окисления и восстановления, процессы дыхания и гниения к общему представлению о присоединении или выделении флогистона. Хотя флогистонная теория была несовершенной и стояла „на голове11, тем не менее она явилась основной предпосылкой „для революционного преобразования тогдашней системы химических пошь тий“, совершенного великим французским химиком Антуаном Лавуазье.
К 70-м годам XVIII в. растущие противоречия между учением о флогистоне и новыми фактами, полученными в результате плодотворного развития аналитической и пневматической химии, привели к коренной реформе в химии.
В классических работах А. Лавуазье было ниспровергнуто учение о флогистоне и утверждена кислородная теория. Факт за фактом собирал Лавуазье для утверждения кислородной теории и для борьбы с теорией флогистона. За изучением процесса горе-
6
ПРЕДИСЛОВИЕ
пия серы, фосфора последовало изучение тепловых явлений, но только после определения состава воды Лавуазье окончательно выяснил центральную роль кислорода в химических процессах. Лавуазье провел количественные опыты по сжиганию серы и фосфора в воздухе, изучил обжигание свинца и олова, как это в свое время делали Бойль и Ломоносов, и пришел к выводу: при обжигании происходит соединение металла с воздухом. Затем Лавуазье поставил новые опыты и показал, что для полного обжигания металла требуется определенное количество воздуха, что «дефло-гистированный воздух» Пристли (т. е. кислород) и есть та часть воздуха, которая соединяется с металлом при обжиге. Вскоре после выхода в свет «Начального курса химии» (1789) кислородная теория Лавуазье совершила свое победное шествие по странам Европы и Америки.
В процессе становления химии как науки формировалась химическая номенклатура. Длительный период случайных названий (по месту нахождения или характерному признаку вещества, по имени автора, его открывшего, например Глауберова соль) сменился периодом рациональной химической номенклатуры, разработанной комиссией Парижской Академии наук в составе А. Лавуазье, К. Бертолле, Л. Гитона де Морво и А. Фуркруа в 1787 г. Предложенные рациональные названия указывали качественный состав тел. Номенклатура, разработанная французскими учеными, получила широкое распространение и в других странах. В России ее последователями стали академики А. И. Шерер, В. М. Севергин, Я. Д. Захаров, Г. И. Гесс и др.
В работах Лавуазье и его последователей была строго доказана непревращаемость химических элементов при всех известных тогда реакциях и процессах; был составлен первый список химических элементов; получил экспериментальное обоснование закон сохранения массы вещества; утвердился количественный метод исследования с применением физических приборов (весы, ареометр, калориметр, термометр и др.); с правильных позиций началось изучение химизма реакций биологического окисления и процессов дыхания; получила признание точка зрения Лавуазье, что твердое, жидкое или газообразное состояние одного и того же вещества зависит только от степени тепла, заключающегося в нем. Созданная Лавуазье кислородная теория сыграла важную роль в развитии не только химии и биологии, но и металлургии. Отжившие представления, связанные с гипотезой флогистона, препятствовали правильному объяснению сложных металлургических процессов. Освободившись от устарелых взглядов на горение и окисление, металлургия достигла значительных результатов в производстве металлов и сплавов различного назначения. К концу XVIII в. после работ К. Бертолле, Г. Монжа, III. Вандермонда и Л. Гитона де Мор-во выясняется, что от различного содержания углерода зависит образование чугуна и стали. Так впервые с помощью химиков
7
ПРЕДИСЛОВИЕ
ПЕРВЫЙ ПЕРИОД
Истоки атомнокорпускулярных представлений
ВТОРОЙ ПЕРИОД
Создание и утверждение атомномолекулярного учения
XVII в. 1661
I I
I I
I I
I I
I
I I
I 1
। Франция ।
1808 1861
Декарт, Гассенди
Англия
Бойль, Ньютон, Дальтон
Италия
Канниццаро
Берцелиус
Англия
Пр аут, Фарадей
Франция
Гей-Люссак, / Ампер, Дюма, / Лоран, Жерар
Кеку ле
Германия
Англия
Купер, Вильямсон, Максвелл
Россия
Бутлеров t / Менделеев
Голландия
1 Вант-Гофф, / Ван-дер-Ваальс
была прочитана первая страница большой книги по металлургии черных металлов.
В конце XVIII в. химия решала важную проблему: могут ли химические элементы соединяться между собой в любых количествах или же они обладают ограниченной способностью к образованию химических соединений, т. е. должно ли соблюдаться строго определенное постоянство процентного весового состава соединений независимо от способа их получения? В связи с этим весы становятся основным орудием при изучении химических превращений. Весовые отношения начинают играть роль основного критерия химического исследования, в свете которого пересматриваются известные до сих нор факты. «Все наши прежние понятия о химических соотношениях теперь изменились»,— говорил Дж. Блэк, работы которого имели важное значение для развития количественных исследований в химии. Количественные методы дали способ сравнения химических явлений, основанный па строгом учете меры и веса, осуществляемом с помощью физических приборов.
Исследования химиков-апалитпков XVIII в. показали, что какие бы бесчисленные изменения не претерпевали простые и сложные вещества в процессе как естественных, так и искусственных, лабораторных, воздействий, вес химических элементов, а также весовые соотношения составных частей соединения остаются ценз-
ПРЕДИСЛОВИЙ
ТРЕТИЙ ПЕРИОД
Создание учения о сложном строении агома
ЧЕТВЕРТЫЙ ПЕРИОД
Развитие современных электронных и квантово-механических теорий
1881 1913
Англия
Аррениус
Д. Томсон, Содди, Резерфорд, Мозли
Германия
Гельмгольц, Нернст, Абегг, ТПтарк
Швеция
Франция
А. Беккерель,
М. и П. Кюри
менными. Научная дискуссия между Ж. Прустом и К. Бертолле в первом десятилетии XIX в. окончательно решила этот вопрос. Она способствовала выяснению и разграничению понятий «химическое соединение» и «механическая смесь». То, что медленно кристаллизовалось в „маточном растворе44 аналитической химии XVIII в., приобрело в начале XIX в. форму четких определений в законе постоянства состава.
Кислородная теория Лавуазье и закон постоянных отношений легли в основу работ химиков-аналитиков и химиков-неоргаников первой половины XIX в., усилия которых традиционно были направлены на анализ и изучение как исходных, так и полученных веществ, на поиски новых элементов, изучение состава минералов, подробное описание свойств открытых элементов и изучение их соединений. Шел интенсивный процесс накопления фактов и их систематизации по классам и типам неорганических соединений.
Учение Лавуазье о химических элементах и его кислородная теория оставляли в стороне решение вопроса о причинах многообразия веществ, о внутренней природе химических элементов, определяющей специфические особенности каждого химического индивида. Но учение Лавуазье о химических элементах и их соединениях имело важное значение в дальнейшем определении весьма ограниченного „набора44 (равного числу химических элементов, известных к тому времени) „сортов44 атомов.
9
ЙРЕДИСЛОВИЕ
В начале XIX в. происходит слияние учения Лавуазье о химических элементах с атомистической теорией. В 1803—1810 гг< Джон Дальтон создает химическую атомистику, открывает закон кратных отношений. В 1811 г. Амедео Авогадро формулирует основные положения молекулярной теории. Начинается новый период развития химии, связанный с возникновением и утверждением атомно-молекулярного учения.
Теоретический способ мышления благодаря атомистике поднялся на новый уровень и определил на многие годы вперед основной путь познания строения вещества. Борьба за утверждение таких основных понятий, как «атом» и «молекула», происходившая в первой половине прошлого века, сыграла огромную положительную роль, ибо она явилась источником новых открытий и способствовала улучшению системы атомных и молекулярных весов, изучению химических и физических свойств соединений, находящихся в прямой зависимости от числа, индивидуальности и расположения атомов в молекуле.
Успехи теории Дальтона исторически были обусловлены в первую очередь способностью объяснить стехиометрические законы, предсказывать на основе закона кратных отношений новые факты, а самое главное, конкретно объяснить качественное и количественное строение веществ на основе атомистических представлений. В атомистике Дальтона впервые вводится представление о различном качестве атомов различных элементов, что характеризуется в первую очередь относительным атомным весом каждого элемента.
На почве атомно-молекулярного учения выросло учение о валентности и учение о химической связи. В 1812—1813 гг. Я. Берцелиус предложил новую функциональную модель атома в виде электрического диполя. Модель атома Берцелиуса внесла новые представления о природе химической связи, объяснила различные химические свойства одного и того же элемента, специфичность и селективность химического сродства различных атомов.
С атомной теорией был связан такой важнейший шаг в развитии химии, как введение химических знаков элементов. Созданная Берцелиусом химическая символика позволяла составлять эмпирические и рациональные формулы химических соединений и химические уравнения. Так возникли предпосылки для изучения строения химических соединений, выяснения порядка расположения атомов в молекуле и распределения в ней химических связей. Исследования в этом направлении привели к созданию теории химического строения и стереохимии2.
Учение о химических элементах, объединенное с атомно-молекулярной теорией, создало широчайшие возможности для изучения свойств химических соединений. Открытие новых химиче
2 История органической химии рассмотрена в одном из томов «Всеобщей истории химии».
10
ПРЕДИСЛОВИЕ
ских элементов и изучение их соединений подготовили почву для открытия периодического закона.
Создание теории химического строения А. М. Бутлеровым (1861) и открытие периодического закона химических элементов Д. И. Менделеевым (1869) венчало становление классической химии как науки.
На предлагаемой схеме мы стремились наглядно представить основные этапы развития атомно-молекулярного учения. Как видно, в 1661—1810 гг.3 лидирующее положение в разработке атомистических представлений занимали английские ученые. Эстафету они передали ученым Франции и Швеции, которые в 1808—1861 гг. внесли наиболее существенный вклад в создание и развитие атомно-молекулярного учения. В 1861—1881 гг. основная роль в разработке этого учения принадлежит ученым России, Голландии и Германии. После открытия электрона (1897) лидирующее положение вновь заняли английские ученые, но в этот период ученые других стран также сыграли важную роль в разработке атомно-молекулярной теории.
Весь сложный и порой противоречивый процесс развития химии как науки был связан с острой борьбой мнений по кардинальным теоретическим проблемам. Появленпе новых гипотез и теорий почти всегда порождало дискуссии, которые помогали преодолевать односторонность той или иной теории, снимать противоречия с теми или иными фактами. Научная дискуссия как важный фактор научного прогресса приобрела в химии современную форму в XIX в. Обсуждение спорных вопросов в научных журналах, участие в дискуссии ученых различных школ и поколений, привлечение эксперимента как беспристрастного судьи — все это присутствовало в научных дискуссиях и обогащало химию новыми идеями и представлениями. Это был процесс движения к истине не по прямой магистральной дороге, а по тернистому пути, через целину незнания, которую надо было пройти и освоить в борьбе и исканиях.
Мы надеемся, что история становления химии как науки, изложенная в этой книге, привлечет внимание широкого круга читателей.
Глава первая написана кандидатом химических наук И. С. Дмитриевым, глава вторая — кандидатом химических наук 3. И. Шептуновой, глава третья — 3. И. Шептуновой, разделы этой главы «Закон сохранения материи» и «Разработка новой химической номенклатуры» написаны доктором химических наук С. А. Погодиным, глава четвертая — кандидатом химических наук А. М. Цукерманом, в гладе пятой «Открытие и классификация
3 1661 год — выход в свет книги Р. Бойля «Химик-скептик»; 1810 год — выход в свет второй части книги Дж. Дальтона «Новая система химической философии».
П
ПРЕДИСЛОВИЕ
химических элементов до середины XIX в.» раздел «История открытия химических элементов» написан А. М. Цукерманом, раздел «Классификация химических элементов до открытия периодического закона» — доктором химических паук Н. А. Фигуровским, глава шестая написана академиком Б. М. Кедровым, раздел этой главы «Стереохимические аспекты химической атомистики в работах Я. Берцелиуса» — И. С. Дмитриевым, разделы «Системы атомных весов Берцелиуса» и «Система химических формул Берцелиуса 1826 г. и „двойные атомы“» — кандидатом химических наук М. Г. Фаерштейном, глава седьмая написана И. С. Дмитриевым и М. Г. Фаерштейном, глава восьмая — доктором химических наук А. А. Макареней. Заключение к книге написано Ю. И. Соловьевым.
В Приложениях I и II впервые на русском языке публикуются фрагменты из двух трактатов Р. Бойля по философии науки (1657) и «Происхождение форм и качеств согласно корпускулярной философии» (1666); в Приложениях III—V — фрагменты из книг Г. Э. Шталя: «Основы зимотехнип, или общая теория брожения...» (1697), «Случайные мысли и полезные размышления по поводу спора о так называемой сере» (1716); «Физические и химические опыты, наблюдения, замечания, числом триста» (1731); в Приложении VI приведена статья А. Лавуазье «О горении вообще» (1777); в Приложении VII впервые па русском языке публикуется раздел из «Учебника химии» Я. Берцелиуса «Опыт теории химических пропорций и химического действия электричества» (1819).
Монографии серии «Всеобщая история химии» не нумеруются, но на корешке и титульном листе каждой книги изображен один из символов, помещенных на переплете. Данный том символизируют весы.
Ю. II. Соловьев
ЧАСТЬ ПЕРВАЯ
ВОЗНИКНОВЕНИЕ ХИМИИ
КАК НАУКИ
ГЛАВА ПЕРВАЯ
в
ОСОБЕННОСТИ РАЗВИТИЯ НАУКИ В XVI—XVII ВВ. МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
ОБЩАЯ ХАРАКТЕРИСТИКА ЭПОХИ
В XVI—XVII вв. в ряде наиболее развитых в технико-экономическом отношении стран Западной Европы (в первую очередь, в Англии, Нидерландах, некоторых итальянских государствах) на арену общественной жизни и политической борьбы выходит новый класс — буржуазия. В этот период создаются и укрепляются национальные и общеевропейские рынки, что приводит к значительному расширению структуры хозяйства, дифференциации производства, общественному разделению труда. В свою очередь, общественное разделение труда обусловило разделение труда техническое, связанное с увеличением многообразия и сложности производственных операций, осуществляемых на основе механических и химических закономерностей, а отсюда — резкое усиление «сциентистской» компоненты экономического развития [1].
«Техника механическая и химическая — подчеркивал В. И. Ленин,— потому и служит целям человека, что ее характер (суть) состоит в определении ее внешними условиями (законами природы) » * 1 (Курсив наш.— И. Д.) и без знания последних успешное развитие производства становится невозможным. Иными словами, производство становится научным, а наука — производительной, что наложило глубокий отпечаток на характер и цели деятельности ученых Нового времени. «Многие явления на производстве,— писал Р. Бойль,— не только составляют часть естествознания, по некоторые из них могут быть причислены даже к наиболее благородным и полезным его разделам. Ведь они показывают нам природу в движении, и притом особенно, когда она благодаря силе и мастерству человека выведена... из своего обычного состояния, что ...является особенно поучительным условием для исследования» (цит. по [2, с. 165]). П. Шоу, ортодоксальный бэконианец, посвятивший свою жизнь, по его собственному признанию, «развитию в Англии истинной химии в ее связи с наукой, искусствами, торговлей и предпринимательством», определил ее как «искусство,
Примечание. Литературу см. стр. 395—398,
1 Ленин В. И, Полы. собр. соч., т. 29, с. 170,
14
МЕСТО ХИМИЙ И ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
.... .. ' ...- 1 1 . ' -Т' - W
суть которого заключена в действии, а цель в пользе» (цит. по [3, с. 221]). Аналогичных свидетельств можно привести великое множество. Тенденция к сближению науки с производством проявляется и в росте числа изобретений. Так, во второй половине XVII в. в Англии число патентов на различные изобретения и открытия увеличивалось следующим образом (см. [4]): .
Годы 1660-1669 1670-1679 1686-1689 [1690-1699
Число патен- 31 51 53 102
тов
Особо следует отметить то обстоятельство, что господствующим видом материального производства в мануфактурах, строительстве, арсеналах, горных разработках и верфях XVI—XVII вв. становится механическое машинное производство, сама природа которого (природа машины) в известной мере предопределяла направление и характер теоретической науки. Действительно, поскольку «внутренняя сущность машины есть... результат планомерного ограничения, сужения движения до полного исключения всякой неопределенности» [5, с. 238], постольку отражение такого «парализованного», по выражению Гегеля, движения в понятиях требовало создания некоторой исходной предельно идеализированной пространственно-временной «схемы» движения, описывающей перемещение изолированного бескачественного материального объекта (в пределе — материальной точки) по определенной траектории в пространственно-временном континууме.
Но это лишь одна сторона взаимосвязи науки и производства, характеризующая путь от развернутого (мануфактурного) механического производства к механическому естествознанию и механической философии. Другой стороной этой взаимосвязи является глубокое внутреннее соответствие, согласованность капиталистически обобществленного труда и механической техники. Эта согласованность есть «не просто хронологическое совпадение. Было бы несерьезно объяснять дело тем, что-де механические процессы составляют самую доступную для естествознания область природы, и потому, мол, с их освоения началось развитие научной техники2. Нет — это глубокая внутренняя адекватность, выражающая тот важнейший закон общественного развития, что общество овладевает природою в той мере, в какой овладевает своими собственными общественными отношениями» [6, с. 421].
2 Вообще, переход к познанию простейших природных форм и структур, как правило, является предметом зрелой науки, а не начального периода ее формирования. В известном смысле, основное содержание периода предыстории какой-либо конкретной науки, как специфического этапа ее развития, сводится к поиску этих простейших структур и к преобразованию эмпирически и социально-экономически заданных реалий в идеализированный объект познания.— И. Д.
15
ГЛАВА ПЕРВАЯ
Таким образом, три важнейших феномена социально-экономической и культурной жизни начала Нового времени — развитие механической техники, становление капиталистического способа производства и зарождение на базе механики современного естествознания — оказываются внутренне глубоко связанными, что в заметной степени определило характер развития практически всех сфер природопознания, в том числе и химии. Каким образом?
Во-первых, капиталистический способ производства коренным образом изменил предметный статус вещей, которые в форме товаров стали основным средством социального общения. В товарном теле «все чувственно воспринимаемые свойства погасли» 3. Качественность вещи уже не является ее единственной и наиболее глубокой характеристикой.
Во-вторых, капиталистическое обобществление труда основано, с одной стороны, на выхолащивании из него творческого начала, на доведении его до абсолютной упрощенности, автоматизма, бес-качественности физиологической затраты, а с другой — на низведении бесконечного многообразия природы до «абсолютной натуральной упрощенности, безразличности и бескачественности механической техники» [ 6, с. 419].
В итоге, и предмет, и труд, и техника при капитализме принимают абстрактный характер, что приводит к организации природных процессов в основном в форме выхватывания механических закономерностей природы, тогда как все прочие закономерности носят подчиненный характер. Иными словами, самостоятельная техническая, а вместе с ней и научная, реальность принадлежит механическим закономерностям, тогда как все прочие (химические, биологические и др.) реализуются лишь постольку, поскольку они связаны с механикой и потребностями механического производства [6].
Отсюда следует, что социально-экономические условия в период утверждения капитализма ставили химию как науку, изучающую природные объекты прежде всего со стороны их разно-качественности, в весьма своеобразное положение. Химия как самостоятельная область знания на заре Нового времени оказалась вне общенаучной программы своей эпохи, программы, ориентированной на механическую модель мира, на девальвацию качества и изучение однородного материала. Это одна сторона социально-экономической детерминации развития химии в XVI—XVII вв.
Другая сторона связана с тем, что ни в природе, ни в производстве механическая форма движения материи не реализуется в чистом, абсолютно изолированном от других форм движения, виде. И в первую очередь «механизм» оказывается неразрывно связанным с «химизмом». Не случайно поэтому К. Маркс и В. И. Ленин ставили рядом технику механическую и химическую.
3 Маркс К., Энгельс Ф. Соч. 2-е изд., т. 23, с. 46.
16
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
Конечно, в эпоху господства механического производства химические процессы носят в основном подготовительный или сопутствующий характер (если не считать чисто химических производств), но в то же время любой механический процесс принципиально неотделим от химического. Сколь бы мала ни была немехапическая ’ компонента механического производства, она принципиально неустранима, а вместе с ней принципиально неустранимым (и со временем все возрастающим) оказывается социальный запрос на развитие химических знаний.
Указанные два фактора, взапмопротивоположпые по своей направленности, в конечном счете определили специфику развития химии (точнее, специфику противоречий, обусловивших ее развитие) в XVI—XVII вв. Именно здесь затаены наиболее глубокие истоки всех кажущихся непоследовательностей, всех «метаний» химической мысли между дедуктивно-механическим и индуктивно-эмпирическим методами познания вещества, между корпускуля-ризмом и преформационпзмом, между Р. Бойлем и Г. Шталем.
С одной стороны, свойственная механике («механической философии») тенденция к универсализации захватила и химию, подчиняя ее методологическим канонам механицизма. Отсюда — глубокий скептицизм по отношению к скрытым качествам и субстанциальным формам, стремление к моделированию химических явлений на основе комбинаторно-механической каузальной схемы, т. е. в терминах сцепления, разделения, группировки и перегруппировки отдельных фрагментов изучаемых объектов. Но, с другой стороны, объяснение «происхождения качеств», т. е. объяснение физико-химической индивидуальности тела, как бы мы сейчас сказали, явилось той задачей, которая вплотную подвела механическое естествознание к границам его объяснительных и предсказательных возможностей. Именно при изучении химической (а вслед за ней и биологической) формы движения материи с особой ясностью обнаружилась узость механического принципа причинности, что неизбежно вело к известному разрыву между логикой теоретизирующего химика-механициста и логикой химика-практика: первый был скептиком, противником субстанциальных форм, схоластических начал п скрытых качеств, тогда как второй все еще находился в плену традиционного перипатетического и алхимического элементарпзма, рецептурной, ремесленной практики и, в лучшем случае, самых общих и неопределенных атомистических реминисценций. В итоге, неадекватность чисто механического объяснения химических явлений и свойств толкала к поискам пе-мехапических объяснений (см. далее).
Говоря о состоянии общественного сознания в XVII в., следует прежде всего отметить, что оно резко отличается от ренессансного, ибо фиксирует острую внутреннюю противоречивость любой ситуации, будь то социальные конфликты пли же природные явления.
17
Мава йеРвай
г-—..,........................ „ , , -----.....
Если в феодальном обществе жесткая прикреплеыность индивида к сословию делала индивидуальность носительницей социальной общности, к которой эта индивидуальность принадлежала («дворянин всегда остается дворянином, разночинец [roturier] — всегда разночинцем, вне зависимости от прочих условий их жизни» 4), то в обществе буржуазном жизненные условия, в которых находится индивид, становятся для него как бы «случайными», что порождает расслоение человеческой личности, «отличие индивида как личности от классового индивида» 5. Это обстоятельство наложило глубокий отпечаток как на обыденное сознание эпохи, так и на ее философское самосознание, а через последнее и па характер научного и художественного мышления.
В XVII столетии формируется новый тип мироощущения, для которого бытие оказывается не чем-то застывшим, замкнутым в себе, но динамичным, постоянно изменяющимся. Гармонически успокоенной и уравновешенной ренессансной картине бытия, XVII век противопоставил в науке и в искусстве картину движущегося, меняющегося, смятенного, драматически взбудораженного мира. Мыслители этой эпохи видели в фундаменте вещей прежде всего противоречия, разорванность, коллизию диаметрально противоположных начал. С какой стороны естествознание должно подходить к моделированию природных явлений — с конкретно-эмпирической или идеально-абстрактной? Это был вопрос XVII века. В следующее же столетие указанные два подхода вступили в противоборство, причем первая «реалистическая», тенденция часто преобладала. Так, в химии склонный к эмпиризму Г. Шталь высту* пает против корпускуляристских абстракций Бойля, подобно тому, как в искусствознании XVIII века реалист III. Сорель выступает против идеализирующей классицистической эстетики. И общность эта, как нам представляется, далеко не случайна, ибо отражает близость мировоззренческих установок и стиля мышления интеллектуалов-современников, вне зависимости от сферы их деятельности и интересов. Но вернемся к науке XVII столетия.
Важной особенностью механико-материалистического понимания природы является его взаимосвязь с атомистическими представлениями о строении материи. Что же послужило источником этой взаимосвязи? В XVI—XVII вв. намечается известная переоценка ремесленной и технической деятельности. Умение сделать вещь рассматривается уже не как искусство, а как знание, более того — как сущность всякого знания о вещи. Иными словами, в Новое время познавательная деятельность отождествляется с конструирующей [7, с. 104—113; 8, с. 164]. В условиях господства механицизма, когда главное место в человеческой практике и в мышлении занимают комбинаторные процессы (сборки—разбор
4 Маркс К., Энгельс Ф, Соч. 2-е изд., т. 3, с. 77.
5 Там же.
18
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
ки, сочленения—отделения и перегруппировки отдельных материальных образований), когда научная теория по сути становится системой потенциальных способов технического конструирования и когда природа уподобляется гигантскому часовому механизму,— в этих условиях наиболее адекватными представлениями о строении вещества и его превращениях оказываются те, которые оперируют с дискретными образованиями, что позволяет распространить метод изучения макрообъектов (наглядные комбинаторные сборноразборные схемы) на микромир (химик, по Бойлю,— микромеханик) .
Семнадцатый век не только воскрешает античную, но и создает свою собственную атомистическую доктрину, призванную «подогнать» учение о веществе под общее механистическое понимание природы и объяснить качественную гетерогенность мира в терминах геометро-кинематических характеристик качественно однородных атомов.
Проблема происхождения качеств становится одной из коренных в науке Нового времени, центром конфронтации спагириков и перипатетиков, с одной стороны, и корпускуляристов,— с другой и «подобно тому как с различием способов производства марксизм связывает различные общественно-экономические формации, так со сменой способов решения проблемы генезиса свойств веществ оказывается возможным связать ступени развития химии» [9, с. 13]. Пафос борьбы с перипатетико-спагирическими теориями, с «бесом субстанциальных форм» (Г. Ольденбург) за установление «истинной химической философии» лежит в основе корпускуля-ризма XVII столетия. Это обстоятельство заставляет нас более детально рассмотреть сущность различных, противостоящих друг ДРУГУ учений, без чего невозможно понять внутреннюю логику процесса, который Ф. Энгельс охарактеризовал краткой и емкой формулой: «Бойль делает из химии пауку» 6.
КРИЗИС КОНЦЕПЦИИ СУБСТАНЦИАЛЬНЫХ ФОРМ И КАЧЕСТВ
Одним из центральных понятий средневековой схоластики являлось понятие о степени бытия — степени его совершенства, его актуализованности. «...Есть нечто, в предельной степени обладающее истиной, и совершенством, и благородством, а следовательно, и бытием... Но то, что в предельной степени обладает некоторым качеством, есть причина всех проявлений этого качества; так, огонь как предел теплоты есть причина всего теплого...» (курсив наш.—If. Д.) учил Фома Аквинский [10, с. 111] 7. Именно здесь * В
® Маркс К., Энгельс Ф. Соч. 2-е изд., т. 20, с. 501.
В XIV—XVI вв. существовало даже особое учение об интенсификации и ремиссии качеств [И, с. 168—171].
19
ГЛАВА ПЕРВАЯ
берет свое начало перипатетическая концепция субстанциальных форм и качеств в ее как философском, так и в химическом (спа-гирическом) преломлении, концепция, которая в течение нескольких веков служила основой для решения проблемы уникальности, качественной определенности предмета, способного, по мысли перипатетиков, существовать только в процессе бесконечного самосовершенствования, постоянного очищения от инородного, имманентно ему не присущего бытия. Сущность вещи с абсолютной полнотой выражена не в самой вещи, по в ее субстанциальной форме, тогда как реальные предметы оказываются ниже своей сущности и форма может выступать в них лишь как скрытое, оккультное, качество.
Будучи спроецированной па химическую проблематику, эта методологическая установка порождала фундаментальный и сквозной для химии Нового времени вопрос о способе образования сложного тела из элементов («ап elementa manent in mixto» — «остаются ли в сложном теле элементы») или —в более широкой постановке — вопрос о соответствии скрытой сущности предмета и его чувственно воспринимаемого, акцидентального, по выражению того времени, бытия.
Парадоксальной казалась сама возможность образования истинного «миксиса» (mixtio secundum veritatem), т. е. однородного, абсолютно совершенного объекта, сложной формы — из элементарных форм, ибо каждая часть миксиса потенциально и актуально неотличима от целого. Это противоречие между генетической гетерогенностью соединения и его реальной гомогенностью разными мыслителями решалась по-разному (подробнее см. [12, с. 126-127]).
Так, согласно Фоме Аквинскому, в процессе образования сложного тела из элементов свойства последних изменяются и они уже не являются более крайним выражением некоего качества. Субстанциальная форма переходит в некую промежуточную, создавая тем самым возможность для совмещения различных форм в едином целом миксиса, а следовательно, и для появления нового качества 8. Исходные качества представлены в новом теле не субстанциально, но виртуально, и соответственно сами носители свойств существуют в материальном субстрате тела только в скрытом виде, обнаруживая себя лишь in virtu, в наличии у тела определенных свойств. Так, если тело подвергается действию огня, то оно становится горячим, принимая на некоторое время «форму тепла», и в зависимости от того, восприняло ли оно эту форму полностью («совершенным образом») или нет, тело охлаждалось при удалении огня или улавливало огонь и таким образом могло передавать эту форму другим телам. Например, вода остается теплой после 8
8 Проблема субстанциальных форм детально рассмотрена в книгах В. П. Зубова [11] (особенно в главах II и III), М. Боус-Холл [13] и Э. Метцжер [14, 15].
20
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
удаления огня, воздух же после захода солнца темнеет. Это, по мнению Аквината, происходит, оттого что вода может удерживать форму огпя (хоть и несовершенным образом), а воздух форму света — пет.
Однако развитие аналитических методик и химического эксперимента в целом, общее изменение характера теоретического мышления под влиянием научной революции Нового времени привели к трансформации старых доктрин. В частности, предположение о том, что в ходе химической реакции субстанциальные формы перераспределяются между реагентами, было бы совершенно адекватным и достаточным при условии, что исследователь довольствовался чисто вербальными определениями. Но как раз этот тип «объяснений» в XVII в. уже безнадежно утрачивал свои позиции, рационалисты говорили о нем с презрением и насмешкой. «Судите сами,— писал Бойль,— что эта за манера — объяснять явления исходя из начала, природа которого открыто признается неизвестной» [16, б, т. 3, с. 38].
Теория субстанциальных форм не могла привести к фактологически значимым результатам, а потому и к сколько-нибудь достоверным и однозначным предсказаниям. Но наряду с отсутствием «внешнего оправдания» все отчетливее выявлялось ее несоответствие формирующимся рационалистическим критериям «внутреннего совершенства», что с наибольшей силой обнаружилось в следующем парадоксе: с одной стороны, свойства тела, не будучи укоренены в нем, оказывались полностью от тела отделенными, тогда как с другой,— они вполне с ним совпадали, служа своего рода онтологической подстановкой под предмет, ибо никакого посредующего звена (в виде состава, структуры или какого-либо иного принципа внутренней организации) между свойством тела и его материальным субстратом не было. Последний (субстрат) оказывался своего рода пассивным вместилищем субстанциальных форм. Иными словами, формы представляли собой метафизически трансформированные свойства в их оторванности от своего материального субстрата, тогда как сам субстрат являл собой метафизически трансформированное тело в его оторванности от своих свойств.
Попытки разрешить это противоречие путем придания формам вещественного статуса с целью вместить их в рамки операциональных определений (Д. Зеннерт, Э. де Клав) только углубляли его (см. далее). В итоге общая картина развития химии в XVII в. оказалась крайне противоречивой, что нашло отражение практически во всех аспектах химической деятельности и, в частности, в поразительной идейной гетерогенности научной и натурфилософской литературы того времени, где рассматривались проблемы природы вещества. Наряду с сочинениями Декарта, Гассендп, Га-лилея, Бойля, большой (нередко большей) популярностью пользовались трактаты Гермеса Трисмегиста, Альберта Великого, Рац-
21
ГЛАВА ПЕРВАЯ
монда Луллия, Василия Валентина, Псевдо-Арнольда из Виллано-вы, анонимный «ТигЬа philosophorum» и др. Или другой аспект — начало формирования химической технологии. Этот процесс оказался результатом сложного взаимодействия химического ремесла и теоретизирующей алхимии [17, с. 266—269].
Становление научной химии происходило в острой борьбе с оккультизмом и пережитками перипатетико-спагирических воззрений. Реакцией на «Химика-скептика» Р. Бойля были не только восторженные отзывы Г. Ольденбурга, но и появление в 1666 г. «Химика-спиритуалиста» У. Спэстоу. Сам Бойль, как и Ньютон, был не только выдающимся естествоиспытателем, но и известным теологом. Но сейчас для нас важны позитивные изменения, происшедшие в химии XVI—XVII вв.
Важнейшее из них состоит в том, что субстанциальные формы начинают рассматриваться как вполне реальные ингредиенты сложных тел, т. е. обретают статус вещественности. Это заметно уже в работах Ф. Парацельса и более отчетливо проявилось у И. Ван Гельмонта, согласно которому сложное вещество состоит из тех простых (или более простых) тел, на которые оно разлагается и из которых образуется [17, с. 264]. Характерный пример,—получение Ван Бельмонтом стекла из песка и поташа с последующим количественным разложением продукта на исходные вещества. Отсюда вывод: стекло не однородное тело, а лишь соединение («seel tantum est appositio») частиц песка и соли [18, с. 35]. Аналогичных примеров можно привести немало [17, с. 265; 19], но нас здесь будут интересовать не частные моменты, а общий строй мышления химика-практика середины XVII в., в основе которого лежал фундаментальный принцип, с наибольшей отчетливостью сформулированный уроженцем Любека, доктором медицины, химиком, математиком и логиком Иоахимом Юнгиусом (1578—1657). В чеканной аксиоматике его посмертно изданной «Doxoscopiae physicae minores» (1662) [20] в формализованном виде отразилась вся логическая структура лабораторного и ремесленного опыта того времени.
Подобно многим своим современникам-химикам, Юнгиус много внимания уделял изучению разнообразных последовательностей химических превращений, образующих замкнутый цикл. В качестве примера, далеко не единственного у немецкого ученого, рассмотрим весьма обстоятельно изученную в его время цепочку превращений, которая в современной записи представлена на схеме (см. стр. 23).
И. Юнгиус ввел специальный термин для переходов подобного рода— «химическая редукция». Тело называется редуцированным, если оно, претерпев некоторые превращения, вновь переходит в исходное состояние.
Ход рассуждений Юнгиуса при рассмотрении указанной редукционной цепочки таков: если в переходах (1)->(2) и (или)
22
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ flOBOfrO ЕРЁМЕЙЙ
(О
(3)
На влажном воздухе В присугсгвии СО2
РЬСОз- РЬ(ОН)2
При нагревании в присутствии восстановителей (в XVII в. — древесный уголь, поташ)
Основной карбонат свинца, старое название: cerrussa
__________________ РЬО
(В XVII в. реализовать не удавалось)
Старое название: sandyx
+ восстановитель или сильное прокаливание (/
2000°С)
Редукционная цепочка Юнгиуса в современной записи
(2) -> (3) свинец разрушается, распадаясь на составные части9, то непонятно, каким образом его удавалось регенерировать из (2) и (3), используя только нагревание и действие неметаллического тела-восстановителя. Источником недоумения служило старое, восходящее к Аристотелю, представление о невозможности полного восстановления свойств исходного тела в условиях материальной изоляции каждого из продуктов его разложения (по аналогии А. Биллиха — «ослепшего не сделать зрячим»). Иными словами, если цикл включает следующие превращения:
разложение
редукция
то редукция от В к А, по Юнгиусу, возможна только путем добавки к В некоторого реагента (скажем, остальных продуктов разложения А), способного наделить В свойствами А (например, вернуть ему горючесть, определенный цвет и т. д.).
Если же А не разлагается, но соединяется с каким-либо телом С
А^В,
-С
то редукция либо сводится к разложению В в условиях материальной изоляции, либо происходит с участием четвертого вещества D путем «обмена» («замещения»):
AC + D-> A + GD (см.10).
Но независимо от способа регенерации А в последнем случае пред
9 Напомним, что, согласно представлениям того времени, металлы могли быть сложными веществами.
В качестве примера укажем па изучавшуюся А. Сала «экстракцию» меди из раствора медного купороса с помощью железа [19, с. 77—78].
23
ГЛАВА ПЕРВАЯ
полагалось, что А входит в состав В в качестве ингредиента. т. е. В^АС (см.11).
Подобные рассуждения привели Юнгиуса к мысли о том, что свинец в переходах (1)->(2) и (2)->(3) пе разрушается («Plum-barn in utraque Mutatione Incorruptum permanere certum est») [20], но входит в (2) и (3) в качестве составной части. Общий вывод немецкого исследователя («синдиакритическая гипотеза», по его терминологии) гласит: «...если несколько реакций (Mutationes) протекают последовательно так, что из образующихся веществ удается посредством редукции получить исходное тело, то последнее в ходе всех этих превращений должно оставаться неизменным» [20] 12.
Эта гипотеза явно пли неявно, но довольно широко использовалась химиками XVII в., о чем красноречиво свидетельствуют труды Джакомо Забарелла, Иеронима Дандини, Ансельма Боэциу-са де Боодта, Анжеле Сала, Антонина Гюнтера Бпллиха, Николя Жибера и многих других. При этом часто применялся ее «ослабленный» вариант: если вещество С образуется из веществ А и В и разлагается на них, причем обе реакции протекают в условиях материальной изоляции, то А и В являются ингредиентами С.
В итоге, заурядная, на первый взгляд, эмпирическая цепочка химических превращений оказалась включенной в преформацио-нистский контекст аналитико-синтетической деятельности химика-практика, выступая эквивалентом доказательства и критерием сложносоставной природы веществ задолго до того, как количественные методы заняли в химии одно из лидирующих мест.
Выраженная в синдиакритической гипотезе Юнгиуса главная идея классической преформационистской традиции13, объединившись спустя полвека с механико-корпускуляристскими концепциями, заявит о себе в трудах Г. Шталя: «Все составное и сложное (omnia Mixta et Gomposita) предполагает простое, что я имею обыкновение называть элементами. Но в индивидуальном виде названные таким образом сущности не встречаются в природе, и все, что существует, сложно и состоит из гетерогенных частиц, отличающихся формой, величиной, положением и движением... Простые тела и элементы, хотя и не существуют в чистом виде и отдельно, могут, однако, мыслиться как отличные друг от друга» [21, с. 1], «начала (по Шталю,— это простые тела или «первичные материальные основания составных тел (mixta)».—Я. Д.) опреде
11 Аналогично в первом цикле можно считать В ингредиентом А.
12 Нетрудно понять, что вывод этот справедлив (насколько он вообще справедлив!) только в случае циклов второго типа.
13 Голландский историк химии Р. Хойкаас назвал эту традицию эмпедокло-вой [19, с. 65—66, 79], отличая ее тем самым как от перипатетико-спа-гирической, отождествляющей свойства предмета с самим предметом, так и от демокрито-эпикурейской, ставящей природу свойства в зависимость от геометро-кинематических характеристик ультиматов.
24
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
ляются как a priori, т. е. как то, что в составном теле уже предсу-ществует, так и a posteriori, т. е. как то, па что это тело в конечном итоге разлагается. Оба эти определения будут правильны, если иметь в виду чистое, естественное разложение, чего, однако, нелегко добиться от современной химии п потому едва ли можно достичь искусством. Поэтому в настоящее время превалируют различия между физическими и химическими началами составных тел (ср. дискуссии о физическом и химическом атоме в XIX в.— и. Д.).
Физическими именуют те начала, из коих составное тело в действительности образовано, но они до сих пор не установлены... Начала, на которые все тела разлагаются известными до сих пор химическими операциями, называют обычно химическими» [22, с. 4].
При сходстве иерархических схем Бойля и Шталя14, отображающих внутреннюю организацию природных тел, налицо глубокое различие между ними в принципах объяснения генезиса свойств веществ: у первого — структурно-механистический, связывающих! изменение свойств макротел с изменением локальных движений микрочастиц и с их всевозможными перегруппировками; у второго — элементаристскпй, акцептирующий внимание на зависимости состав—свойство.
Поэтому коллизия «перипатетизм—корпускуляризм», анализу которой столько сил было отдано историками науки, лишь внешним образом выражает всю специфику и остроту ситуации в химии XVII столетия, ибо отказ от перипатетических тетрад и спа-гирических tria prima элементов-первокачеств к середине века был практически предрешен и реально действующее противоречие, послужившее внутренним двигателем идейного и эмпирического развития химии в последующие двести лет, лежало в совсем иной области — в области конфронтации «торжествующего» корпуску-ляризма и преформационистской («эмпедокловой») традиции. Перед глубиной этой конфронтации многие другие контрасты эволюции химической мысли XVII—XX вв., если и по меркнут, то выявляют между собой известное родство.
АТОМИСТИЧЕСКИЕ КОНЦЕПЦИИ XVII В. И ИХ ВЛИЯНИЕ ИА ФОРМИРОВАНИЕ КОРПУСКУЛЯРНОЙ ТЕОРИИ ВЕЩЕСТВА БОЙЛЯ
Было бы ошибкой думать, что взгляды Р. Бойля* представляют собой замкнутую и в своей оригинальности полностью изолированную от других натурфилософских представлений доктрину.
** См- ниже в данной главе и главу вторую.
Роберт Бойль родился 25 января 1627 г. в замке Лисмор (Ирландия). Б го отец Ричард Бойль (граф Корка) — богатый аристократ — дал сыну раз-
25
ГЛАВА ПЕРВАЯ
Наоборот, свою главную цель английский ученый видел в «иллюстрации некоторых понятий корпускулярной философии разумными экспериментами и доказательстве того, что вещи могут быть, по крайней мере, правдоподобно объяснены без обращения к непостижимым формам, реальным качествам, четырем перипатетическим элементам или же к трем химическим началам» [16, а, т. 1, с. 356]. Он неоднократно указывал на органическую связь своей теории с воззрениями других ученых и философов.
Фактически речь идет об истоках и характерных особенностях научно-методологической традиции корпускуляризма Нового времени. Выше мы уже говорили о соотнесенности механицизма и атомистики в историко-логическом аспекте. Теперь обратимся к конкретно-историческому, «поименному» рассмотрению различных атомистических концепций.
Фрэнсис Бэкон Веруламский (1561—1626). По удачной характеристике Р. Каргона, «Бойль был бэкониапцем, приведшим в действие теоретические построения Декарта и Гассенди» [23, с. 94].
Три аспекта в учении английского философа оказались особенно близкими Бойлю: во-первых, пафос отрицания схоластики, выводившей законы природы «из общих понятий и тому подобных заскоков ума за пределы природы» [24, т. 2, с. 305], во-вторых, утверждение идеала новой, опытной науки, состоящей «в применении рационального метода к чувственным данным», где «индукция, анализ, сравнение, эксперимент суть главные условия» этого метода [7, т. 2, с. 142], и, в-третьих, настойчивое стремление к установлению «скрытого схематизма тел» [24, т. 2, с. 85] и к открытию законов и определений «чистого действия, которые создают какую-либо простую натуру, как, например, теплоту, свет, вес, во всевозможных материях и воспринимающих их предметах» [24, т. 2, с. НО].
Уже в ранних работах Бойль ставит своей целью реализовать в конкретных экспериментальных исследованиях бэконовскую
постороннее образование. В возрасте восьми лет Роберт свободно владел французским и латынью. Годы его учебы прошли в Итоне и Женеве. Здесь у юноши проявился первый интерес к естествознанию. После путешествия по Европе Роберт Бойль вернулся в свое имение Столбридж, где в тиши библиотеки изучал многочисленные труды по естествознанию, медицине и религии. В 1654 г. он переселился в Оксфорд, где провел четырнадцать самых плодотворных лет своей жизни. Он сразу активно вошел в жизнь Оксфордского университета и стал близким другом многих ведущих профессоров. Здесь он организовал свою лабораторию, в которой выполнил исследования по физике, химии и минералогии. Он собрал богатую коллекцию руд и минералов, которую оставил по завещанию Лондонскому Королевскому обществу, основанному при его активном участии в 1660 г. С 1668 г. Р. Бойль жил в Лондоне. Здесь он продолжал активную научную деятельность до конца своей жизни. Умер Бойль в Лондоне 30 декабря 1691 г.
О жнзттп и деятельности Р. Бойля см.: More L. Т. The Life and Works of the Honourable Robert Boyle. London, 1944; Boas M. Robert Boyle and Seventeenth Century Chemistry. Cambridge, 1958.— Прим. отв. ped.
26
МЕСТО ХЙМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
программу «великого восстановления паук». «Многие подробности,— писал он в „Физиологических опытах44,— были по моему замыслу собраны с целью продолжения Sylva Sylvarum, пли естественной истории лорда Веруламского» [2, с. 124] 15.
Однако за пределами общности философско-методологических установок, в сфере конкретных естественнонаучных исследований между Бэконом и Бойлем начинаются существенные расхождения, корни которых следует искать в особенностях исторической эволюции механистического материализма. К. Маркс, говоря о судьбе бэконовой философии, подчеркивал, что «в своем дальнейшем развитии материализм становится односторонним... Физическое движение приносится в жертву механическому или математическому движению; геометрия провозглашается главной наукой» 16. Маркс связывал этот поворот в истории материализма с именем Т. Гоббса. Но аскетизм мехапико-материалистической картины мира не менее отчетливо проявился и в творчестве Бойля. Именно здесь, на наш взгляд, затаены наиболее глубокие различия в восприятии и в оценке атомистики автором «Химика-скептика» и создателем «Нового органона наук».
Начиная примерно с 1605 г. симпатии Бэкона к атомному учению проявляются все отчетливее, видимо, не без влияния атомистов так называемого Нортамберлендского круга — группы естествоиспытателей, философов и поэтов, объединившихся на рубеже XVI—XVII вв. под покровительством Генри Перси, графа Нортамберленда17. При всех различиях во взглядах, жизненных позициях, интересах и стремлениях, этих людей объединяло новое мироощущение, переданное в прекрасных стихах Дж. Допна:
И в сфере звезд, и в облике планет На атомы Вселенная крошится, Все связи рвутся, все в куски дробится, Основы расшатались, и сейчас Все стало относительным для нас 18.
В контексте пашей темы наибольший интерес представляют взгляды интеллектуального лидера группы — Т. Хариота. В его полемике с И. Кеплером [23, с. 26—27] о причинах отражения и преломления света (1606—1608) можно увидеть зародыш будущей коллизии между перипатетической традицией и корпускуляризмом
15 «Sylva Sylvarum or a Natural History in ten centuries» («Лес лесов, или естественная история в десяти центуриях») — одна из последних работ Ф. Бэкона, написанная около 1624 г. (опубликована посмертно в 1627 г.) и посвященная естественнонаучным проблемам.
*6 Маркс К,, Энгельс Ф. Соч. 2-е изд., т. 2, с. 143.
В группу входили: математики и физики — Т. Хариот (1560—1621), у. Уарнер (1550—1640), Н. Хилл (ок. 1570 — ок. 1610), Н. Тополи (1564— 1632), философы и поэты — Дж. Донн (ок. 1572—1631) и К. Марло
18 (1564—1593).
Перевод Б. В. Томашевского.
27
ГЛАВА ПЕРВАЯ
Нового времени. По Кеплеру, способность тела отражать и поглощать световые лучи всецело определяется соотношением в нем двух субстанциальных качеств — прозрачности и непрозрачности [25, т. 16, с. 32], тогда как, по Хариоту, все свойства вещества зависят от величины, формы и движения атомов или составленных из них корпускул [25, т. 15, с. 367; т. 16, с. 172—173].
Но вернемся к Бэкону. В трактате «О принципах и началах» 19 20 (написай около 1612 г.) он вполне определенно высказался в пользу философии Демокрита, «глубоко и тонко» проникшей в тайны природы [24, т. 2, с. 307]. Однако принятие английским философом античной атомной доктрины, сведения о которой он черпал главным образом из поэмы Лукреция Кара2, было далеко пе безоговорочным.
Бэкон отстаивал принцип гетерогенности атомов, согласно которому последним следует приписать специфические свойства и тем самым «сохранить особенность и исключительность атома как в отношении субстанции, так и в отношении движения» [24, т. 2, с. 307]. Все известные качества (тяжесть, твердость, плотность и т. д.) должны проявляться в атомах иначе, чем в макротелах, «самобытные свойства» которых есть результат соединения разнокачественных атомов. Атом Бэкона наделен внутренней активностью и особым материальным субстратом — в его теле «есть элементы всех тел, а в его движении и силе — начала всех движений и сил» [24, т. 2, с. 306], поэтому Демокриту «следовало бы приписать атому особого рода движение, подобно тому как он наделил его особой материей и особенными силами» [24, т. 2, с. 307].
Взгляды Бэкона пе получили признания, большинство атомистов XVII в. придерживались концепции качественной гомогенности атомов, объясняя индивидуальность природных тел в терминах геометро-кинематических свойств ультиматов. Да и сам Бэкон к 1620 г., т. е. ко времени выхода в свет «Нового органона наук», стал относиться к атомной теории более сдержанно: «не меньшее (по сравнению с перипатетизмом.— И. Д.) зло состоит и в том, что в философии и в размышлениях своих люди направляют усилия на исследования начал вещей и последних оснований природы, в то время как вся польза и практическая действенность заключается в средних аксиомах. Отсюда и получается, что люди продолжают абстрагироваться от природы до тех пор, пока не приходят к потенциальной, бесформенной материи; и не перестают рассекать природу до тех пор, пока не дойдут до атома. И если бы даже это было истинно, то немногим могло бы содействовать благосостоянию людей» [24, т. 2, с. 32],—взгляд, во многом близкий позиции Г. Шталя.
19 В русском переводе «О началах и истоках» [24, т. 2, с. 301].
20 Так что речь у Бэкона шла, по сути, не столько о Демокритовой, сколько об эпикурейской атомистике.
28
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
По дело не только в соображениях практической полезности. Ф. Бэкон критикует атомистику еще и за то, что опа предполагает два ложных, по его мнению, допущения — «пустоту и петекучую материю» [24, т. 2, с. 87]. Абстрагирование от красочного многообразия мира было органически чуждо английскому философу,-материя у Бэкона, по словам К. Маркса и Ф. Энгельса, «улыбается своим поэтически-чувственным блеском», опа наполняет собою весь мир, и ее первичные формы «суть живые, индивидуализирующие, внутренне присущие ей, создающие специфические различия сущностные силы» 21 *. Унылая комбинаторика механической атомистики не вписывается в пантеистическую картину мира (ие отсюда ли указанное выше стремление Бэкона к максимальной индивидуализации атома?). В природе, как опа представлена в «Иовом органоне...», существуют лишь «грубая материя» и абсолютно легкий, подвижный, материальный дух — «activating spirit». Задача натуралиста — «исследовать, сколько есть во всяком теле от духа и сколько от осязаемой сущности, а также обилен ли и тучен этот самый дух или тощ и беден» [24, т. 2, с. 86] и т. д. и т. и.
В итоге критика Бэконом столь ненавистного ему схоластического перипатетизма в сфере конкретного изучения природы оборачивается наихудшимп реминисценциями натурфилософских спекуляций схоластов. Не случайно поэтому уже в XVII в. многие рационалисты, подобно Т. Гоббсу, отвернулись от Бэкона, а его «Лес лесов...» был назван дремучей чащей хаотических сведений и нелепостей, вроде того, что алмазы образуются при иотепии •_» 9 9 камней .
Иной подход у Бойля. Поэтическая чувственность бэкоиовской материи превращается у него в абстрактную чувственность механистической атомистики, где первичными свойствами бескачест-венных корпускул оказываются их размеры, форма и механическое движение. И это ие просто односторонняя, а потому и наиболее последовательная, трансформация бэконианства; здесь начинается принципиально новое — декларация экспериментальной науки сталкивается с другим мощным идейным течением века — с декартовой апологией метода.
Рене Декарт (1596—1650). Если для Аристотеля и схоластов общее, выходящее за пределы родовой принадлежности, было лишь абстракцией, но никак не реальной сущностью, то для рационалистов XVII столетия и, в первую очередь, для Декарта, подобные онтологические разграничения полностью сняты. Природное неравенство вещей, их качественная разнородность и определенность Уничтожены и совершенно растворены в чисто количественных различиях. В результате знадие о мире приобретает «очевидность,
2* Маркс К., Энгельс Ф. Соч. 2-е изд., т. 2, с. 142.
А. Ремюз даже приписывал влиянию натурфилософской деятельности Бэкона относительную неудачу исследований Бойля, что, конечно, было сильным, хотя и весьма показательным, преувеличением [26, с. 29].
29
глава Первая
равняющуюся математическому доказательству» [27, с. 505], где в центре внимания исследователя оказываются не отдельные числовые значения величин, а тип их изменения, т. е. функциональная зависимость. Только универсальный математический метод и дедукция, а не аристотелевский силлогизм23 и не сенсуальный опыт признаются способными дать адекватное знание о мире.
«Метод необходим для отыскания истины»,—гласит четвертое картезианское правило для руководства ума [27, с. 88]. Поиски истины без метода приводят к бесконечным блужданиям во мраке, но именно «так,— сетует Декарт, и Бойль соглашается с ним,— трудятся почти все химики, многие геометры и немалое число философов» [27, с. 88].
Для реализации своей методологии, состоящей в поиске переменных величин как элементов бытия, французский философ мысленно устраняет из реального природного тела все конкретное (твердость, тяжесть, цвет, запах и т. д.), не оставляя в нем ничего «внутреннего», никакого второго плана бытия (скрытых качеств, стремлений, напряжений и т. д.), исчезает все разнообразие свойств, остается лишь однородная геометрическая протяженность, рассматриваемая «не как акциденция, а как истинная форма и сущность» материи [27, с. 196]. Тождество тела и его качественной (физико-химической) определенности уступает место тождеству тела и его пространственного положения. Субстанция отождествляется со своим атрибутом — чистой протяженностью.
В итоге, в картезианской физике не остается места пустоте, вакууму — бочка может быть пуста лишь в том смысле, что в ней нет воды или другого вещества, воздействующего на наши органы чувств, но в ней есть воздух, а если его откачать — останется matiere subtile, т. е. эфир. Аналогично сгущение и разряжение вещества сопровождаются изменением размеров пор, заполненных другим веществом. «...Пространства, в которых мы ничего не чувствуем,— подчеркивал Декарт,— заполнены той же самой материей и содержат ее по крайней мере столько же, сколько и пространства, занятые телами, которые мы чувствуем» [27, с. 186].
Но если нет пустоты, то нет и атомов. Материя, единственным атрибутом которой оказывается протяженность (характеристика непрерывная), потенциально бесконечно делима. «Я не мыслю мелкие частицы земных тел в виде атомов или неделимых частиц,— писал Декарт,— напротив, считая их составленными из одной и той же материи, я полагаю, что каждая из них может быть делима бесконечным множеством способов» [28, с. 197]. Но актуально «материя телесных вещей» существует в виде относительно стабильных качественно однородных корпускул, отличаю
23 Борьба с силлогистикой, с «терминологической гидрой», по выражению
М. Монтеня, сопровождалась поисками реального силлогизма (Syllogis-mus realis), который выражал бы не связь терминов, а объективные закономерности [26, с. 210—217].
30
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
щихся друг от друга формой, размерами, характером движения. Их дальнейшее дробление возможно только при наличии достаточной силы [27, с. 504].
Развивая идею об относительно стабильных корпускулах, Декарт допускает существование трех элементов (по сути, трех типов частиц): 1) элемента огня — его частицы очень подвижны и не имеют определенной формы и величины, «ибо стремительность движения огня способствует разнообразному делению» [27, с. 188], 2) элемента воздуха, частицы которого, обладая определенной величиной и круглой формой, наделены более медленным движением, и 3) элемента земли, медленно движущиеся частицы которого характеризуются наибольшими размерами. При этом Декарт совершенно не пользуется субстанциальными качествами (теплом, холодом, сухостью, прозрачностью), ибо «эти качества сами нуждаются в объяснении», тогда как, по его теории, все свойства тел «можно объяснить, основываясь только на понятиях движения, величины, фигуры и расположения частиц материи» [27, с. 189].
Мы привели здесь некоторые важные детали картезианской физической картины мира ввиду ее существенного, идущего через Бойля, влияния на химические воззрения, господствовавшие в XVII—XIX вв. Далее мы увидим, сколь близки взгляды Декарта и Бойля на строение материи и в этом отношении последний стоит ближе к французскому мыслителю, чем к Ф. Бэкону. Это отмечал и сам Бойль: «Хотя атомистические и картезианские гипотезы во многом отличаются друг от друга, тем не меиее при сопоставлении их с учением перипатетиков и с другими вульгарными доктринами их можно рассматривать как одну философию... И картезианцы, и атомисты объясняют одни и те же явления с помощью разнообразно движущихся малых частиц различной формы» [16, б, т. 1, с. 355-356].
Однако общность эта оказалась не только конструктивной, но и роковой. Подобно тому как картезианский рационализм не смог стать универсальной философией природы (и, не в последнюю очередь, в силу ограниченности картезианской физики), хотя и не смог совершенно игнорироваться последующими поколениями философов-рационалистов, так и органически связанная с картезианской механистическая философия Бойля, глубоко войдя в сознание естествоиспытателей XVIII в., не смогла стать универсальной теорией вещества и надолго заняла почетное, но мало к чему обязывающее, место в предисловиях и вводных замечаниях к химическим трактатам. Философская однозначность принятой Бойлем картезианской картины качественно гомогенного механического мира обернулась конкретно-научной (физической и химической) неоднозначностью частных констатаций, о чем не без иронии писал I. Шталь: «...если бы кто-либо захотел приготовить селитру и начал бы размышлять о всевозможных фигурах и их сочетаниях, он никогда бы ничего не добился; ио если взять обыкновенную
ГЛАВА ПЕРВАЯ
соль, жирную землю и проделать определенные операции, то селитру получить нетрудно... Абстракции в химии пользы не приносят» [29, с. 29].
В итоге механистической атомистике было вменено в вину то, в чем она сама ранее обвиняла схоластику,— склонность к риторическим определениям и объяснениям одного неизвестного через другое, еще менее известное: «то, что частицы имеют некую форму,— обратимся снова к Шталю,— справедливо, но не в той степени, чтобы мы могли быть в этом уверены так, как это свойственно риторической точке зрения» [30, с. 51]. И действительно, под определенным углом зрения начинает выявляться некая методологическая общность перипатетического и картезианского подходов к изучению природы, а именно — гносеологический примат абстрактно-общего (элементы-стихии, субстанциальные формы, бескачественные корпускулы и т. д.) перед частным (конкретными физическими и химическими явлениями), что служит характерной чертой всякой «риторической культуры».
Наше рассмотрение идейных истоков корпускулярной философии Бойля будет неполным, если не остановиться на собственно атомистических концепциях первой половины XVII в., в частности на взглядах П. Гассенди и ряда английских атомистов так называемого Ньюкаслского круга.
Пьер Гассенди (1592—1655) 24. В отличие от Бэкона и Декарта, он не создал новаторской системы мира, но именно ему в первую очередь европейская культура обязана возрождением физики и этики Эпикура и Лукреция Кара. Вместе с тем учение Гассенди не было безжизненным слепком с античного наследия. Творчески осмысленное и переработанное им, оно стало важным источником философских и естественнонаучных поисков Дж. Локка, И. Ньютона, Р. Бойля.
По словам К. Маркса и Ф. Энгельса, «материализм выступил против Декарта в лице Гассенди, восстановившего эпикурейский материализм. Французский и английский материализм всегда сохранял тесную связь с Демокритом и Эпикуром» 25.
Действительно, в основе натурфилософии Гассенди лежит твердое убеждение в материальном единстве природы. «Материя есть общая матрица, из которой происходят все тела и все их превращения и преобразования» [31, т. I, с. 229].
Материальной основой мира, по Гассенди, являются неделимые и непроницаемые атомы, между которыми — пустота (Vacuum coacervatum) [32, т. 1, с. 163—165]. «Атомы называются так не потому, что они математические точки, как это обычно думают..., а потому, что не существует силы в природе, способной их рассечь или разъединить...» [31, т. III, с. 466-467].
24 Правильнее Гассенд (Gassend), традиционное Gassendi — родительный падеж латинизированного варианта фамилии.
25 Маркс К., Энгельс Ф. Соч. 2-е изд., т. 2, с. 140.
32
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
Хотя все атомы мыслились Гассенди «однообразными и одинаковой природы», они, однако, обладали, по его мнению, рядом характерных свойств и «внутренних особенностей». Каковы же эти особенности?
Во-первых, атомы телесны, им свойственны определенные плотность, твердость и масса, а значит, и некая, хоть и незначительная, протяженность; во-вторых, они наделены характерной и разнообразной формой (фигурой); в-третьих, атомы обладают тяжестью или весом (gravitas, pondus), а вследствие этого стремлением (impetus) к самодвижению и к агрегации, в результате чего образуются сначала маленькие сращения (minimulas concretiuncu-las), или молекулы (moleculae — уменьшительное от moles — маленькая масса), а затем, в ходе последующих сращений, молекулы укрупняются и в итоге становятся доступными для наблюдения.
Атомы, по Гассенди, «активны и подвижны», «они «сообщают движение всем вещам и служат источником и первопричиной всех природных движений» [31, т. I, с. 337]. Свойства макротел определяются указанными выше геометрическими и механическими качествами атомов и молекул, а всякая трансформация свойств веществ в конечном счете обусловлена перемещением атомов: «всякое изменение... происходит от перестановки частей или увеличения либо уменьшения их числа» [32, т. 1, с. 183].
В обширной литературе, посвященной французскому философу, всегда особо отмечается двойственный, компромиссный характер его воззрений,— с одной стороны, ему принадлежит несомненная заслуга освобождения эпикурейского учения «от интердикта, наложенного на него отцами церкви и всем средневековьем». Тогда как с другой — налицо стремление «как-нибудь примирить свою католическую совесть со своим языческим знанием, Эпикура — с церковью, что было, конечно, напрасным трудом» 26.
Эти слова К. Маркса в полной мере применимы и к Бойлю, с той оговоркой, что если философское двоедушие Гассенди отчасти обусловлено необходимостью искать окольные пути для обхода поставленных инквизицией запретов на пропаганду науки и делать «приемлемые смягчения» в пунктах, касающихся веры (разум стоит обедни!) 27, то искренность компромиссной позиции Бойля не вызывает сомнений28. Бойль был сыном своего века, и, хотя в какой-то мере к нему применимы слова декартовского девиза: bene vixit, qui bene latuit («хорошо живет тот, кто хорошо спрятался»), бурное революционное время в Англии, когда разногласия по поводу богословских тонкостей доводили людей до мученичества, до костра, до тюрьмы, наложило на его взгляды глубокий отпечаток. Англичане, по словам К. Маркса, «восполь
8 Маркс К., Энгельс Ф. Соч. 2-е изд., т. 40, с. 152.
Подробно этот вопрос разбирается в книге Б. Э. Быховского [33, с. 168—
28 См. интересные исследования Дж. Джакоба [34—36].
2 Всеобщая история химии
33
ГЛАВА ПЕРВАЯ
зовались для своей буржуазной революции языком, страстями и иллюзиями, заимствованными из Ветхого завета. Когда же действительная цель была достигнута, когда буржуазное преобразование английского общества совершилось, Локк вытеснил пророка Аввакума» 29.
Теперь сопоставим взгляды Гассенди и Бойля в аспекте учения о веществе. В небольшом черновом наброске английского ученого, посвященном атомной философии, с пометкой: «...эти бумаги должны быть непременно сожжены» *, есть такие слова: «...атомная философия, изобретенная или введенная в употребление Демокритом, Левкиппом, Эпикуром и их современниками, а впоследствии — после того, как с нашествием варваров и варварства из романского мира было изгнано все, кроме случайно уцелевшей перипатетической философии,— либо совершенно игнорировавшаяся европейскими схоластами, либо упоминавшаяся как упраздненная система нелепостей, в наше, менее пристрастное и более любознательное, время была столь счастливо воскрешена и широко прославлена в различных частях Европы благодаря трудам Гас-сендуса, Магненуса30, Декарта и его последователей...» [38]. Документ не датирован, но, по единодушному мнению исследователей, его следует отнести к началу 1650-х годов, т. е. к периоду формирования мировоззрения Бойля31.
29 Маркс К., Энгельс Ф. Соч., 2-е изд., т. 8, с. 120. Но до того как в умах людей Дж. Локк окончательно вытеснил пророка Аввакума, отношение между рационализмом и теологией было далеко не однозначным. Здесь уместно вспомнить одно замечание К. Маркса о генезисе философии, которая «сначала вырабатывается в пределах религиозной формы сознания и этим, с одной стороны, уничтожает религию как таковую, а с другой стороны, по своему положительному содержанию сама движется еще только в этой идеализированной, переведенной на язык мыслей религиозной сфере» {Маркс К., Энгельс Ф. Соч. 2-е изд., т. 26, ч. 1, с. 23). Mutatis mutandis — это относится и к процессу возникновения современного естествознания в XVI—XVII вв. Отдельные религиозные течения (например, основанная на принципах практицизма и утилитаризма пуританская этика), не оказывая прямого влияния па научную методологию, тем не менее санкционировали определенный образ мышления и действия, направленный на экспериментальный подход и познание «божественного механизма» природы [37].
30 О Жане-Кризостоме Маньяке (Магненусе) см. [И, с. 201—203].
31 В сохранившемся списке «трактатов, уже начатых или написанных» Бойлем к началу 1650 г., фигурируют следующие названия: «О натуральной философии и философах», «О химии и химиках», «Об атомах», «О механике» и т. д. [39, с. 64]. Другой список включает тематику запланированных исследований, среди которых значатся: «Наблюдения о некоторых химических различиях солей», «Против перипатетической четверки и химической троицы элементов» и др. [39, с. 76].
* Эта помета отражает опасения благочестивого христианина тех последствий, которые вытекали из атомизма для религии. Ии Бойлю, никому другому пе хотелось прослыть атеистом в Англии XVII в. Отсюда желание примирить пауку с религией, выразить преданность христианству. Здесь кроются причины затяжной душевной борьбы многих английских ученых той нетерпимой эпохи.— Прим. отв. ред.
И
Место химии в естествознании нового времени
Таким образом, уже в юности он смог познакомиться с сочинениями Гассенди, в первую очередь, по-видимому, с его «Animad-versiones in decimuin librum Diogenis Laertii...» (1649), в которых содержался систематический свод эпикурейской философии «Syntagma philosophiae Epicuri» [40], в том числе и атомное учение. Кроме того, обращает на себя внимание поразительное ’(иногда дословное) сходство в рассуждениях Р. Бойля и У. Чарлтона (1619—1707) 32, английского последователя Гассенди, автора обширного труда «Эпикуро-гассендо-чарлтониапская физиология, или Фабрика естественной науки, возведенной на гипотезе атомов...» 33 [41], штудировавшегося Бойлем и молодым Ньютоном. Чарлтон был некоторое время близок так называемому Ньюкаслскому кружку, о котором здесь следует сказать несколько слов.
Такое название в историко-научной литературе получила сформировавшаяся в 1630-х годах в Англии группа ученых и философов, любителей науки, в которую входили: выдающийся английский философ Томас Гоббс (1588—1679); Уильям Кэвендиш, впоследствии герцог Ньюкасла (1592—1676) — богатый покровитель группы, его жена —леди Маргарет (1623—1673), известная в то время своими поэмами на манер Лукреция Кара, где излагалось атомное учение Эпикура; его брат —Чарлз Кэвендиш (1591— 1654) — профессиональный военный, математик и литератор; Дж. Пелль (1610—1685) — математик и священнослужитель; Уильям Петти (1623—1687) — выдающийся экономист, родоначальник классической буржуазной политэкономии, в молодости увлекавшийся атомистикой, и др. Группа просуществовала до середины 1650-х годов. В 1640-х годах многие ее члены жили в эмиграции в Париже, где сблизились с П. Гассенди, Р. Декартом и другими философами и естествоиспытателями.
Практически все члены кружка были атомистами. Типичным примером могут служить взгляды Гоббса, согласно которому Вселенная состоит из видимых тел, невидимых «малых атомов, рассеянных по всему пространству между Землей и звездами», и эфира, «заполняющего всю остальную Вселенную, так что в ней совсем не остается пустого места» [42, т. 1, с. 426]. Атомы, по Гоббсу, представляют собой малые, практически неделимые, находящиеся в постоянном движении, образования различной твердости. Соединяясь, они дают по-разному движущиеся тела разнообразного состава, формы и величины.
Движение в натурфилософии Гоббса, как и у Декарта, играет особенно важную роль — «многообразие всех форм происходит из многообразия тех движений, из коих они образованы» [42, т. 1, С. 63].
32 Соответствующие текстологические сопоставления см. в [23, с. 97—98].
33 О Чарлтоне см. [23, с. 77—92] и [33, с. 129—130].
35
2*
ГЛАВА ПеГВАЙ
Т«Г|.~ • ... — , ,, , . ,, ! , , — I •»
Аналогичные мысли высказывали и другие члены группы. В поэтической форме атомистические идеи были выражены в поэмах М. Кэвендиш [43, с. 5]:
Small Atonies of themselves a World may make
As being subtle, and of every shape
And as they dance about, fit places finde
Such Formes as best agree make every kinde...34
В англо-американской историко-научной литературе роль Ньюкаслского кружка в развитии атомистики оценивается довольно высоко. Так, например, Р. Каргой характеризует деятельность этой группы как «поворотный момент в судьбе атомной философии», «фактор, способствовавший ее открытому признанию в Англии» [23, с. 42] и приводит слова У. Петти, называвшего группу «неофициальным университетом механической философии».
Бесспорно, деятельность Ньюкаслского кружка во многом способствовала укреплению позиций атомного учения, ослаблению влияния на науку схоластических доктрин и религиозного фанатизма и, в известной мере, подготавливала почву для широкого признания корпускуляризма Бойля35 и Ньютона. Но в то же время, в силу своего подражательно-декларативного характера, публикации членов этого «неформального колледжа» не сыграли сколько-нибудь заметной роли в развитии и углублении атомной доктрины.
Итак, мы рассмотрели основные идеи и учения, в контексте которых создавал и развивал свою корпускулярную теорию вещества Р. Бойль. Между ними нетрудно уловить поразительную общность, которая проявляется в том, что:
1) в основе механистической атомистики лежал абстрактный рассудок с его представлениями об изолированных телах и внешних связях между ними;
2) атомы и пустота рассматривались как первоначала и с их помощью объяснялся весь мир в целом;
3) законы механики признавались инвариантными относительно произвольного изменения размеров тела;
34 «Малые атомы, будучи тонкими и обладая различной формой, двигаясь друг около друга, сами по себе могут создать Мир, они располагаются в пространстве (fit places), обнаруживая такие Формы, которые наилучшим образом отвечают каждому типу [свойств тел]...».
Следует заметить, что английская поэзия XVII в. в целом оказалась очень чуткой к новым научным и философским течениям своего времени.
35 Известно, что Бойль был знаком со многими членами Ньюкаслского кружка — Дж. Пеллем, У. Петти [16, б, т. 1, с. LVIII; т. 6, с. 137]. М. Кэвендиш [39, с. 129, 192, 236]. Вместе с тем он решительно выступил против «тех современных почитателей Эпикура», которые изгнали Бога из мира, полагая, что все вещи образованы случайным сочетанием движущихся атомов» [16, б, т. 2, с. 40—43]. Очевидно, Бойль имел в виду Т. Гоббса и М. Кэвендиш (вспомним, например, ее приведенную выше . строчку: «Small Atomes of themselves a World may make)».
36
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
4) все явственнее наблюдалась уходящая своими корнями в практику машинного производства тенденция к абстрагированию от качественного многообразия природных тел, от их чувственно воспринимаемых качеств, которые, удаляясь из предметного субстрата, соотносились с перцептивными особенностями человека*;
5) атомы и корпускулы рассматривались как качественно однородные ультиматы (т. е. как образованные из одного и того же материального субстрата);
6) свойства макротел интерпретировались в рамках наглядных, комбинаторно-кинематических схем, т. е. в терминах соединения-разъединения и перегруппировки мельчайших частиц вещества, отличающихся по своим формам, размерам и характеру механического движения, что знаменовало собой начало в естествознании этапа аналитического восхождения к абстрактным определениям, от «конкретного, данного в представлении, ко все более и более тощим абстракциям» 36;
7) наметился путь к построению иерархической градации природных объектов по степени их усложнения («мпогосоставности»), причем более сложные образования понимались как механическая совокупность, конгломерат менее сложных частиц;
8) механистическая картина мира соотносилась с теологическим представлением о «вселенском часовщике», который «не только сообщил движение материи, но изначально направлял разнообразные движения ее частей так, чтобы они образовали предначертанный им мир» [16, б, т. 4, с. 68].
Глубоко и язвительно точно особенности механистической атомистики раскрыл Гегель: «Атомы неразличимы, одинаковы в себе, или, другими словами, их сущность положена, как чистая непрерывность, так что они, скорее, непосредственно сливаются в одни ком. Представление, правда, удерживает их раздельность, сообщает им чувственно представляемое бытие, но раз они одинаковы, то, как чистая непрерывность, они суть то же самое, что и пустота. Но все, что существует,— конкретно, определенно. Как же можно понять различность исходя из этих начал? Откуда происходит эта определенность,—например, цвет, форма...? Главное же затруднение состоит в том, что, как только мы признаем, что эти атомы в качестве маленьких частиц существуют самостоятельно, их соединение станет совершенно внешним и случайным складыванием» [44, с. 269—270].
Однако, критикуя абстрактный механический атомизм, нельзя, антиисторично, забывать о его прогрессивной роли в эпоху научной революции XVII в. и становления научной химии. Атомистика в это время выступала мощным орудием борьбы со схоластикой и пережитками средневекового миросозерцания. Благодаря ей в химии была по-новому осмыслена проблема генезиса свойств веществ.
36 Маркс К.} Энгельс Ф. Соч. 2-е изд., т. 12, с. 726,
37
ГЛАВА ПЕРВАЯ
Г1 III и. . I. . Д . I . - - - - - , -- — -- - Ч - - — -
«Споря о происхождении и различных видах вещей,— писал Ньютон,— они (перипатетики.— И. Д.) приписывали причинам, изменяющим существование и различие вещей, различные формы, но о причинах и основаниях какой-либо отдельной формы или о том, насколько она отлична от других,— об этом никто никогда не говорил. Итак, они пропускали то, объяснение чего есть высшее дело философов и что единственно может удовлетворить разум...» [45, с. 142].
Сам факт переформулировки химических проблем (особенно проблемы происхождения свойств веществ) в терминах «кинематического корпускуляризма» (Бойль), т. е. в рамках декартовой научной программы, выявляет важный аспект взаимосвязи картезианской методологии (картезианского метода поиска переменных величин как элементов бытия) и атомистики. Действительно, переменная величина Декарта — не бесконечное множество пространственных координат, а бесконечность значений одной, себе тождественной, координаты. Аналогично, в механистической атомистике, подчиненной кинематическим закономерностям, нашла выражение идея бесконечного многообразия взаимных пространственных положений субстратно-однородных себетождественных ультиматов, т. е. идея бесконечности значений координаты в конфигурационном пространстве структурированной системы.
И подобно тому, как в математике сразу стало необходимым дифференциальное и интегральное исчисление,— в учении о веществе (т. е., в первую очередь, в химии) возникла сходная потребность — установить характер изменения свойств в зависимости от изменения движения и геометрии частиц и, наоборот, по данному изменению свойств макротела сделать выводы о характере движения, а также о геометрических особенностях ультиматов. Это один — кинематический — вариант механистической программы в химии, вариант Бойля, в основе которого лежала, повторяем, картезианская методология. Второй вариант — динамический,— восходящий к Ньютону, основан на трех концепциях — сложной иерархической внутренней структуры вещества, невесомых флюидов и короткодействующих (short-range) сил, доступных количественному определению. На концепции Ньютона мы остановимся далее, а сейчас обратимся к более детальному рассмотрению корпускулярной теории Бойля.
КОРПУСКУЛЯРНАЯ ТЕОРИЯ БОЙЛЯ
И ЕЕ ЗНАЧЕНИЕ ДЛЯ ХИМИИ
Сам Р. Бойль, как правило, скромно называл свое учение «корпускулярной или механической гипотезой» (иногда — «философией»), а себя представлял лишь как популяризатора («spokesman») атомно-корпускулярных представлений, который «пишет,
38
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
скорее, для всех атомистов в целом, чем для какой-то их партии» [16, а, т. 2, с. 455]. Критически сопоставляя возрожденную к середине XVII столетия античную атомистику с современными ему теориями строения материи, в первую очередь с картезианской, английский ученый подчеркивал в них прежде всего общие моменты, а не то, что их разделяло. Бойль видел свою главную задачу в разработке единой обобщенной атомно-корпускулярной концепции в ее связи с физическими и химическими экспериментами, концепции, построенной на рационалистических началах и стоящей в оппозиции перипатетическим доктринам субстанциальных форм и качеств.
«Когда, среди других исследований,— писал Бойль,— я направил свои усилия на развитие натуральной философии, то вскоре понял, что понимание химических операций было если и не абсолютно необходимо, то в высшей степени благоприятно для истинного познания природы... Я занимался химией не столько ради нее самой, сколько ради натуральной философии, поэтому большинство задуманных и осуществленных мною экспериментов было, вообще говоря, направлено не на увеличение числа реакций и не на то, чтобы стяжать себе славу обилием искусно проделанных трудных опытов, а на то, чтобы служить основанием... для экспериментальной истории природы» 37 [16,6, т. 1, с. СХХХ]. Мысль о союзе химиков и корпускуляристов проходит лейтмотивом через все натурфилософские произведения Бойля, чего, подчеркнем, не было ни у Декарта, ни у Гассенди, ни, подавно, у атомистов Ньюкаслского кружка. Именно здесь, в точке пересечения атомистической и химической традиций, рождалась наука химия. Другое дело, что становление научной химии шло далеко не простым путем и химик постоянно сталкивался с драматической коллизией между исторически необходимыми требованиями и практической невозможностью их осуществления, ибо со времени Бойля и до появления квантовой механики химия (в тех ее аспектах, которые связаны со строением вещества!) развивалась в контексте неадекватной (или не вполне адекватной) физической теории, что в значительной степени и определило специфику ее развития. Не случайно поэтому Ф. Энгельс, отмечая, что в XVII в. «Бойль делает из химии науку» 38, в то же время отцом современной химии называл не Бойля, и даже не Лавуазье, а Дальтона39, в химической атомистике которого имел место стихийно-диалектический синтез идей химического элементаризма, механической атомистики и рационалистической методологии. В этой исторической перспективе мы и будем далее рассматривать корпускулярную теорию Бойля.
37 В этой связи показателен упрек, сделанный в адрес Бойля картезианцем Б. Фонтенелем, который, сравнивая его с К. Дюкло, высказал сожаление по поводу того, что для настоящего химика Бойль слишком рационалистичен п недостаточно мистичен [14, с. 267—268].
38 Маркс К., Энгельс Ф. Соч. 2-е изд., т. 20. с. 501,
39 Там же, L 608.
39
ГЛАВА ПЕРВАЯ
С наибольшей полнотой эта теория была изложена им в пространном трактате «Происхождение форм и качеств согласно корпускулярной философии...» (1666) [16,6, т. 3, с. 1—137], а также, в кратком виде, в работе «Преимущество и основания корпускулярной философии» (1674) [16,6, т. 4, с. 68—78; 2, с. 187—209] 40. Рассмотрим основные положения этого учения, условно сгруппировав их в следующие пять пунктов.
1. Умозрительной предпосылкой, лежавшей в основании корпускулярной теории Бойля, была идея о том, что все природные тела «состоят из одной всеобщей материи (Catholick Matter)» [16, а, т. 2, с. 513; 16,6, т. 3, с. 93], представляющей собой «субстанцию протяженную, делимую и непроницаемую» [16, б, т. 3, с. 15].
Идея единой, качественно-однородной материи, генетически связанная с формирующейся в XVI—XVII вв. механистической картиной мира (см. выше), имеет принципиальное значение для истории химии, а в известной мере — и физики. Это особенно хорошо просматривается в ретроспекции Дальтон—Бойль (оба имени в этом абзаце, скорее, нарицательные, символизирующие соответственно классическую химическую атомистику XIX — начала XX в. и корпускуляризм XVII—XVIII столетий). В глубоком контексте химической атомистики лежит идея о качественной гетерогенности атомов, индивидуальность которых определяется их «внутренней механикой» (Д. И. Менделеев) и поэтому не сводится, а лишь считается сводимой к чисто количественным различиям по массе, размерам и тому подобным свойствам. Эта идея, как уже отмечалось, органически чужда корпускуляризму XVII—XVIII вв., объяснявшему качественное разнообразие веществ различием геометро-кинематических или динамических параметров, однородных по своей химической природе ультиматов.
В еще более широкой, неклассической, ретроспекции, захватывающей современную кваптовомеханическую теорию вещества, последняя представляет собой диалектический синтез обоих предыдущих подходов к решению проблемы индивидуализации: химическая индивидуальность атома обусловлена в первую очередь его электронным строением (включающим в себя энергетический, пространственный, динамический и прочие аспекты), тогда как структурные единицы неядерной части атома — электроны — абсолютно тождественны, отличаются друг от друга только спин-орбитальные состояния.
Будучи принятой и ориентированной на объяснение физико-химических явлений, идея Catholick Matter с логической необходимостью ведет к корпускуляризму.
40 Указаны произведения, в которых корпускулярная философия Р. Бойля излагается в наиболее цельном виде, ссылки же на нее Бойль делает практически во всех своих работах.
40
ЙЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
Д_ ---- - 1
2. Действительно, «так как материя по своей природе едина, то наблюдаемое разнообразие тел с необходимостью должно следовать из чего-то иного, но пе из природы материи, эти тела образующей. Если допустить, что все ее действительные или мыслимые части пребывают в постоянном покое друг относительно друга, то * непонятно, как вообще могут иметь место какие-либо изменения. Поэтому, чтобы из универсальной материи получить все разнообразие природных тел, остается принять, что некоторые или же все ее части пребывают в движении» [16, б, т. 3, с. 15].
Материя и движение, по мысли Бойля, суть две ипостаси внутренней природы тела, «два его великих и наиболее общих начала» [16,6, т. 3, с. 16]. Вместе с тем движение не является прирожденным свойством материи, как это полагал Эпикур, ибо оно не возникло вместе с ней, а имеет своим источником божественный промысел.
3. В силу отличий в характере и скорости пространственных перемещений отдельных фрагментов Catholick Matter последняя дробится на множество «различным образом движущихся частиц многообразных форм и размеров... которые (частицы.— И. Д.) слишком малы, чтобы быть доступными для ощущения» [16,6, т. 3, с. 16-17].
4. Дробление материи — процесс конечный, приводящий в пределе к первичным частицам — minima или prima naturalia, наделенных определенной величиной (size), формой (shape) и движением (или покоем) 41. Эти частицы «делимы лишь мысленно или всемогуществом бога, но в силу их малости и прочности их природы в действительности они вряд ли когда разделяются и в этом смысле могут быть названы наименьшими (minima) или первичными (prima) природными образованиями» [16,6, т. 3, с. 29].
Сам термин prima naturalia широко использовался и до Бойля, в частности греческими и средневековыми комментаторами Аристотеля, но английский ученый вложил в него новый смысл. В его понимании «первоприродные», в отличие от качественно-гетерогенных перипатетических начал, субстратнооднородны и отличаются друг от друга перечисленными выше механическими свойствами.
5. Prima naturalia универсальной материи способны объединяться в отдельные совокупности (Бойль употреблял термины «cluster», «primitive cluster», «concretion», «coalition», «prima mixta» и др.) [16, б, т. 3, с. 15-17]42.
41 В дополнениях ко второму изданию «Физиологических опытов» (1669) Бойль отметил, что в природе существует лишь относительный, но не абсолютный покой, т. е. полного отсутствия движения не бывает [16, б,
42 т. 1, с. 443—457].
См. также Приложение II.
41
глава Первая
.р. ........................ - .±
Эти вторичные образования играют в его учении важнейшую роль, поэтому на них следует остановиться особо.
Во-первых, корпускулярные кластеры (prima mixta) являются основными структурными единицами тел и при образовании последних способны входить в их состав, как «не теряя своей собственной природы или структуры», так и «претерпевая нарушение сцепления (cohesion) между своими частями» [16, б, т. 1, с. 474-475].
Во-вторых, каждая первичная частица (prima naturalia), входящая в состав кластера, кроме упомянутых в п. 4 свойств, характеризуется также: а) определенным пространственным положением (posture) относительно некоторой системы отсчета (вертикальным, горизонтальным, наклонным) и б) порядком (order) или местоположением (situation), т. е. положением относительно других частиц. Указанные параметры определяют, по Бойлю, «некоторое расположение (disposition or contrivance) частей в целом, которое можно назвать структурой (texture) целого» 43 [16, а, т. 2, с. 461; 16, б, т. 3, с. 76].
В итоге четыре параметра кластеров — их величина, форма, движение и структура — определяли физико-химическую индивидуальность образованного ими тела.
В-третьих, незримые «локальные движения» (как самих кластеров, так и составляющих их фрагментов) играют особо важную роль в понимании природы свойств. Размеры, форма и структура корпускул «в сравнении с их движением часто кажутся следствием, а еще чаще — не более чем условиями, предпосылками, необходимой причиной, лишь модифицирующей то воздействие, которое одна часть материи оказывает на другую посредством движения» [16,6, т. 3, с. 15]. Таким образом, одному и тому же типу корпускулярного движения всегда отвечает определенная совокупность свойств тела и соответственно изменение характера движения неизбежно влечет за собой трансформацию последних. Следует подчеркнуть, что движение рассматривается Бойлем не только как причина модификации свойств вещества, но и, наряду с другими характеристиками микрочастиц, как фактор, определяющий природу и происхождение присущих телу свойств.
Изложенные выше основные положения корпускулярной теории Бойля показывают, что его представления о содержании по
43 Слово «texture» первоначально имело значение: «ткацкое искусство, плетение», а также обозначало любую природную вещь, которая представляется сплетенной, сотканной из чего-либо. Впоследствии этот термин
стал употребляться в расширенном смысле — конституция, структура вещи. Существительное «structure» обозначает процесс или способ построения чего-либо, а также взаимное отношение составных частей пли элементов целого, определяемое свойственной им природой или характером [46, т. 10, с. 1165; т. И, с. 240].
Р. Бойль употреблял оба термина (texture и structure) как синонимы,
предпочитая, однако, первый из них.
42
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
нятия «свойство» («качество») принципиально отличались от принимаемых перипатетиками и спагириками. Согласно последним, субстанциальные формы суть особые сущности, из коих ни одна не требует специального разъяснения и интерпретации ни в терминах объект-субъектных, ни объект-объектных отношений. Поэтому совокупность форм образует экспланас перипатетико-спа-гирической концепции вещества, т. е. ту, не требующую дальнейшего обоснования, совокупность понятий, в терминах которых объясняется физико-химическая индивидуальность данного тела. Это, в свою очередь, исключало необходимость всякой рефлексии над природой форм и качеств, над тайной их происхождения.
Между тем, в условиях интенсивно развивающегося эмпирического знания о свойствах веществ и их превращениях подобные взгляды оказывались уже неадекватными и физически бессодержательными, и не только по причинам, указанным в приведенной на стр. 38 цитате из Ньютона. Определение состава сложного вещества только по совокупности его свойств, как правило, противоречило данным химического анализа, что в общем случае можно представить так: пусть сложное тело К обладает неким набором из N свойств {qi} (&=1,2,..., 2V), причем свойства (gi,..., qk) можно сопоставить некоторому первоначалу А (скажем, горючесть и желтизну связывали с серой), свойства (g^+i,..., qi) — началу В, а свойства (gz+i,..., — началу С:
(ffi*...
со A co В co C
Тогда качественный состав тела К, выраженный через совокупность материальных инкрементов свойств К, можно представить как К^АВС.
Однако это далеко не всегда согласовывалось с данными химического анализа и синтеза, интерпретируемыми в преформационист-ском духе. Реальный материально-субстратный состав мог не совпадать с предполагаемым субстанциалыю-квалитативистским, так как ингредиентами сложного вещества зачастую оказывались тела с другим, отличным от {^}, набором свойств (обозначим его {<?j}) и с иной их соотнесенностью:
(^1» . .
со X
(2р+г • • •» ^)» со Y
(qq+v •••><) coZ
И Т. д.
и т. д.
Например, вещество К могло быть горючим и желтым, но не содержать в себе серу, понимаемую в данном контексте как конкретное вещество, а не как абстрактный синоним горючести и жедтизны.
В этой ситуации Бойль решительно выступил против мнения, согласно которому «все, что люди имеют обыкновение называть качеством, с необходимостью требует некой реальной физичв’
43
ГЛАВА ПЕРВАЯ
ской сущности» [16, б, т. 3, с. 21]. По мнению английского ученого, в телах «нет ничего реального и физического», кроме величины, формы и движения составляющих их корпускул, а потому любое свойство вещества не есть нечто первичное, наперед заданное, но представляет собой результат действия двух факторов — геометрии и локального движения микрочастиц44.
В учении Бойля находит свой конец стоявшая за перипатетическим понятием «миксиса» мировоззренческая установка на рассудочное сведение конкретного факта к универсалиям. Там, где перипатетизм видел ответ на вопрос о причинах наличия определенной совокупности качеств у сложного тела, там Бойль усматривал лишь проблему происхождения самих качеств.
Корпускуляристская постановка этой проблемы имела принципиальное значение для развития химии, так как она способствовала ее глубокой рационализации и физикализации на базе механики, а также определяла функции, замысел, постановку и интерпретацию химических экспериментов. Не так называемое «первое научно обоснованное определение понятия химического элемента» [47], якобы данное Бойлем (о чем см. далее), но утверждение нового понимания проблемы свойств, введение понятия «свойство» в экспланандум химической теории составляют едва ли не главную заслугу английского ученого перед химией.
Однако заслуга эта имеет и свою оборотную сторону, которую на исходе XVII столетия отметил Г. Шталь, а за ним и другие химики XVIII в.
Конечная цель Бойля — объяснить свойства макротел, их физико-химическую индивидуальность, опираясь только на знание механических характеристик корпускулярных кластеров и prima naturalia,— оказалась недостижимой. По справедливому мнению В. П. Зубова, корпускулярно-механическое исследование химических явлений практически осталось лишь «на стадии программы» [11, с. 248]. Для реализации этой программы необходимо было найти такое измеримое аддитивное свойство атома, которое позволяло бы связать макро- и микроуровень химической организации вещества,— таким свойством стал атомный вес. Но для этого потребовалось не только известная модификация самой атомистики, но и глубокая разработка таких физических понятий, как вес и масса тела, а также установление зависимости между числом частиц в образце и его массой или объемом. Развитие же атомно-корпускулярных представлений, как они сложились в XVII в., в итоге привело к объяснительным процедурам, обратным изначальному замыслу,— «форма частиц каждого тела
44 При этом Бойль полностью принимает допущение об инвариантности механических законов относительно изменения размеров тела: «...законы движения имеют место не только в больших массах и в кусках средних размеров, но и в наименьших осколках материи» [2, с. 197—198].
44
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
непосредственно выводилась из свойств этого тела и каждое вновь открытое свойство объяснялось посредством ранее не замечавшегося усложнения в конфигурации корпускул» [14, с. 299], что с наибольшей отчетливостью проявилось, пожалуй, в «философии остриев и крючочков» Н. Лемери [14, с. 281—338].
Рассмотрим теперь, как в контексте механистического кор-пускуляризма интерпретировались некоторые фундаментальные понятия химии XVII в. Остановимся на трех из них —понятиях «химическая трансмутация», «химический элемент» и «химический анализ».
ПРОБЛЕМА ТРАНСМУТАЦИИ МЕТАЛЛОВ
Проблеме трансмутации металлов и перипатетических (спаги-рических) начал-элементов Бойль посвятил многие страницы «Физиологических опытов», «Химика-скептика» (особенно его второй части), последней главы «Происхождения форм и качеств» и других трактатов, не говоря уже о специальных чисто, казалось бы, алхимических его работах: «О прокаливании ртути с золотом» [48] и «Деградация золота» [16,6, т. 4, с. 371—379], где описаны опыты, якобы показывающие возможность трапсму-тации металлов друг в друга.
В часто цитируемом письме к Джозефу Глэнвилю (от 18 сентября 1677 г.) Бойль подчеркивает, что «хотя большая часть этих историй (о трансмутации неблагородных металлов в золото.—Я. Д.) — неправда, однако не все из них таковы», и сообщает о том, как некий Венцель «в присутствии императора, его свиты и хороших химиков несколько раз демонстрировал трансмутацию обычных металлов в золото». «Если все это так,— заключает Бойль,— то, полагаю, один этот позитивный пример доказывает реальность того, что они (химики.—Я. Д.) называют философским камнем, лучше, чем все мошенничества и уловки, с помощью которых химики-обманщики вводили в заблуждение неискушенных и легковерных людей» [16, а, т. 5, с. 244].
Известно также, что Бойль был одним из тех, чьи усилия способствовали отмене в 1689 г. указа Генриха IV английского, запрещавшего алхимические штудии и опыты по трансмутации. В архиве Бойля имеются также многочисленные свидетельства тесной связи его трансмутационных изысканий с теологическими исследованиями [39, с. 166—178].
Все это дало основание некоторым исследователям (см., например, [23, 39]) говорить о влиянии на Бойля алхимической традиции [49, с. 91] и даже характеризовать его как алхимика [50].
Однако внимательное изучение вопроса показывает, что Бойль отнюдь не был сторонником реликтовых алхимических доктрин и на алхимию смотрел глазами химика-механициста XVII в. Его
45
ГЛАВА ПЕРВАЯ
THE SCEPTICAL CHYMIST-. O R CHYMICO-FHYSIC AL Doubts & Paradoxes, Teaching the
EXPERIMENTS WHEREBY
VULGAR SPAGYRISTS Are wont to Endeavour to Evince their SALT, SULPHUR AND
MERCURY, Tobe
The True Principles of Things.
jam tenerentw omnia, & imperia ac Wfifejfe Verity effet I Nihil ex Desreth tnutaremta. Nunc Veritatem cum cis qui docent, qu^rimt^ Sen.
LONDON, Printed for J, Croske, and are to be fold ar rhe Shipin'Sr.P^Z? Church-Yard. r66i.
Титульный лист книги P. Бойля «Химик-скептик» (1661)
понимание трансмутации принципиально отличалось от традиционно алхимического, а сама убежденность в осуществимости такого превращения веществ отличала его взгляды от мнений многих иатрохимиков и спагириков.
Напомним, что, согласно алхимической, ртуть-серной, теории металлов, панацеей трансмутации любого металла в золото должно быть некое определенное соединение отцовского (ртуть) и материнского (сера) начал. «В природе встречаются тела, в которых оба начала соединены уже в нужной пропорции, сгущены и связаны по надлежащим правилам... Ищи эти тела,— призывал Р. Бэкон,— подвергай огню, очищая, проверяй отношение, улучшай и совершенствуй состав...» (цит. по [51, с. 237]). Состав, определенное соотношение элементов-качеств, понятых одно-
временно и как элементы-вещества,— вот что в итоге определяло индивидуальность получаемого тела и в потенции открывало путь к желанному камню философов.
Иначе у Бойля: «...полагая, что все металлы, а также другие тела сделаны из одной Всеобщей Материи, для всех для них одинаковой, но отличаются друг от друга формой, размерами, движением и структурой составляющих их малых частиц, от чего зависят все действия материи (Affections of Matter) и качества отдельных тел,— я не нахожу ничего невозможного в природе вещей для того, чтобы один тип металлов мог бы трансмутироваться в другой...» [16, 6, т. 3, с. 93—94]. По Бойлю, изменение свойств вещества обусловлено либо внутренней перестройкой корпускулярных кластеров (prima mixta) — без их соединения с какими-либо другими частицами, либо вторичным объединением их с другими корпускулами, либо частичной деструкцией этих кластеров.
Превращениям первого рода Бойль придавал особое значение, так как внутри- и межкорпускулярные перегруппировки, по его мнению, являются обязательной стадией любого механического процесса, а в своем чистом виде составляют суть процесса транс
Место химий ё ёсФествознанйи нового времени
мутации45. «...Если путем простого механического изменений внутренней диспозиции или структуры тела удастся уничтожить какое-либо его постоянное качество, которое, как обычно полагают, обусловлено субстанциальной формой или внутренним началом (inward principle) тела, и тут же создать механическим путем новое качество... то такое явление в немалой степени, будет говорить в пользу гипотезы, согласно которой качества зависят от некоторой структуры (contexture) и других механических действий малых частиц тел...»46 [16, а, т. 3, с. 567]. Отсюда явственно видно принципиальное значение трансмутации как аргумента в пользу корпускуляристского решения проблемы происхождения качеств.
Два обстоятельства необходимо учитывать, говоря о концепции трансмутации Бойля. Во-первых, prima mixta — лишь относительно стабильное образование, сохраняющееся неизменным только в ограниченном числе химических превращений и лабораторных манипуляций, где они ведут себя, «как одна цельная корпускула (as one entire corpuscle)» [16, а, т. 3, с. 610]. Во-вторых, что пи одно природное тело, по Бойлю, по может состоять непосредственно из minima naturalia, ибо в противном случае все свойства таких тел были бы совершенно тождественны, так как дробление вещества до практически возможного предела (т. е. до minima naturalia) привело бы не к элементарным компонентам тела, каждому из которых отвечал бы свой тип «первоприродных», по к качественно абсолютно однородной субстанции, к своего рода «плазме», в свойствах которой индивидуальные различия между исходными веществами были бы сняты, вырождены. «Если при химических разложениях,— писал Бойль в «Химике-скептике»,— продукты разделения оказались бы чистыми и простыми телами совершенно элементарной природы, то ни одно из них не могло бы обладать какими-либо специфическими свойствами в большей мере, чем другое» 47 [16, а, т. 1, с. 354].
45 Такое понимание трансмутации сходно с описанным И. Юнгиусом процессом метасинкризиса [52]. Однако, по мнению немецкого энциклопедиста, подобные превращения представляют второстепенный интерес, главными же остаются реакции распада и соединения.
46 Наряду с описанным выше механизмом трансмутации путем изменения механических характеристик prima mixta, Бойль допускал также, что некоторые свойства тела могут определяться наличием или отсутствием в нем особых «благородных и тонких корпускул», с которыми, в частности, он связывал особые свойства золота [16, б, т. 3, с. 95—96], и даже заявил, что нашел растворитель, способный избирательно экстрагировать этот вид корпускул и превращать тем самым золото в серебро. Однако впоследствии эта идея не получила развития в его работах.
47 Заметим, что, в отличие от ошибочного мнения, высказанного в работе [53, с. 54—55], в этом и в аналогичных ему по смыслу фрагментах речь фактически идет об определяющем влиянии характера механических свойств корпускулярных кластеров на индивидуальные свойства соединения, а не об однородности и чистоте вещества как необходимом условии его элементарности.
47
ГЛАВА ПЕРВАЯ
Из сказанного ясно, почему Бойль столь много внимания уделял превращениям, происходящим под влиянием «действующей, а не материальной причины», т. е. «без добавления сколько-нибудь значительного количества простых ингредиентов, участию которых они (оппоненты Бойля.— И. Д.) хотят приписать... качества» вновь образующегося вещества [16, а, т. 3, с. 600]. К таким процессам ученый относил трансмутацию воды в растениях, зарождение птенца в яйце, образование окислов металлов в замкнутых сосудах при нагревании, образование осадка, полученного после многократной перегонки воды, и т. д. И хотя впоследствии результаты наблюдений и экспериментов Бойля либо были признаны неправильными, либо получили иную трактовку, сама идея внутрикорпускулярных перегруппировок содержала в себе рациональное зерно.
Таким образом, путем трансмутации, т. е. путем последовательной перестройки prima mixta данного вещества, возможно, по мнению Бойля, не только получить золото из свинца, землю из воды и т. д., но и, вообще, «посредством незначительной добавки или отнятия материи... и регулярной серии изменений, постепенно предрасполагающих материю к превращению, в конце концов можно из любой вещи создать любую другую (anything from anything)» [16, а, т. 2, с. 474]. Возникающие при этом трудности имеют технический, но не принципиальный характер.
В итоге убежденность Бойля в возможности трансмутации следует расценивать не как результат влияния на него алхимической традиции и не как идейный атавизм средневековья, а как прямое следствие дискретно-комбинаторного характера его корпускулярной теории вещества.
Говорить о Бойле как об алхимике (как это делал, например, Л. Мор [50, 54]), даже при наличии глубинной субъективной связи его физических воззрений с его теологическими работами и миссионерской деятельностью, столь же неправомерно, как рассуждать всерьез о влиянии алхимической традиции на Э. Резерфорда, ибо Бойль понимал трансмутацию в совершенно ином, философском и научном, контексте, чем алхимики, и формальное (терминологическое) сходство проблематики не должно вводить в заблуждение.
ПРЕДСТАВЛЕНИЯ БОЙЛЯ О ХИМИЧЕСКИХ ЭЛЕМЕНТАХ
Подавляющее число историков химии, от Г. Коппа [55] до Дж. Партингтона [56] и Б. М. Кедрова [53], по-разному трактуя те или иные аспекты химических представлений английского ученого, сходились в одном: последовательно проводя концепцию химического анализа, Р. Бойль дал первое научно обоснованное определение химического элемента как предела разложения ве
48
МЁС^О &ИМЙЙ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЁМЕЙЙ
щества, положив его в основу объяснения всех химических процессов, в результате чего «впервые стал определяться предмет химии как науки, изучающей в первую очередь химические элементы и их соединения» [53, с. 56—57]. Данное Бойлем определение химического элемента, по мнению Н. А. Фигу ров ского’, {{впервые поставило перед химиками реальную задачу: искать те последние, далее не разложимые, составные части сложных тел (а не понимать их только философски, как это делали схоласты-перипатетики и спагирики), познание которых должно в конце концов решить стоявшую в течение многих веков задачу о природе сложных тел в связи с их свойствами (курсив наш.— И. Д.)» [57, с. 209].
Иные взгляды изложены в работах Э. Мейерсона [58], Э. Метцжер [14, 15], М. Боус-Холл [2, 13], Т. Куна [59, с. ISO-181; 60], Л. Ланжевен [61], В. П. Зубова [И, с. 257], ГО. И. Соловьева [62], Р. А. Вихалемма [63] и др.
«Почти всегда,— пишет Кун,— когда это понятие (химического элемента.— И. Д.) вводится, его происхождение приписывается химику XVII века, Роберту Бойлю, в книге которого «Химик-скептик» внимательный читатель найдет определение «элемента», вполне соответствующее определению, используемому в настоящее время... В качестве части педагогического арсенала, который делает из человека ученого, такой возврат к прошлому оказывается чрезвычайно успешным. Тем не менее все это иллюстрирует еще раз образец исторических ошибок, который вводит в заблуждение как студентов, так и непрофессионалов относительно природы научного предприятия.
Согласно Бойлю, который был в этом совершенно прав, его «определение» элемента не более чем парафраза традиционного химического понятия', Бойль предложил его только для того, чтобы доказать, что никаких химических элементов не существует (курсив наш.— //. Д.)» [59, с. 180—181].
Кто же прав? Действительно ли номинация элемента углубляется у Бойля до новаторской проблемы или остается реликтом вербальных спагирических определений?
Уже из сказанного выше о корпускулярной теории Бойля и о его взглядах на трансмутацию ясно, что концепции английского физико-химика по существу исключали всякую возможность существования в природе каких-либо химических элементов как качественно-гетерогенных, мельчайших, химически неделимых материальных образований, инвариантных относительно любых химических превращений сложных тел, в которые они {элементы) входят в качестве ингредиентов. Но чтобы окончательно убедиться в логической последовательности Бойля, обратимся к контекстуальному смыслу его знаменитого «определения».
Первоначально свои соображения о химических элементах Ьойль изложил в небольшом отрывке, названном «Размышления
49
tЛАВА ПЕРВАЙ
Г------------- . .. ......................,л
по поводу экспериментов, на которые обычно ссылаются, когда утверждают существование четырех перипатетических элементов или трех химических начал сложных тел», написанном им между 1654 и 1657 гг.48 Текстологические сопоставления позволяют рассматривать эту работу как подготовительную к созданию «Химика-скептика». Уже в начальных абзацах «Размышлений...» Бойль обращается к проблеме элементов.
«Эксперимент, на который, как правило, опираются сторонники обычного мнения о четырех элементах, состоит в том, что если сжечь на открытом огне зеленую ветку, то сначала улетучится дым, который полагают воздухом, затем станет выкипать до конца жидкость, которая считается водой, огонь обнаруживает себя по характерному свечению, а остающаяся в конце негорючая часть есть не что иное, как элемент земля.
Прежде чем рассмотреть этот эксперимент, я предварительно разъясню, что под элементом я здесь понимаю те простые тела, из которых составлены тела сложные и на которые они (т. е. сложные тела.— И. Д.) в конечном счете разлагаются. Замечу далее, что из некоторых тел эти четыре элемента не могут быть выделены...» [64, с. 158]. В качестве примеров Бойль приводит золото, серебро, кальцинированный тальк. Впоследствии эти мысли вошли в «Химик-скептик», но уже в более развернутом виде и в рамках иного композиционного построения.
Как известно, «Химик-скептик» — трактат-диалог. После того как один из участников диалога Карнеад, выступающий от лица самого Бойля, высказывает критические замечания в адрес перипатетиков и спагириков, его оппонент — спагирик Элевтериус — заявляет: «Я ожидаю, Карнеад, что после того, как Вы столь непринужденно (freely) изложили свои сомнения в том, существует ли какое-либо определенное число элементов, Вы обратитесь к вопросу о том, существуют ли элементы вообще» [16,6, т. 1, с. 561]. На что Карнеад отвечает: «Не будет абсурдным сомневаться в этом, ибо надо еще доказать, столь ли уж необходимо допускать существование каких-либо элементов или гипостатиро-ванных начал вообще» [16,6, т. 1, с. 562]. И далее, чтобы быть правильно понятым, Бойль-Карнеад формулирует традиционное, «химическое», понимание элемента, которое мы приводим в нарочито расширенном контексте.
«Теперь, как и ранее,— отвечает Карнеад,— чтобы избежать ненужных затруднений от ведения спора по отдельности с перипатетиками (Aristotelians) и с химиками, я буду выступать против и тех, и других (одновременно.—Я. Д.), поскольку их доктрина
48 При жизни ученого эта работа не была опубликована. В архивном отделе библиотеки Королевского общества (Лондон) сохранилась ее копия, сделанная непременным секретарем общества Г. Ольденбургом. В 1954 г. рукопись была опубликована М. Боус-Холл [64].
50
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
относительно элементов все еще одобряется современниками, так как она представляется им основанной в значительной степени на эксперименте» [16, а, т. 1, с. 356]. И далее, чтобы «вести дело не только беспристрастно, но и благожелательно» по отношению к своим оппонентам, Бойль-Кариеад предлагает сосредоточить внимание на двух элементах — земле и воде, ибо воздух «не входит в состав сложных тел как один из элементов, но лишь располагается в их порах, или, вернее, наполняет в соответствии со своим весом и текучестью, все полости тел...»49 [там же]. Поэтому воздух нельзя считать элементом. Естественно, в этом месте возникает необходимость договориться о терминологии, что Бойль и делает:
«А чтобы предупредить ошибки, я должен уведомить Вас, что я теперь подразумеваю под элементами, как и те химики, которые говорят, будто своими началами они вносят полнейшую ясность 50, некоторые первоначальные (primitive) и простые или совершенно несмешанные тела, которые, не будучи образованы из каких-либо других тел или друг из друга, являются ингредиентами, из которых непосредственно составляются все так называемые совершенно смешанные тела и на которые эти тела в конечном счете распадаются. Теперь я сомневаюсь в том, может ли существовать такое тело, которое постоянно встречается [во всех и в каждом из тех тел], о которых говорят как о составных» 51 [16, а, т. 1, с. 356; 16,6, т. 1, с. 562]. И далее: «Я не понимаю, почему мы должны верить, будто существуют некие первозданные (primogeneal) и простые тела, из коих, как из предсуществующих (pre-existent) элементов, природа вынуждена строить все другие. Я также не могу понять, почему нельзя думать, что природа способна производить тела, считаемые сложными, составленными (mixt out) одно из другого, по-разному меняя и сочетая их мельчайшие части, без разложения материи на такие простые или гомогенные субстанции, как это утверждают» [16,6, т. 1, с. 583].
Итак, как явствует из приведенных фрагментов «Химика-скептика», Бойль лишь в систематическом виде изложил элемен-таристские идеи своих предшественников52, не давая никакого
49 Огонь же, по словам Бойля, «еще раньше воздуха рассматривался (в подлиннике более сильное выражение: exploded, т. е.. был. дискредитирован.— И. Д.) здравомыслящими людьми как мнимая (imaginary) вещь» [16, а, т. 1, с. 356].
Выделенные нами курсивом слова обычно опускаются теми, кто видит в Бойле основателя современных (или близких к таковым) представле-ний об элементах.— И. Д.
1 В собрании сочинений Бойля, изданных под редакцией П. Шоу в 1738 г., приведенные нами в скобках слова исключены и вместо них стоит про-сто «в телах» [65, т. 3, с. 336].— И. Д.
Ср., например, с определением французского медика и химика XVII в. Э, де Клава; ««Элементы — суть простые тела, из коих изначально состоят
51
ГЛАВА ПЕРВАЯ
своего определения элемента; более того, его взгляды в корне расходятся не только с перипатетико-спагирической, но щ вообще, с любой концепцией элемента. Ценность этой концепции для химика -—и в XVII в., и в наши дни — состоит, во-первых, в относительной инвариантности в ходе химических превращений определенной совокупности качественных и количественных характеристик наименьших частиц того гомогенного материального образования, которое полагается элементарным, и, во-вторых, в неразложимости этого образования на качественно новые субстанции 53. Но именно эти два момента были неприемлемы для Бойля-теоретика, по мнению которого, «в общем понятии mixtio не подразумевается с полной определенностью, что ингредиенты его в виде мельчайших частиц сохраняют свою природу и существуют раздельно внутри соединения» 54 [16,6, т. 1, с. 506].
Анализ других работ Р. Бойля полностью подтверждает сделанные выводы. Так, во втором английском издании «Химика-скептика» Бойль в специальном приложении, озаглавленном «Эксперименты и замечания относительно образования химических начал», писал: «Если тела, именуемые началами, могут получаться de novo, то насколько доказательным будет утверждение о том, что природа, образуя сложное тело, использует те начала, которые у нее уже есть в наличии» [16, а, т. 1, с. 376]. Подобных высказываний английского ученого можно привести немало. Все они говорят о том, что Бойль пе остановился на отрицании элементов Аристотеля и Ф. Парацельса. Он поставил вопрос шире и глубже,— надо ли, вообще, для описания химических явлений вводить представление об элементах? Как убежденный последователь кинематического корпускуляризма Бойль склонялся к анти-элемептаристской позиции: кластеры (prima mixta), определяющие свойства тел, лабильны и разложимы. Процессы образования или разрушения соединений связаны, как правило, с существенной перестройкой prima mixta, так что само понятие ингредиеп-
все сложные тела (composition des mixtes) и на которые эти последние в конечном счете разлагаются или могут быть разложены» [66, с. 39 (см. также, с. 27, 40, 260)]. «Никто и никогда,— настаивает де Клав,—не сможет лучше узнать состав вещей, иначе как путем их разложения, которое есть не что иное, как сведение сложных тел к их началам» [66, с. 39].
53 В атомистической терминологии это означает признание существования в природе определенного конечного числа различных видов атомов, так что атомы каждого вида отличаются своей совокупностью коренных свойств, остающихся относительно неизменными при переходе атомов данного вида из одного соединения в другое.
54 В этой связи характерно замечание П. Шоу: «Химики ... говорят об элементах как о самых первичных корпускулах, из которых составлены сложные тела,— представление, ведущее их к бесконечным трудностям и являющееся основанием большей части возражений против них (химиков.— II. Д.) со стороны мистера Бойля» [67, с. 158].
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
та сложного тела оказывается в теории Бойля весьма расплывчатым.
Если сложное вещество образуется из вещества А и В, то в силу лабильности кластеров этих А и В нельзя сказать, что состав сложного вещества есть АВ. Аналогично, при анализе тела могут в одних условиях получаться продукты А и В, а в других — С и D и т. д. Можно, конечно, возразить: если А и В неразложимые далее тела, которые получаются при анализе данного вещества в наиболее жестких условиях, то концепция элементов вполне правомерна. Но в том-то и дело, что практически неразложимыми, по Бойлю, является не prima mixta, a minima naturalia, которые, однако, в силу своей качественной однородности (см. выше), не могут рассматриваться как элементы. Но точку на этом ставить рано, ибо действительность эпохи оказалась много богаче и диалектичнее ее сознания.
Как правило, историки пауки, исследующие творчество Р. Бойля, не обращают внимания па глубокое противоречие между теми выводами, которые логически вытекали из общих положений его корпускулярного учения, и теми, к которым он пришел в результате своей практической деятельности у горна или у лабораторного стола. Бойль-теоретик был решительным противником идеи об элементах, тогда как Бойль-практик (причем, практик наблюдательный и искусный) вынужден был признать существование инвариантов состава *. Поэтому на деле запрет на элементы оказался не абсолютным (как и картезианский запрет на неделимые атомы) — практически, отрицание и скепсис затронули не столько саму идею элемента, сколько модальность этой идеи, одностороннее сведение ее к классифицирующему каталогу абстрактно мыслимых сущностей. Как бы химик-практик, современник Бойля, ни относился к корпускулярным концепциям, представления о существовании неких простейших образований, о неизменных (пусть, относительно неизменных) компонентах сложных тел, о некоторой иерархии составов и о связанном с ней генезисе свойств веществ составляли необходимый минимум химического мышления, то, без чего в XVII столетии обойтись было уже невозможно55.
* Мнение, что Бойль был скорее экспериментатором, чем теоретиком, поддерживалось работами самого ученого, в которых неоднократно подчеркивалась первостепенная важность экспериментальных фактов. Бойль, однако, не считал, что теории возникнут сами собой из совокупности экспериментальных данных. Он видел необходимость гипотез, которые могут направить эксперимент, указать путь к целенаправленному научному исследованию и объяснить данные опыта (см. Приложение I).— Прим. отв.
55 Ред'
5 По словам В. П. Зубова, «у химиков первой половины XVII в., да и позднее, термин «субстанциальная форма (как и термин «элемент».— И. Д.) приобрел еще иное, так сказать, лабораторно-техническое, значение. «Субстанциальной формой» стали наделять лишь практически неразложимые или неподдающиеся искусственному синтезу вещества» [11, с. 261—262].
33
ГЛАВА ПЕРВАЯ
Но для последователя кинематического корпускуляризма, каким был Бойль, ситуация осложнялась следующим противоречием: с одной стороны, сплошь и рядом эксперимент показывал, что многие тела, пройдя редукционную цепочку превращений, аналогичную тем, которые описывал И. Юпгиус, оставались в итоге неизменными (например, кислота может растворить один металл в сплаве и оставить нетронутым другой и т. д.) 56, но с другой стороны, свойства промежуточных сложных тел оказывались порой диаметрально противоположными свойствам ингредиента (например, растворяя сурик в уксусной кислоте, получают сладкую соль, весьма отличную от обоих ингредиентов, и уксусная кислота как таковая, «по-видимому, разрушается» [16,6, т. 1, с. 507]). Иными словами, «контроль по составу» (точнее, по «генеалогии» соединения) и «контроль по свойствам» приводили к несовпадающим результатам. Разработанное Бойлем представление о прочных и непрочных корпускулах было далеко не универсальным решением вопроса, а данное в рамках этого представления понятие об элементе («...собрания корпускул, способных крепко прилипать друг к другу или могущих быть дополненными одни другими, составляют прочные, трудноразложимые скопления частиц... [которые] могут быть названы первичными кластерами или элементами вещей» [16, а, т. 3, с. 77]) оказалось парафразой па этот раз картезианской идеи относительно стабильной корпускулы.
Таким образом, понятие о химическом элементе у Бойля в полной мере не отвечает ни одной из приведенных выше историко-научных трактовок, так как оно, вообще, не умещается в одностороннюю схему анализа, а требует учета скрытых за этим понятием реальных противоречий в эволюции теоретических представлений химии.
ХИМИЧЕСКИЙ АНАЛИЗ
Говоря о научных заслугах Бойля, обычно отмечают его вклад в создание качественного химического анализа, называя английского ученого «зачинателем» этой области химии [53, с. 55]. Однако такая оценка тоже требует некоторой конкретизации.
Действительно, Бойль одним из первых начал систематическое использование в химической практике кислотно-основных индикаторов 57. Им же стал разрабатываться сероводородный метод качественного анализа*. Однако в своих лабораторных исследо-
56 См. Приложение II, пункты 2 и 4.
57 До Бойля индикаторы применяли английские химики Э. Жордан (1631) и Р. Уитти (1660).
* Более подробно о химическом анализе см„ главу четвертую,— Прим, отв, ред,
54
МЕСТО ХИМИЙ В ЕСТЕСТВОЗНАНИЙ ЙОЁОГО ВРЕМЕЙЙ
Баниях ученый не ограничивался чисто эмпирической стороной дела, а стремился связать полученные результаты с той или иной теоретической проблемой, в данном случае с проблемой интерпретации химико-аналитических данных.
Что дает анализ, в частности анализ, проводимый с помощью огня (analysis by fire), т. е. методом термодеструкции (наиболее распространенным во времена Бойля [68, 69]),—истинные компоненты соединения или артефакты, т. е. продукты вторичных процессов, происходящих в анализируемом теле под действием огня и сводящихся к внутри- и межкорпускулярным перегруппировкам? Иными словами, является ли огонь или другой анализатор тел пассивным инструментом, позволяющим «аккуратно» расщепить соединение, не изменяя его внутреннюю структуру и характер локальных микродвижений в нем, или же результаты анализа оказываются неинвариантными относительно выбора аналитического метода и способа его реализации? Эти вопросы имели для Бойля принципиальное значение.
По мнению ученого, «когда тело разлагается огнем па свои ингредиенты, предполагаемые простыми, то образующиеся субстанции на самом деле не являются подлинными и настоящими элементами, а произведены акциденталыю огнем, который, рассыпая тело на мельчайшие части (особенно если эти части находятся в замкнутых сосудах), вынуждает их после разложения вновь соединяться друг с другом, но уже иначе, чем раньше... [16,6, т. 1, с. 583]. Огонь как инструмент анализа, «даже тогда, когда он разделяет тело на субстанции различной консистенции, в большинстве случаев не разлагает тело на гипостатические начала (как это утверждали большинство химиков и особенно спагири-ки.— И, Д.\ по лишь образует из этих частей новые структуры» [16,6, Т. 1, с. 485].
Эти соображения толкали Бойля к поискам других, «мягких», методов анализа — экстракционных, индикаторных и т. д. Но важнее, на наш взгляд, сама постановка вопроса — мысль о влиянии аналитического метода на результаты анализа, столь актуальная для теоретических основ современных физических методов исследования строения вещества58.
Таким образом, химия обязана Роберту Бойлю не только исследованием многочисленных качественных реакций, разработкой аналитических методик и т. д., но и глубокой постановкой вопроса о характере аналитических процедур с позиций корпускулярной микромеханики, вопроса, в котором ясно просматривается исторический антецедент современной концепции приборного эксперимента.
Эта мысль нашла свое выражение в принципе относительности к средствам наблюдения В. А. Фока, в соответствии с которым наблюдаемые свойства системы зависят от метода наблюдения и могут меняться от метода к методу.
55
ftjIABA ПЕРВАЯ
Подытожим наше рассмотрение теоретических проблем химии XVII в. и творчества Р. Бойля.
Во второй половине XVII столетия в химии функционировали и противоборствовали между собой три концепции генезиса свойств химических соединений:
1. Субстанциалистская — свойства тела, {PJ, определяются особыми сущностями, субстанциальными формами и качествами, {sk}, способными соединяться с косной, бескачественной материей (субстратом) тела:
{^} = /(Ы)
(здесь и далее индекс I нумерует свойства макрообъекта, а индекс к — определяющие свойство факторы).
2. Преформационистская — свойства тела определяются свойствами входящих в него компонентов (как правило, имелись в виду компоненты элементарной природы), {efe}, т. е. качественным составом соединения:
3. Корпускуляристская — свойства тела определяются характером локальных движений составляющих его корпускул, а также их структурой, размерами и формой, т. е. кинематико-геометрическими параметрами кластеров (prima mixta), {mh}:
Конфронтация указанных пониманий природы свойств веществ определила практически все существенные особенности развития химии на долгие годы, вплоть до начала XIX в.
Два обстоятельства — невозможность последовательного проведения корпускулярной программы и архаический, не отвечающий состоянию развития экспериментальной химии второй половины XVII столетия, характер теории субстанциальных форм —заставили химиков обратиться в итоге к преформационистским концепциям, в рамках которых удалось провести первую систематизацию соединений по их составам и свойствам. По словам Э. Метцжер, «корпускулярная система в целом, как о ней сказал Юнкер, правдоподобна и даже неоспорима. Но она оказывалась недостаточной всякий раз, когда ученый намеревался объяснить какие-либо материальные явления. В этом случае он вынужден был обращаться к вспомогательным гипотезам, которые не противоречили системе, но были не более чем добавкой к ней... И наиболее умеренные возражения, которые XVIII век адресовал методологии Бойля и Лемери, представляются как амплификация к словам Паскаля: «Вообще надо сказать, что все происходит посредством форм и движений, ибо это так, но говорить о том, из чего и как построена машина (Вселенной.—Я. Д.),— нелепо, ибо это бесполезно, неопределенно и трудно» [14, с. 450].
56
МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ
Но вместе с тем, при всех негативных моментах корпускуля-ризма Бойля, его учение сыграло в истории химии выдающуюся роль. Из недр корпускулярной философии в XVII в. вырастала новая структура химического мышления. В конце XVII в. было, однако, более значительным влияние Бойля-экспериментатора, нежели Бойля-теоретика. Так, в физико-химической диссертации Бона (1685) изложены корпускулярные идеи Бойля и его идеи об элементах. Но основное внимание автор уделяет экспериментальным исследованиям Бойля. В книге преподавателя философии и медицины Барнера «Философская химия» (1689) упоминаются эксперименты Бойля, имеющие значение для медицины.
Химия в конце XVII — начале XVIII вв. не пошла по пути, указанному Бойлем, причина заключалась в том, что перед химиками были поставлены новые задачи, решение которых взяла на себя теория флогистона.
ГЛАВА ВТОРАЯ
РАЗВИТИЕ ХИМИИ
В КОНЦЕ XVII—XVIII ВВ.
В XVI-XVII вв. начался процесс выделения химии в самостоятельную науку. В то время химия владела множеством практических сведений, успешно применяемых в металлургии, медицине, фармации и ремеслах.
Однако теоретические представления в химии отставали от ее экспериментальных результатов, поскольку, несмотря на возрождение атомистических воззрений, формирующих мысль о вещественности всего существующего в мире и, следовательно, о реальности химических элементов, атомы представлялись тогда, как и в Древнем мире, вечными и неизменными, а исчезновение их качеств в химических соединениях было необъяснимо. В этом, возможно, скрывается причина сосуществования учения о невещественных элементах-стихиях с атомистическими воззрениями. Такое положение в химии продолжалось до конца XVIII в. Появившиеся в XVII в. важные для химии идеи лишь постепенно способствовали становлению химии как науки.
ОПРЕДЕЛЕНИЕ ПОНЯТИЯ «ХИМИЯ» В УЧЕБНИКАХ ХИМИИ ВО ВТОРОЙ ПОЛОВИНЕ XVII-XVIII ВВ.
Процесс превращения химии в самостоятельную науку, начавшийся в XVII столетии, теснейшим образом связан с определением предмета и задач химии, с определением понятия «химия».
Представление о химии как практическом искусстве, обслуживающем медицину, фармацию, металлургию, в середине XVII в. начинает трансформироваться в связи с новым подходом к химии, который четко сформулировал Р. Бойль, утверждавший, что химию следует рассматривать с позиций философа, а пе с позиций врача или алхимика. Подобный взгляд нашел отражение в «Курсе химии» Николя Лефевра (1616—1669), члена Лондонского Королевского общества, опубликованном во Франции в 1660 г., за год до появления труда Р. Бойля «Химик-скептик» *.
Примечание. Литературу см. стр. 398—401.
1 Известно шесть французских изданий «Курса химии» Н. Лефевра и одно английское. В 1751 г. этот курс был издан также Н. Лангле-Дюфренуа (см.: Daumas М. Lavoisier tlieorecien et experimen late цг. P., 1955, p. 94)
59
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII вв.
Н. Лефевр посвятил несколько страниц в своем двухтомном «Курсе химии» обоснованию утверждения, что химия — это наука. Цо мнению Лефевра, теоретическая и практическая химия существует в трех видах: «...Совершенно научная и умозрительная (химия.— 3. Ш.) может называться философской химией, которая не имеет другой цели, как созерцание и познание природы и результатов, так как она выбирает в качестве объекта вещи, которые никоим образом не находятся в нашей власти. Так, эта философская химия стремится познать природу неба и небесных светил, начало элементов, причину метеоров, происхождение минералов и пищу животных и растений... рассуждая пока о различных впечатлениях. Второй вид химии может называться иатрохимией, что означает химическая медицина (La Medicine chimique), имеющая целью действие, которое может быть достигнуто все-таки методом умозрительной и научной химии, так как медицина состоит из двух частей — теории и практики» [1, с. 6-7].
Задача практической химии в применении к медицине сводилась к изучению организации и жизненных отправлений животных. Наконец, третий вид химии — фармацевтическая, цель которой нахождение методов получения лекарств,— также состоит из теории и практики.
Таким образом, химия, согласно Лефевру, основывается на теории, т. е. размышлении о природных телах, и практике, с помощью которой она изменяет природные тела.
Отделяя химию от других наук, Лефевр рассматривает вопрос о различии между физикой и химией. При общем предмете изучения, химия, в отличие от физики, не довольствуясь только умозрениями, с помощью эксперимента испытывает соотношения (рго-portiones) природных тел, основываясь на чувствах (имеются в виду чувства осязания, обоняния, вкуса и т. п.) [1, с. 8]. Лефевр обращает внимание еще на одно различие — между «химическим физиком» (Le Pliysicien chimique) и физиком. Это различие касается их подхода к первичным «началам», составляющим сложные тела. Одна из задач, которую всегда ставит перед собой химик,— отмечал Лефевр,— это получение составных частей тел разложением смеси. Он подчеркивал, что химия оставляет вне поля зрения малые части, «делимые или неделимые», и устремляется только к сущностям видимым и осязаемым. Для химии важно то, «что мы видим в работе, так, если мы говорим, что такое-то тело состоит из кислого спирта, горькой соли и сладкой земли, то вам необходимо видеть, трогать, обонять части, извлекаемые нами...» [1, С. 8],.
Н. Лефевр, таким образом, придерживался точки зрения И. Ван Гельмонта и др., что предметом изучения химии должны быть составные части тел, па которые эти тела могут быть разложены.
59
ГЛАВА ВТОРАЯ
В теоретической части «Курса химии» Н. Лефевр изложил представления об элементах Аристотеля, однако сомневался в их существовании. Основу философской химии он видел в учении о пяти началах Парацельса, которое возникло из наблюдений качественного превращения веществ. При перегонке веществ растительного и животного происхождения всегда получалось пять продуктов: спирт или ртуть (легкая горючая жидкость), флегма (водянистая негорючая жидкость), масло или сера (густая маслянистая и вязкая горючая жидкость); нелетучий остаток при выщелачивании водой давал еще два продукта: растворимый в воде (соль) и не растворимый (земля или «мертвая голова») [2, с 29]. Из этих пяти начал спирт, масло и соль Парацельс считал «активными» началами, воду и землю — «пассивными» [3, с. 41]. Лефевр разделял представления Парацельса, разъясняя, что его начала не следует принимать за действительные химические элементы. Лефевр предполагал, что, кроме этих начал, в природе существует рассеянная всюду субстанция (универсальный спирт, или универсальный дух — L’esprit universel), способствующий переходу металлов в нелетучее состояние. Повторив опыты Г. Поп-пия по увеличению веса металлов при обжиге, Лефевр заключил, что сурьма, которую он обжигал при помощи собирательной линзы, прибавляла в весе, поглощая универсальный дух. Впоследствии Л. А. Чугаев, как и ранее Ж. Дюма, увидел в этом предположении «смутное предчувствие кислорода» [4, с. 48].
Н. Лефевр жил в эпоху, когда вырабатывались и складывались основные методологические концепции, па которых позднее строилось здание науки о неживой природе. Схоластическому методу мышления натурфилософии противостояли новые воззрения о необходимости опытного исследования природы. По натурфилософские представления перипатетиков, поддерживаемые католической церковью и университетами, все еще продолжали существовать, несмотря на критику со стороны выдающихся мыслителей.
«Курс химии» Лефевра, в значительной мере отражая эклектические представления того времени, содержит, однако, новый методологический подход к химии как самостоятельной области науки, способной, как и физика, решать стоявшие перед ней задачи не только экспериментальным путем, но и теоретическим (философским).
Влияние воззрений Лефевра заметно в «Курсе химии» (1663) К. Глазера (1628—1673), сменившего Лефевра в должности преподавателя химии в Королевском саду в Париже. Глазер рассматривал химию как самостоятельное «научное искусство», а пользу, доставляемую «этой прекрасной наукой», охарактеризовал как «ключ, могущий открыть естествоиспытателям врата тайн природы» [5, с. 3].
В опубликованных позднее химических руководствах в определениях химии подчеркивается практический аспект в изуче-
но
ЙАЗёМЙЕ ХИМИИ В КОНЦЕ XVII—XVIII вв.
нии химических явлений: химия представляется «искусством», необходимым для доказа-
D Е
тельства истин в других науках. Химию как искусство трактовали французский химик Н. Лемери (1675) [6, с. 2], немецкий профессор И. Бон (1685) [7] и польский врач
Я. Барнер (1689) [8].
Искусством «разложения тел смешанных... на их составные части, а также искусством «соединения составных частей в тела» считал химию Г. Шталь (1723) [9, с. 1]. В 1729 г. немецкий ученый Г. Тейхмейер в своем определении химии предлагал рассматривать ее как действенную науку (что, по его мнению, более правильно), которая учит «посредством огня и растворителей разлагать сме-
CONTENANT -
LA MANIERE DE FAIRE les Operations qui font cn ufage dans la Mcdecine , par une Methode facile.
АГЕС DES RAISONNEMENS fur chaque Operation 9pour I’tnfiruciion de ceux qut 'veulent stypliquer a cette Science.
Par M. NICOLAS LEMERY, de Г Academic Royale des Sciences, Dofteur en Medecine.
ONZIEME EDITION.
Rev cue, ccrrigfe & augment?? par Г Auteur.
Л LET D E,
Chez
।
THEODORE HA AK, Marchand Librairc, 1716.
шанные тела, отделять, очищать и вновь соединять начала тел» [10, с. 3]. Его соотечест-
Титулъный лист книги Н. Лемери «Курс химии» (1716)
венник И. Юнкер в 1730 г.
полагал, что химия философская есть искусство...» [11, с. 1]. Это определение во многом повторяет определение Шталя.
Голландский ученый Г. Бургаве (1668—1738) в 1732 г. также
определил химию как «искусство, каким образом производить
надежные химические операции, посредством которых при помощи соответствующих инструментов можно открывать или обнаруживать чувствительные тела и собирать их в сосуды с тем, чтобы познать отдельные полученные продукты и причины действий, а также применение этих продуктов в различных искусствах» [12, с. 30]. В этом определении, сформулированном Бургаве в его книге «Элементы химии», в отличие от определения Лемери, заметно стремление автора изъять химию из-под опеки медицины, представить ее как самостоятельную науку [13, с. 244].
В XVIII в. в химии распространяется термин «физическая химия» — понятие, идентичное понятию «умозрительная, или теоретическая химия» [2, с. 41]. Термин «физико-химический» нстречается и ранее у немецкого алхимика Г. Купрата (см. [14, с* ^]), а также у Н. Лефевра и Р. Бойля, В 1701 г. Г. Горис цодразделял химию на четыре вида: физическую, медицинскую, пробирную и златоделательную [15, с. 18]. Под понятием «фи-
61
ГЙАВЛ ВТОРАЯ
ELEMENTA
ОУ А Е ANNIVERSARIO LABORE DOCUIT 1NPUBLICIS, PRIVATISQ.UE
S С Н О I. I S, HERMANNUS BOERHAAVE Т о м и $ Primus.
I СО N Т I NS Т HISTORIAN E T A RT I S T H F О R I A M.
cum tab ulis aeneis.
Cura Privifegio Pot. Regis Pol. & El. Sax.
L I Р S I А Е,
А р v л Casparvm Fritsch.
W ОСС XXXII
Титульный лист книги Г. Бургаве «Элементы химии» (1732)
знческая химия» в различных химических руководствах подразумевался не только теоретический аспект химии, но и практический, дающий материал для построения теорий. Физическую химию называли также «чистой» химией (Лефевр в XVII в. под чистой химией подразумевал философскую химию) в отличие от прикладной химии.
В 1729 г. Г. Тепхмейер, выделив несколько разделов химии: физическую, медицинскую металлургическую, философскую (алхимию), ремесленную и хозяйственную, подчеркнул отличие физической химии от философской, вложив другой смысл в понятие «философская химия». В своем определении физической химии он стремился отграничить какую-то часть химических знаний, необходимых для теоретической химии: «Физическая, или чисто умозри-химия — та, в которой естество-частью соотношения и
с. 35]).
тельная, вернее созерцательная испытатель изучает частью начала тел сочетания начал по степеням прочности, постоянства и летучести и таким образом строит себе теории» (цит. по [2, с. 32]).
Немецкий химик и металлург И. Генкель (1679—1744) под физической химией понимал «искусство основательно исследовать тела и разумно разлагать их вообще» (цит. по [2, с. 35]). Подобные формулировки понятия «физическая химия» встречаются в руководствах немецких химиков первой половины XVIII столетия —И. Картейзера и К. Неймана [2, с. 41].
В приведенных определениях физической химии XVIII в. заключена мысль о необходимости разграничения чистой химии, какой является физическая химия, от прикладной (имеется в виду химико-технологический аспект химии).
Определение химии, данное М. В. Ломоносовым * более широкие перспективы в развитии этой науки. «Химия
, открывало
* Михайло Васильевич Ломоносов родился в семье помора 8(19) ноября 1711 г. в деревне Мишанинской, расположенной у устья Северной Двины. Уже в юности у Ломоносова пробудился интерес к естественнонаучным
62
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
наука об изменениях, происходящих в смешанном теле, поскольку оно смешанное»,— писал он в работе «Элементы математической химии» [16, с. 67] и позднее в «Курсе истинной физической химии» (1752—1754). Русский ученый выдвинул перед химией задачу изучения состава, свойств, превращений веществ и сущности этих превращений.
М. В. Ломоносов четко разграничил цели теоретической и практической химии. «Практическая часть химии состоит в историческом познании изменений смешанного тела»,—писал он,— ...Теоретическая часть химии состоит в философском познании изменений смешанного тела...» [16, с. 68—69].
Называя химию наукой, в отличие от большинства химиков, считавших химию искусством, М. В. Ломоносов для обоснования своего положения предложил новую формулировку основной задачи химии — познание изменений, происходящих в смешанных телах (химических соединениях). Термин «познание изменений» подразумевает изучение не только химических явлений, но и химических процессов [17, с. 24].
знаниям. В конце 1730 г. Ломоносов поехал в Москву учиться. Здесь он поступил в Московскую славяно греко-латинскую академию, где изучал латынь, риторику и философию. В 1735 г. в числе 12 московских семинаристов Ломоносов был переведен в Петербург в Университет при Петербургской Академии наук. Здесь он пробыл 8 месяцев, показав «отменную склонность к экспериментальной физике, химии и минералогии». Осенью 1736 г. Ломоносов вместе с двумя товарищами (Виноградовым и Райзером) поехал в Германию для изучения горного дела. Первоначально Ломоносов слушал лекции по физике и химии в Марбургском университете, а затем изучал горное дело, металлургию, маркшейдерское дело во Фрейберге. Летом 1741 г. Ломоносов вернулся па родипу. 8 января 1742 г. Ломоносов был назначен адъюнктом, а в 1745 — профессором химии (академиком) Петербургской Академии наук.
В 1742 г. Ломоносов обратился в академическую канцелярию с предложением организовать химическую лабораторию. Это обращение было безрезультатным. Через год он написал вторично прошение, которое также не было удовлетворено «за неимением при Академии денег», как гласила резолюция канцелярии. Однако этот отказ не остановил Ломоносова. В начале 1745 г. он вновь обратился с прошением о постройке лаборатории. В 1748 г. химическая лаборатория была построена — это была первая в России научно-учебная химическая лаборатория. В ней Ломоносов проводил опыты по обжигу металлов, делал анализы минералов и читал лекции. Он стоял в центре научной деятельности Академии, ведя борьбу за честь и самостоятельное развитие отечественной науки. Научная деятельность Ломоносова была весьма разнообразной, но основное внимание было направлено на развитие химии и физики. В 1760 г. Ломоносов был избран членом Шведской Академии наук, в 1764 г.— почетным членом Болонской Академии. Умер М. В. Ломоносов в Петербурге 4 апреля 1765 г.
О жизни и деятельности М. В. Ломоносова см.: Меншуткин Б. Н. Жизнеописание М. В. Ломоносова. *3-е изд. М.; Л.: Изд-во АН СССР, 1947; Морозов А. М. В. Ломоносов. 2-е изд. Л.: Изд-во АН СССР, 1952; Рас-кин Н. М. Химическая лаборатория М. В. Ломоносова. М.; Л.: Изд-вр АН СССР, 1962.— Прим. отв. ред.
ГЛАВА ВТОРАЯ
В 1749 г. Ломоносов, отмечая, как важно взаимодействие физических и химических знаний для понимания «скрытой природы тел», утверждал, что «когда химические истины будут объединены более строгим методом и будет ясно, насколько одна истина может быть объяснена или выведена из другой, то химия сама по себе будет наукой» [16, с. 223].
С этих позиций Ломоносов подошел и к определению физической химии: «Физическая химия есть наука, объясняющая на основании положений и опытов физики то, что происходит в смешанных телах при химических операциях. Она может быть названа также химической философией, но в совершенно другом смысле, чем та мистическая философия, где не только скрыты объяснения, но и самые операции производятся тайным образом» [16, с. 483].
Сформулировав новые задачи химии, Ломоносов отверг положение о том, что химия — искусство разделения и соединения веществ [17, с. 26]. Но именно этого определения в сущности продолжали придерживаться химики и после Ломоносова, считая разложение и соединение веществ главной целью химии.
Последователи И. Ньютона пытались внести в определение химии дополнения, связанные с идеей притяжения между частицами тел. И. Лудольф в Германии (1752), определяя химию как искусство и науку, которая «учит искусным способом как разлагать природные тела, так и соединять, или производить то и другое одновременно, и именно так, чтобы сделать все понятным с помощью размышлений», отметил, что разложение и соединение одних тел происходят «посредством других тел, которые действуют при помощи свойственной им природной силы» [18, с. 1].
Развитие аналитических исследований нашло отражение в определении химии, предложенном французским ученым-флоги-стиком А. Боме в 1773 г.: «Химия — наука, основанная на эксперименте: она имеет объектом изучения анализ или разложение всех природных тел и соединение всех этих тел или их принципов, одних с другими для образования новых соединений». И далее: «Цель химии... распознать сущность и свойства природных тел» [3, с. 1—2].
Содержание понятия «химии» как самостоятельной науки утвердилось в конце XVIII в. после реформы в химии, проведенной А. Лавуазье.
УЧЕНИЕ О ФЛОГИСТОНЕ
Природа процессов горения, окисления и восстановления, с которыми химики постоянно имели дело в конце XVII —начале XVIII вв., особенно привлекала внимание исследователей [19, с. 134]. Одной из попыток объяснить эти процессы была флоги-
64
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
стопная теория, разработка которой принадлежит немецкому естествоиспытателю Г. Шталю *.
В основу теории флогистона было положено представление немецкого химика И. Бехера (1635—1682) о жирной земле terra pinguis, присутствие которой в телах наделяло их свойствами • горючести. Традиционное признание аристотелевских и парацель-совских «начал» не удовлетворяло Бехера. Он принял существование только двух элементов-качеств — земли и воды. Земля, по его мнению, состояла из трех начал: земли стекловидной (находящейся в песке, камнях, земле и кристаллах горных пород), олицетворяющей принцип сухости, твердости и прочности, затем земли воспламеняемой, заключающей принцип горючести, который Шталь позже назвал флогистоном, и земли ртутной, т. е. принципа летучести и одновременно тяжести (находится в ртути, металлах и других телах). Горючесть тел связана по только с наличием в них земли воспламеняемой, пли жпрпой, по и с присутствием серы и частиц соли. Соединение всех трех земель, по мнению Бехера, приводило к образованию металлов.
Учение Бехера о составных началах тел — землях — представляло, по существу, видоизменение представлений Аристотеля и Парацельса и трех алхимических начал — соли, серы и ртути. Увеличение в весе металлов при прокаливании на воздухе Бехер, как и его современники, считал результатом присоединения к ним огненной материи, не тождественной видимому огню, но аналогичной тому, что приписывалось алхимиками сере. Бехер применял термин «горючая» сера для обозначения начала горючести, придающего телам способность к горению.
Развив па базе представлений Бехера теорию горения, Шталь ввел понятие о флогистоне как некоем абстрактном принципе горючести, который становится «горючей субстанцией» только тогда, когда находится в сложном теле в сочетании с другими веществами. Флогистон (термин происходит от греческого слова qAoyicFTov — горючий), по мнению Шталя, находится в большей или меньшей степени во всех трех царствах природы — расти-
* Георг Эрнст Шталь родился 21 октября 1659 г. в Ансбахе. Уже в ранние годы проявился его интерес к химии и медицине. С 1679 по 1684 г. он изучал химию и медицину в Йенском университете. В 1715 г. был возведен в королевские лейб-медики и переехал в Берлин. Будучи придворным врачом прусского короля Фридриха Вильгельма I, Шталь широко содействовал развитию химии и медицины в Пруссии. По его инициативе в Берлине была основана Медико-хирургическая коллегия для подготовки военных врачей. Кафедрой химии руководил сам Шталь. Г. Шталь скончался в Берлине 4 мая 1734 г.
О жизни и научной деятельности Г. Шталя см.: Strube I. Die Phlo-gistonlehre Georg Ernst Stahles (1659—1734) in ihrer historischer Bedeu-tung.— Ztschr. Geschichte der Natur., Technik und Medizin, 1961, Jg. 1, Hf. 2. Список его многочисленных работ по химии, горному делу, металлургии п пробирному искусству приведен Дж. Партипгтоном. См.: Partington J. R, A History of Chemistry. Vol. 2. L., 1961, p. 659—662.— Прим. отв. ped.
3 Всеобщая история химии 65
ГЛАВА ВТОРАЯ
GEORG. ERNEST. STAHLII,
Confifiar. AuIic.&Archiatri Regii,
FUNDAMENTA
CHYMIAE
Dogmaticae& cxperimcntalis,
& quidcm turn communions phyficx mechanic# pharmaceutics ar medicx
turn (ubhrniorif ficdi&z hermetic* * atcfw Mymic*.
Olim
in privates Auditorum ufuspofita, jam vero
Indultu Autoris publics ]uci expofita, Anncxus eft ad Coronidis confirmationem Traflatus Ilaaci Holland! de Sahbus & Olets
Metallorum. ____
———N о К UIBERC
Sumptibus WOLFGANG I MAURITH endteri h/ered.
Typn JOHANNIS IRNEST1 ADEEBULNERI.
An. MDCCXXIH.
@rnfl6taW/
Mbntgl. <preuHdw uno
£cib «MEDICI,
CHYMIA
RATIONALE
E T
EXPERIMENT ALIS;
£Wcr:
(yrunbhdx z bee ur unb ^ernunfft (jcmdfje unb nut Expenmeritcn enviefene
Ginkituns jiicCh ymie;
Sjannnen (wupfficfjlirf, фк Mixtion bertr Sublunarifcben (ГбгреГ/ nebft bertn З^гкд'ипд unb Relation gogtn einontw unterfudjet z unb mit bitten E*ptnmentcrt gtjttgtt roirb.
5ltb(l tin» Sugebc
ЗДоп bcncttMercuriisMetalloruni,Mercurio animate, unbLap/dePhiioIbphorum*
Jnxytt 2tnfl<ige.
’йМфг ben bcrigenjbrud jfcepinngtrtinigct,, unb mit 3f<W £0Uan$$ Traflat
Q5on ben <2>n|f}tn unb Oeblen ber ^etallett t rrnebrtt rnctofti.
□ P J T ©7 “
Jaipur 17^
Титульный лист книги Г. Шталя «Основания догматической и экспериментальной химии» (1723)
Титульный лист книги Г. Шталя «Химия теоретическая и экспериментальная» (1729)
гелыюм, животном и минеральном — и присутствие его в телах обусловливает их запахи и цвета [20]. Автор учения о флогистоне предполагал, что при нагревании сложных тел флогистон улетучивается и, соединяясь с воздухом, воспламеняется. Невозможность уловить этот горючий принцип объяснялась его способностью растворяться в воздухе.
Впервые Шталь назвал флогистоном жирную землю Бехера в 1697 г. в работе «Основания зимотехнии, или общая теория брожения... вместе с новым опытом искусственного получения настоящей серы» 2 (в 1715 г. эта работа была переиздана в сборнике «Небольшие химико-физико-медицинские работы»). Представление о флогистоне и его роли при химических превращениях Шталь наиболее полно изложил в 1716 и 1731 гг.3
Большим числом опытов Шталь доказывал восстановительную способность флогистона, нагревая металлы до образования «известей» (окислов), а затем вновь получая металлы перемеши
См. Приложение III.
•3 См. Приложения IV п V.
66
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
ванием полученных «известей» с углем или с другими «жирными» веществами.
Исходя из существовавшего еще в древности представления, что процесс горения сопровождается разложением тел па более простые, химики-флогистики полагали, что металлы — вещества сложные, извести — простые. Им казалось справедливым положе- * ние об уменьшении веса при разложении, а поэтому металл, по их мнению, должен обладать большим весом, чем образовавшаяся при его сгорании земля.
Теория флогистона была первой, объясняющей с единой позиции различные химические процессы. Возможность просто объяснять эти процессы, по словам Э. Мейера, «ослепляла как самого Шталя, так и последовавшее за ним поколение химиков, так что никто из них не замечал вопиющих противоречий между флогистической теорией и фактами» [21, с. 93].
Среди химиков XVIII в. были и такие, которые сомневались в правильности теории флогистона. В 1728 г. Г. Штабель в курсе «Догматико-экспериментальная химия» отмечал, что объяснения увеличения веса окалин при их разложении присоединением флогистона неверны, поскольку прибавление материи должно увеличивать вес, а уменьшение ее количества — уменьшать его» (цит. по [22, с. 36-37]).
Г. Бургаве в своем учебнике «Элементы химии» (1732) не упоминает слова «флогистон». Но Бургаве использует термин «пища огня» (pabulum ignis), также признавая существование некоего другого горючего принципа, входящего в состав серы.
«Современники, упоминая об анализе и составе металлов, говорят только о флогистоне и стеклообразной земле, которые, согласно их взглядам, участвуют в образовании металлов. Но... Бургаве, который много работает с этими веществами, не придерживается такого мнения»,— свидетельствовал учившийся у Бургаве Ж. де ля Метри (1709-1751) [23, с. 59].
Проводимые Бургаве наблюдения горения в закрытом пространстве, за поведением птицы под стеклянным колпаком приводили его к мысли о скрытой силе, находящейся в воздухе и играющей роль в процессах горения и дыхания. «Счастлив тот человек, кто откроет это»,—писал Бургаве [12, с. 500].
Г. Бургаве обладал исключительным умением систематизировать факты и знания, и изданный им учебник «Элементы химии» долгое время служил одним из лучших руководств в преподавании химии. Как химик-практик Бургаве известен меньше, однако его практические работы представляли определенный интерес Для своего времени. Он выступал против утверждения алхимиков, что ртуть можно без добавления каких-либо веществ превращать в твердый металл. В течение 15 лет Бургаве нагревал ртуть в закрытом сосуде и доказал, что она не изменилась. Он 500 раз перегонял ртуть и, вопреки утверждениям алхимиков, нашел,
67
3*
ГЛАВА ВТОРАЯ
что она не превращается в летучее вещество с особыми свойствами. Наконец, Бургаве доказал, что растворением азотнокислого свинца в воде нельзя получить ртуть, как думали алхимики.
Современником Шталя и Бургаве был немецкий врач Ф. Гофман (1660—1742). Его сближали с Бургаве взгляды на химию как самостоятельную науку. Как и Бургаве, Гофман не принадлежал к сторонникам теории флогистона [24].
В Германии почти все химики были приверженцами учения о флогистоне. Так, И. Юнкер (1683—1759) в двухтомной работе «Конспект теоретической и практической химии, представленный в форме таблиц» (1730) впервые высказал положение об «абсолютной легкости» флогистона, из которого следовало, что «горючее начало» имеет отрицательный вес [И]. Используя это понятие, он объяснял увеличение веса металла при кальцинации, когда из металла удаляется флогистон, а остаток обжигаемого металла становится более тяжелым.
Ученик Шталя К. Нейман (1683—1737) исследовал ряд природных продуктов и написал несколько трудов по различным проблемам прикладной химии [25]. Его преемник по кафедре в Медико-хирургической коллегии в Берлине И. Потт (1692— 1777) известен усовершенствованиями в области фарфорового производства. Будучи сторонником учения Шталя, он трактовал флогистон как разновидность серы [26].
И. Эллеру (1689—1760) одному из первых принадлежат исследования в области растворимости солей в воде в зависимости от температуры [27]. В сочинении «О происхождении руд и металлов» (1753) Эллер указывал, что в металлургической практике есть факты, которые противоречат теории флогистона [28].
Одним из выдающихся представителей периода флогистонной химии в Германии был А. Маргграф (1709—1782), основавший в 1754 г. химическую лабораторию в Берлинской Академии наук. Будучи прекрасным экспериментатором, Маргграф оставил большой след в развитии аналитической химии *, он широко применял количественные исследования [29, с. 103—104] и сделал важные открытия, сыгравшие большую роль в развитии сахарной промышленности. Примечательно, что кристаллы сахара в корнеплодах свеклы им были обнаружены с помощью микроскопа |3(), с. 70-86].
Сторонниками теории флогистона были также немецкие ученые, известные более своей литературной деятельностью в области химии: Л. фон Крелль (1744—1816), издававший ряд химических журналов, в частности «Химическую летопись для друзей естествознания, фармакологии, домоводства и мануфактуры» («Chemische Annalen fur die Freunde der Naturlehre, Arzneyge-lehrtheit, Haushaltungskunst und Manufacturen») [31], Ф. А. Грен
* Более подробно см. главу четвертую.— Прим. отв. ред.
68
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
L —— ——
(1760—1798), опубликовавший «Систематическое руководство по общей химии» [32], И. Виглеб (1732—1800),^известный исследованиями по минералогической химии и работами в области истории химии (см. [33, с. 111]).
Во Франции к числу видных сторонников учения о флогистоне • принадлежали Э. Жоффруа-старший, известный исследованиями в области химического сродства, А. Дюамель де Монсо, Г. Руэль и П. Макер.
Г. Руэль развил интересные представления о солях как соединениях кислот с различными основаниями, подразделил соли на нейтральные, кислые и щелочные [34]. Прекрасный экспериментатор, Руэль был одновременно незаурядным педагогом, учениками его были А. Лавуазье, Ж. Пруст, Н. Леблан.
Г. Руэль рассматривал флогистон не столько как элемент, сколько как «инструмент» огня, игравший роль в составлении материи. Он называл «флогистоном» вещественные частицы огня, осуществлявшие соединение частиц тел одних в другие [35, с. 73-77].
Одним из самых ревностных сторонников теории флогистона являлся П. Макер. «Из всех теорий,— писал он,— она наиболее ясна и наиболее согласна с химическими явлениями. Отличаясь от систем, порожденных воображением без согласования с природой и разрушаемых опытом, теория Шталя — надежнейший путеводитель в химических исследованиях; многочисленные опыты, проводимые ежедневно, не только далеки от того, чтобы ее опровергнуть, ио, наоборот, становятся доказательствами в ее пользу» [36, с. XXXIII—XXXIV]. Макер много сделал для распространения химических знаний. Среди его значительных экспериментальных работ следует отметить и те исследования, которые относятся к технологии крашения [37].
Среди английских ученых приверженцами флогистона были Дж. Блэк, Г. Кавендиш, Дж. Пристли, Р. Кирван и др. Почти все они занимались пневматической химией.
Т. Бергман (1735—1784) в Швеции прославился работами о химическом сродстве и внес большой вклад в развитие аналитической химии [38]. Его ученик К. Шееле (1742—1786) обогатил химию открытием большого числа новых веществ, ввел новые экспериментальные методы их исследования [39, с. 205—234]. И Бергман, и Шееле были сторонниками учения о флогистоне.
М. В. Ломоносов рассматривал флогистон как материальное вещество, состоящее из корпускул: «Если корпускулы флогистона Движутся одни, то дают безвредный огонь, что доказывает зажженный винный спирт и самые чистые эфирные масла» [40, с- И5]. В диссертации «О металлическом блеске», опубликованной в 1751 г., Ломоносов писал, что при растворении неблагородных металлов в кислотах (кислотных спиртах у Ломоносова) ньхделяется «...горючий пар, который представляет собой не что
69
ГЛАВА ВТОРАЯ
иное, как флогистон...» [40, с. 399], отождествляя таким образом флогистон с «горючим воздухом» (т. е. водородом).
Сторонники флогистонной теории искали причину увеличения веса металлов при прокаливании, стараясь познать сущность самого флогистона, и поэтому вопрос о природе этого горючего начала так или иначе занимал химиков в течение длительного периода. Попытки выделить флогистон оказывались тщетными, поэтому мнения химиков о сущности флогистона были разноречивыми и нередко отличались от мнения Шталя. Так, в XVIII в. флогистон отождествлялся со «световой материей» (П. Макер, А. Рюдигер), с «теплотворным веществом» (К. Шееле), «угольной кислотой, соединенной с теплотворным веществом» (А. Вольта), «соединением теплотворного вещества со световым» (Ф. Грен), с «огненной материей, соединенной со стихийной землей» (А. Боме). М. В. Ломоносов, ранее наблюдая в опытах выделение «горючего газа», полагал, что это флогистон. Г. Кавендиш и И. Ф. Веструмб (1751—1819) считали, что флогистон — это водород, выделенный Кавендишем в 1766 г. и названный им «горючим воздухом» [41]. Однако Кавендиш вынужден был признать, что его «горючий воздух» — не чистый флогистон, а соединенный с парами воды, так как обнаружилось, что этот воздух имеет вес.
Накопление фактов об увеличении веса металлов при прокаливании требовало объяснения, и сторонники теории флогистона прибегали к различного рода гипотезам, не согласующимся с фактами.
С развитием количественных исследований флогистон наделялся вещественными качествами, однако рассматривался невесомым или имеющим отрицательный вес. Такой точки зрения придерживалось большинство химиков середины XVIII в.—С. Ринман, Г. Шеффер, Л. Гитон де Морво, X. Даниэль, Ж. Шарденон, Ф. Грен и др.
Теория флогистона просуществовала сто лет, несмотря на то, что уже в XVIII в. были известны факты увеличения веса металлов при прокаливании, установленные Г. Поппием, Н. Лефевром, Ж. Реем, Р. Гуком, Дж. Мейоу. Однако подобные факты также в основном трактовались в пользу теории флогистона.
Оценивая период флогистона, А. Лавуазье отмечал, что теория Шталя получила столь широкое распространение потому, что на ее основе были сделаны два важных открытия. Одно из них состояло в установлении факта, что металлы — тела горючие и превращение их в окалины представляет явление горения, второе — в том, что свойство гореть, или быть воспламеняемым, может передаваться от одного тела к другому. При накаливании металлы теряют способность горючести, но, соприкоснувшись с углем или другими веществами, способными гореть, металлы возрождаются, воспринимая от горючего вещества свойство горючести [42, с. 624-625].
70
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
В эпоху господства теории флогистона вырисовываются три основных этапа, отражающих характер развития химии того периода.
Для первого этапа (конец XVII в.—30-е годы XVIII в.) характерно победное шествие теории флогистона, первой химиче-* ской теории, объяснившей с единой точки зрения многие химические явления.
На втором этапе (1730—1770) ученые занимались усиленными поисками флогистона, выявлением фактов, не согласующихся с теорией, в частности фактов увеличения веса металлов при горении. В этот период получили развитие пневматическая химия, количественный анализ и другие важные направления. Накопившиеся факты углубляли разрыв между экспериментальными данными и их теоретическими объяснениями.
Третий этап (1770—1790-е годы) характерен острой борьбой сторонников учения о флогистоне с новой кислородной теорией, созданной Л. Лавуазье, и крушением флогистонной теории.
Флогистонная теория рассматривается в истории химии как «переходная эпоха» [13, с. 249]. Она, по существу, возникла из старых представлений, унаследованных от Аристотеля, алхимии и иатрохимии. В основу этой теории были положены взгляды Аристотеля на горение как процесс распада тел, а не соединения. Большинство химиков-флогистиков, как и основатель этой теории Г. Шталь, были врачами и видели в химии науку, подчиненную медицине.
Однако период теории флогистона заметно отличается от предшествующих периодов.
«Химия только что освободилась от алхимии посредством флогистонной теории»,—писал Ф. Энгельс4. Действительно, многие химики периода господства учения о флогистоне уже порвали связь с алхимическими традициями. Теория флогистона, несмотря на ошибочные положения, была первой химической теорией, которая приводила в определенную систему известные в то время многообразные, но разрозненные факты. Она дала толчок многим новым исследованиям. В этом проявилась положительная роль теории флогистона, но в определенный период она стала тормозом в развитии химии, поскольку многие химики того времени не могли с позиции представлений о флогистоне правильно понимать и объяснять новые открытия.
Маркс К.} Энгельс ф, Cqh. Изд. 2-е, т. 20, с. 348,
71
ГЛАВА ВТОРАЯ
РАЗВИТИЕ ПРЕДСТАВЛЕНИЙ О ХИМИЧЕСКОМ СОЕДИНЕНИИ И АТОМНО-КОРПУСКУЛЯРНЫЕ ТЕОРИИ В КОПЦЕ XVII-XVIII ВВ.
Большое влияние на развитие химии XVIII в. оказала динамическая атомистика И. Ньютона (1643—1727). Согласно Ньютону, «первичные твердые корпускулы неизменяемы и неделимы, подобно атомам древних атомистов»... «Мельчайшие частицы материи могут сцепляться посредством сильнейших притяжений, составляя большие частицы, но более слабые; многие из них могут также сцепляться и составлять еще большие частицы с еще более слабой силой — и так в ряде последовательностей, пока прогрессия не закончится самыми большими частицами, от которых зависят химические действия и цвета природных тел; при сцеплении таких частиц составляются тела заметной величины» [43, с. 299].
И. Ньютон, так же как и многие его предшественники — атомисты, рассматривал атомы всех тел физически идентичными состоявшими из некоей единой первоматерии, всякие же качественные изменения, ведущие к возникновению химически индивидуальных веществ, по мнению Ньютона, зависимы от форм, твердости и прозрачности атомов. Главная роль в возникновении новых индивидуальных качеств отводилась сочетанию этих атомов посредством особых сил притяжения. В эпоху теории флогистона учение Ньютона стало основополагающим в формировании представлений о химическом соединении и связанного с ними учения о химическом сродстве.
Идеи Ньютона получили развитие в двухтомном издании Г. Бургаве «Элементы химии». Согласно взглядам Бургаве, «все изменения, которым тела подвергаются в химических процессах и операциях, сводятся... к движению мельчайших частичек, из которых состоят тела. Когда два тела соприкасаются, маленькие частички приводятся в активное движение, при этом соприкосновении появляются новые качества, которыми до этого момента тела не обладали» (цит. по [44, с. 329]).
Химики эпохи флогистона, пытаясь объяснить химические явления с позиции корпускулярных идей Ньютона, в то же время опирались на схоластические представления об элементах-качествах. Так же как ученым XVII в., им не удавалось конкретно связать эти идеи с химическими превращениями.
Г. Шталь был далек от понимания «начал», входящих, по его мнению, в смешанные тела, как элементов. Он рассматривал металлы как сложные тела, состоящие из окалины и флогистона, принимая его за «начало», содержащееся во всех горючих телах.
Исследования воздуха в первой половине XVIII столетия, позволившие выделить «воздухообразные» вещества нз многих
72
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIH ВВ.
*
химических соединений, казалось, подтверждали идеи Аристотеля. Один из видных химиков XVIII в. П. Макер в 1749 г. писал в своем «Курсе теоретической химии»: «...Раздробление и разделение тел не беспредельно... Как бы мы за пего ни принимались, однако... неразделимые существа всегда будут нам препятство-. вать... Сии существа должны мы назвать началами или. стихиями... таковые... земля, вода, воздух и огонь». [45, с. 2]. И хотя Макер отмечает, что не все химики убеждены в этом, но поскольку опытом нельзя установить, «какие суть первые начала, из коих они (тела.—3. III.) сами составлены», то думает, что «гораздо разумнее на них утвердиться и рассуждать о них как о простых и единосложных началах других тел, нежели утруждать себя угадыванием, из каких они частей или стихий составлены, не имея притом ни единого средства к уверению себя, справедливо ли мы узнали или не мечтания ли суть наши мнения» [45, с. 2—3]. Однако далее Макер опровергает свои утверждения, отмечая существование экспериментального доказательства об отсутствии в металлах воздуха и воды.
Представители эпохи флогистона, следуя за Р. Бойлем, ставили перед химией задачу, которая, согласно формулировке Г. Шталя, состояла, как и у Р. Бойля, в том, чтобы «разделять тела, как смешанные, так и составные, и агрегированные на их начала (принципы), а также таковые составлять из начал» [9, с. 1]. Для экспериментальной химии такая постановка задачи была плодотворной.
Сторонники учения о флогистоне трактовали образование смесей и химических соединений в самом широком смысле, опираясь на натурфилософские представления о том, что в мире существует только сложное и составное. Предполагаемые простые тела образуют смеси (mixtum), сочетание которых дает «составные тела» (corpus compositum). Шталь вводит также понятие скопления (agregatio), образуемого сочетанием однородных смесей» [9, с. 65].
Несмотря на ошибочную систематизацию веществ, при которой окислы металлов считались веществами более простыми, чем металлы, учение Шталя оказалось новым в трактовке представлений о химическом соединении по сравнению с предшествующим периодом.
В теоретической химии первой половины XVIII в. особое место занимает научное творчество М. В. Ломоносова, развившего корпускулярные и атомистические воззрения Р. Бойля. Ломоносов поднял корпускулярные представления до научного уровня, Достигнутого химиками только в XIX в. Частички, обладающие фигурой, массой, имеющие конечное протяжение, согласно Ломоносову, подчиняются принципу действия и противодействия. От природы этих частичек, обладающих движением, зависят качества тел, а также физические явления — тепло и холод, элек-
73
ГЛАВА ВТОРАЯ
а »»> ». * । -,-- , '- - - >-> - - - ---
тричество и т. п. «Цементом, превращавшим атомистический хаос в нечто стройное, является у него, как у новейших физиков, представление об энергии»,— подчеркивал Б. Н. Меншуткин близость механистических теорий Ломоносова к современным представлениям [46, с. 151].
Атомно-корпускулярное учение Ломоносова основывалось на его материалистическом мировоззрении: «Материя есть то, из чего состоит тело и от чего зависит его сущность» [40, с. 173]. Он определенно высказывался о конечной делимости материи и пришел к теоретическим выводам, которые впоследствии легли в основу развития физики и химии.
По М. В. Ломоносову, «элемент есть часть тела, не состоящая из каких-либо других меньших и отличающихся от него тел» [40, с. 79]. И далее: «Начало есть тело, состоящее из однородных корпускул» [40, с. 81].
Атомно-корпускулярное учение давало ключ к решению проблемы химического соединения. Ломоносов впервые разграничил понятия элемент, простое и сложное вещество. Основную задачу химии он видел в том, чтобы исследовать как состав «доступных чувствам» тел, так и «начала» тел, из которых они состоят. Ломоносов определил химическое соединение (он называл его смешанным телом) как «симилярное тело, состоящее из других си-милярных тел, разнородных в отношении частных качеств, так что в любой нечувствительной частице составляющие части находятся в том же количественном отношении друг к другу, в каком они находятся во всем смешанном теле» [40, с. 301].
В этом определении понятия химического соединения (наименьшая «нечувствительная» частица является носителем свойств вещества) Ломоносов указывает на индивидуальность каждого из химических соединений, определяемую «частными качествами». В 1745 г. Ломоносов высказал понятие о химически индивидуальном веществе как химически чистом: «Нужные и в химических трудах употребительные натуральные материи сперьва со всяким старанием вычистить, чтобы в них никакого постороннего примесу не было, от которого в других действиях обман быть может» (цит. по [47, с. 330]).
М. В. Ломоносов утверждал, что для познания того, что химик доказывает, нужно прежде всего приобрести знания об изменениях смешанного тела. Мысли Ломоносова о необходимости теоретического объяснения химических явлений, которое невозможно, по его мнению, без знания физики, тесно связаны с его представлением о физической химии как теоретической науке, объяснявшей посредством «положений и опытов физики» изменения, происходившие в смешанных телах при химических опытах.
Химики первой половины XVIII столетия, несмотря на определенные трудности в разложении веществ, при изучении реак
74
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
ций наблюдали выделение неизвестных веществ, к примеру газообразных, и пытались изучать их свойства. Более того, химикоаналитические исследования приобретали все более широкий размах в связи с поисками новых ресурсов и видов природного сырья, необходимого для быстро развивавшегося мануфактурного и других видов производства.
Развитие практической химии, других наук, в частности минералогии, кристаллографии, позволило ученым уже в середине XVIII в. сформулировать ряд теоретических основополагающих положений в науке.
П. Макер, описывая явления кристаллизации (1766), широко пользуется атомной (корпускулярной) теорией, хотя и не употребляет слова «атом». «Чтобы понять,—писал он,—механизм кристаллизации, мы должны заметить: 1. Что составные части всех тел имеют стремление друг к другу, при котором они приближаются, соединяются и слипаются вместе... 2. Что в простых телах это стремление составных частей является более очевидным, чем в более сложных. 3. Что, хотя не знаем образ (figure) первичных составных молекул какого-либо тела, мы не можем сомневаться, что молекулы каждого отличного тела имеют постоянную однородность и особую форму. 4. Что эти составные части не могут иметь равного стремления к соединению любой из их сторон, при этом условии такое стремление более обширно и непосредственно у тех сторон, которыми эти молекулы касаются» [48, с. 488]. У Макера имеются мысли о геометрических формах молекул, об упаковке молекул в кристаллах и постоянстве состава химических веществ. Но следует отметить, что Макер и его современники середины XVIII в. считали, что молекулы как составные части, полученные разложением веществ, состоят из различных смесей воздуха, воды, огня и земли.
А. Фуркруа, современник А. Лавуазье, указывал, что еще до эпохи химической революции некоторые химики, принимая сущности Аристотеля, отличали принципы, более простые, чем элементы Аристотеля. «Они их назвали,—писал Фуркруа,—частицами составляющими (parties constituantes). Это мнение приближалось к мнению древних философов о монадах и атомах...» [49, с. 51].
С эклектическими представлениями связана и своеобразная номенклатура, применявшаяся при описании химических явлений, которая, по словам Фуркруа, отражала такое положение в химии, когда «идеи были перепутаны и перевернуты вверх дном... всякого рода различиями» [49, с. 52].
Химики XVIII в. принимали существование ряда принципов: первичных принципов, имея в виду элементы Аристотеля, вторичных, образованных соединением первичных, принципов ближайших и отдаленных, когда речь шла о растительных сложных веществах. Такое разнообразие принципов связано, конечно,
75
ГЛАВА ВТОРАЯ
D Е
CH YMIЕ THEORIQJJE;
Рar М. ЛA(Q UЕR,de ГAeadeniie Royaledes Sciences, Cenfeur Royal, Docleur- Regent de la Faculte' de Medecine en PUniverJite de Paris, У ancien Profeffeur de Pharmacie,
NOUVELLE EDITION.
A PARIS,
Chez Jean-Thomas Hebtssant, rucS. Jacoues, aS. Paul & aS. Hilaire.
M. DCC. LVI.
Avec Approbation & Privilege du Rot»
Титульный лист книги П. Макера «Основания теоретической химии» (1756)
с невозможностью применения элементов Аристотеля в эксперименте.
П. Макер, стремившийся яснее разграничить понятия, выделял принципы первого, второго, третьего порядка. По мнению Фуркруа, эти названия должны были бы обозначать не принципы, а реальные вещества.
В общем хотя названия веществ основывались на процессах соединения и разложения веществ, однако отражали ошибочные идеи об элементах, а также смешение понятий о простых и сложных телах.
Развитие аналитических исследований во второй половине XVIII в. повлияло на представления об элементе. В трудах приверженцев учения о флогистоне Т. Бергмана и К. Шееле отразилась «аналитическая концепция элемента» [50]. К первичным простым телам они относили не только «принци-
пы» — флогистон, воду и «соляной принцип», которые они принимали вместо аристотелевских,— но и не разложенные тогда земли (т. е. соли) — баритовую, магнезиальную, алюминиевую и др. В некоторых пунктах между взглядами Шееле и Бергмана были расхождения. Взгляды Шееле были ближе к традиционным представлениям Г. Шталя, но, если Шталь рассматривал газы, земли и кислоты, подобно металлам, как более или менее прочные смешанные тела, Шееле защищал их индивидуальность. Бергман жп уже в 1774—1775 гг. проявлял более скептическое отношение к первичным принципам вообще, являясь предшественником «аналитической концепции, предложенной Лавуазье в 1787— 1789 гг.» [50, с. 447]. Таким образом, концепция элемента видоизменялась на основе представлений о составе веществ.
Последователи Шталя, в первую очередь во Франции, много сделали для систематизации химических знаний и определения основных понятий. Используя терминологию Шталя, Макер предпочел вместо «смешанного тела» употреблять термин «соединение» (combination) или «состав» (composition), когда речь шла, по его мнению, о химическом соединении.
76
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
А. Боме, в течение 25 лет работавший «демонстратором» опытов в Парижском Ботаническом саду, в изданном им в 1766 г. «Учебнике химии» изложил представления о соединении и составе тел: «Когда соединяются несколько незначительных по своей массе молекул, они образуют более крупную массу; когда * это соединение образовано частицами одного и того же рода, масса становится особенно чувствительной. Это — форма, которую Бехер и Шталь назвали агрегатом (скоплением). Но когда соединяются два тела разной природы, тогда это соединение дает новое смешанное тело (corps mixte), в свойствах которого участвуют свойства обоих тел, послуживших для его образования; это — то, что Бехер и Шталь называют смешением (mixtion) и что мы будем называть соединением (combinaison) или химическим составом (composition chimique) тел. Мы назовем сложными телами (corps composes) те, которые образуются соединением частиц разного рода. Эти неодинакового рода частицы называются началами (principies), частицами составляющими (parties consti-tuantes), которые следует отличать от частиц составных (parties integrantes)». Боме обратил внимание на то, что «физики, напротив, называют parties integrantes более малые молекулы, которые нельзя далее разделить без разложения» [51, с. 3].
А. Боме, используя терминологию Макера, сложные тела называл «химическими соединениями». Это название отражало не только состояние тела до превращения, но и участие его в процессе химического взаимодействия.
Проблема химического соединения неразрывно связана с вопросами индивидуальности вещества, которая определяет качество, присущее данному веществу. Расширение объектов химического исследования в XVIII в., вызванное развитием качественного и количественного анализа, пневматической химии, выдвигало проблему индивидуальности как одну из основных. Решению этой проблемы способствовало внедрение самых первых простейших физико-химических методов исследования, раскрывающих новые критерии химической индивидуальности.
В XVIII в. основной характеристикой, отличавшей истинное химическое соединение от смеси, была однородность и невозможность разделения его на неоднородные фазы. Уже Г. Бур-гаве в 1732 г. отчетливо указал на признаки, отличавшие химические соединения от смесей: 1) химическое соединение однородно во всех своих частях; 2) составные части химического соединения в состоянии покоя не разделяются; 3) при образовании химического соединения выделяется тепло и исчезают свойства тел, образующих соединения [52, с. 71]. В то же время все химические явления трактовались преимущественно как явления растворения, которые рассматривались тем же Бургаве как Результат образования бесконечного множества «новых видов Материи»,
77
ГЛАВА ВТОРАЯ
В дальнейшем М. В. Ломоносов, затем сторонники теории флогистона отличали два вида растворения: растворение металла в кислоте и разведение соли в воде.
В XVIII в. становится общепризнанным постоянство физических свойств химически индивидуальных веществ, причем постоянство не только удельных весов, установленное много раньше, но и температур кипения, замерзания, а также теплот растворения и смешения [15, с. 18]. Кроме этих, существовали другие критерии, определявшие химический индивид, а именно сокращение объема при реакции (для газообразных веществ), изменение температуры во время реакции, прозрачность и т. п.
Большую роль в развитии представлений о химическом индивиде и формировании этого понятия во второй половине XVIII в. играли другие науки, прежде всего минералогия и кристаллография, положившие в основу изучения минералов и других веществ простейшие физико-химические методы исследования [53].
Однако сторонники теории флогистона, следуя традиционным представлениям, предполагали, что свойства веществ, даже их физические свойства (вес, твердость, ковкость, прозрачность), являются промежуточными между свойствами составляющих компонентов [54, с. 64]. Против этой точки зрения в 1782 г. выступил А. Фуркруа, который, критикуя концепцию химических «начал» приверженцев флогистона, предполагал, что два или несколько веществ, соединенных посредством «сродства составления», образуют химические вещества, свойства которых совершенно отличны от свойств каждого из составляющих их веществ. Он перечислял свойства, определяющие новые качества химических веществ: вкус, цвет, запах, кристаллическая форма, ковкость. Окончательно решить проблему истинности химического соединения помогает одно из его свойств — однородность, однако растворы солей в воде представлялись Фуркруа как менее истинные соединения, поскольку свойства их изменяются менее резко [54, с. 64—65].
Вопросы, связанные с природой химического соединения, в XVIII столетии решались на макроуровне. Химическое соединение обычно рассматривалось как взаимное насыщение составных частей, и на этом представлении основывалось изучение его состава и свойств.
Вопрос о конечных продуктах химического анализа на протяжении почти всего XVIII в. оставался открытым. И все же некоторые сторонники корпускулярной теории пытались пайти ответ на этот вопрос на микроуровне, т. е. на основе представлений о мельчайших частицах.
В то же время, когда основной задачей химического анализа рассматривалось определение состава и свойств веществ, наблюдения их превращений при химических реакциях наводили на мысль о зависимости этих превращений от структуры тел.
78
\ РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
Во второй половине XVIII в. Б. ле Саж во Франции, Б. и В. Хиггинсы в Англии пытались связать атомистику с химическими явлениями.
В. Хиггинс (1763—1825), исходя из весовых соотношений составных частей соединения, предполагал? что мельчайшие предельные частички соединяются в аналогичных цифровых пропор-* циях. Если анализы показывают, что двуокись азота '(nitrous air) образуется из двух частей дефлогистированного воздуха (кислорода) и одной части флогистированного воздуха (азота), следовательно, по мнению Хиггинса, «первичная частичка флогистированного воздуха соединилась с двумя дефлогистированного воздуха, и... эти молекулы (двуокиси азота.— 3. III.) окружены общей атмосферой огня» [55, с. 132].
Различие в соотношениях азота и кислорода зависело от различной величины сродства, действующего между ними [56, с. 133-134].
Атомистические представления В. Хиггинса, однако, не оказали заметного влияния на развитие атомизма в химии, поскольку его атомы были лишены химической индивидуальности и прежде всего весовой характеристики.
Изучая состав и свойства веществ, химики неизменно обращались к важнейшим вопросам химии — химической индивидуальности и химическому взаимодействию. Эти вопросы составляли существо основной проблемы — проблемы химического соединения в ее двух аспектах, один из которых — определение природы химического соединения как индивидуального вещества, другой — сущность процесса соединения химических индивидов. Изучение этих двух аспектов привело, как мы увидим далее, к открытию на рубеже XVIII—XIX вв. двух типов химических соединений — определенных (постоянного состава) и неопределенных (переменного состава).
ПЕРВЫЕ КЛАССИФИКАЦИИ ХИМИЧЕСКИХ ВЕЩЕСТВ
Из словаря, изданного П. Макером (1766), можно составить общее представление о классификациях веществ, предложенных к середине XVIII в. Кислоты, например, были классифицированы как минеральные, растительные и животные. Растительные кислоты, получаемые из фруктовых соков, считались по свойствам отличными от минеральных кислот — серной, азотной, соляной, получаемых из минеральных соединений — квасцов, купоросов, «азотной земли» (нитратов), морской соли (NaCl) и др.,—и прежде всего по силе и резкости свойств.
Менее выраженные кислотные свойства растительных кислот объяснялись присутствиелМ в них «некоторого количества масла» [48, с. 489]. В то время предполагалось, что «животные» кисло
79
ГЛАВА ВТОРАЯ
ты, полученные из животных тканей, содержат «масляные частицы», однородные с теми, которые содержались в растительных кислотах. Макер, например, по этому поводу писал, что «...эти кислоты слишком мало исследованы, чтобы мы имели право судить, существенно ли они отличны от тех, которые получены из растений» [там же].
Четыре вида веществ составляли в середине XVIII в. класс щелочей: минеральные, растительные, ископаемые и летучие. За исключением летучей щелочи (имеется в виду аммиак), все другие могли быть получены в твердом состоянии.
В 60-е годы XVIII в. значительное число веществ было отнесено к классу солей: сульфаты, нитраты, морские соли (хлориды), тартраты, ацетаты, ортофосфаты, борат аммония, арсенат калия и другие мышьяковые соли; известные к этому времени соли, содержащие воду, например стальная соль (FeSO4-7H2O), свинцовый сахар (гидрат ацетата свинца). Некоторые вещества, включенные в этот класс, не обладали характерными для солей свойствами. Они лишь хорошо растворялись в воде. Так, к солям причислялись многие окислы (извести металлов, окислы свинца РЬО2 и РЬ3О4, основные соли, в частности порошок альгарота (SbOCl), и многие другие.
Таким образом, классификация указанных выше химических соединений не была строгой. Она проводилась на основе определения некоторых физических свойств, наблюдений над реакциями, ио аналитическая химия не была еще в состоянии точно характеризовать индивидуальные свойства тех или иных веществ.
В середине XVIII в. были известны и некоторые органические соединения, как, например, отдельные спирты, эфиры, жиры и ряд кислот. Химики отмечали, что винный спирт соединяется со всеми кислотами, при этом кислотность последних уменьшается. Они осуществляли не только реакции спиртов с неорганическими кислотами, но и с органическими, получая эфиры [48, с. 490].
Тринадцать металлов, известных в то время, подразделялись па три категории: 1. Совершенные металлы — золото, серебро, платина — имеют свои специфические свойства, а именно не подвержены действию огня, протягиваются в проволоку и куются в листы. 2. Несовершенные металлы — железо, медь, свинец и олово — менее ковки и тягучи и «уничтожаются огнем» (т. е. окисляются). 3. Полуметаллы — цинк, кобальт, сурьма, висмут п мышьяк — легко превращаются в «извести» (т. е. окислы) под действием огня и обладают слабой ковкостью и тягучестью.
Ртуть находилась вне категории, поскольку имела все общие свойства металла, по чистоте и тяжести была сходна с совершенными металлами, а по летучести принадлежала к полуметаллам [48, с. 489].
80
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
ПРЕДСТАВЛЕНИЯ О ХИМИЧЕСКОМ СРОДСТВЕ В КОНЦЕ XVII-XVIII ВВ.
По мнению известного историка химии Г. Коппа, «почти вся химия может рассматриваться как цепь применений и примеров учения о сродстве» [57, с. 285]. Первые представления о сродстве появились еще в V в. до н. э., когда Гиппократ утверждал, что соединение двух тел происходит только при наличии в них общего принципа. А. Магнус, заметив, что сера соединяется со всеми расплавленными металлами, кроме золота, употребил выражение affinita в книге «De rebus metallicis» («О загадке металлов»), а позже оно встречается у Р. Глаубера (1648) и Сильвия де ле Бое (1659). Р. Бойль, как писал Копп, употреблял слово affinia (1661), обозначающее «сродство», Р. Гук —выражение affinitas chemica — «химическое сродство». И. Бехер провозгласил мнение Гиппократа важнейшим законом природы.
В XVI в. возникли первые идеи связать химическое сродство с корпускулярными представлениями. Mixtio,— писал Ю. Скали-гер (1484—1558),—это «движение мельчайших тел к взаимному контакту, чтобы получилось объединение (unio) [58, с. 696].
В XVII в., когда многие ученые уже отвергали учение Аристотеля и утверждали корпускулярные воззрения, соединения материальных начал при образовании сложных веществ, т. е. проблему химического сродства, объяснялись не с помощью уже отживших оккультных представлений, а с позиций нового, механистического, способа мышления. Наиболее видный представитель механистического направления в химии И. Юнгиус трактовал химические процессы как сочетание и разъединение атомов. Он впервые предположил, что в химическом соединении должна существовать связь, и называл это понятие то сцеплением (cohae-sio), то сродством (affinitas) [59, с. 186].
Объяснение Р. Бойлем химического сродства основывалось также на механистическом представлении о совпадении форм частиц, подобно ключу к замку. Как уже отмечалось в первой главе, Бойль применил корпускулярную философию для объяснения образования сложных тел.
Наиболее ярким выразителем картезианских представлений был Н. Лемери. Так же как Р. Декарт, он признавал у частиц многообразие форм, определяющих свойства вещества. В своем «Курсе химии» (1675) Лемери объяснял способы соединения частиц на примерах взаимодействия кислот с содой, металлами и др. Частички кислот, оканчиваясь остриями, о чем свидетельствует их кислый вкус и кристаллическая форма получавшихся веществ, при взаимодействии с другими телами, способны проникать в вещества, содержащие поры, например щелочи.
В первой половине XVIII в. механистические теории подобного рода постепенно стали исчезать из трудов английских и
81
ГЛАЙА ВТОРАЯ
!! - —-^^— ...... _ “^ —
французских химиков. На их смену пришли новые идеи о йри-тяжении, с которых началась новая эпоха в учении о сродстве.
В 1704 г. в своей «Оптике» И. Ньютон, рассматривая химические явления, подверг критике механистические теории. Он сводил химические процессы, например действие кислот на металлы и щелочи, вытеснение одних веществ другими, к взаимодействию веществ за счет сил притяжения различной интенсивности.
«Части всех однородных твердых тел,— писал Ньютон,— вполне прикасающиеся друг к другу, сцепляются очень сильно вместе. Для объяснения этого некоторые изобрели атомы с крючками, оставляя вопрос без ответа; другие говорят нам, что тела связаны покоем, то есть таинственным качеством, или, скорее, ничем; другие — что частицы связаны согласованными движениями, то есть относительным покоем между ними. Я бы, скорее, заключил из сцепления частиц о том, что они притягивают одна другую некоторой силой, которая очень велика при непосредственном соприкосновении...» [43, с. 294]. «Не обладают ли малые частицы тел,— размышлял Ньютон,— определенными возможностями, способностями или силами... Ибо хорошо известно, что тела действуют друг на друга при помощи притяжений тяготения, магнетизма и электричества; эти примеры показывают тенденцию и ход природы и делают вероятным существование других притягательных сил, кроме этих... То, что я называю притяжением, может происходить... способами, мне неизвестными... Притяжения тяготения, магнетизма и электричества простираются на весьма заметные расстояния и, таким образом, наблюдались просто глазами, но могут существовать и другие притяжения, простирающиеся на столь малые расстояния, которые до сих пор ускользают от наблюдения, и, может быть, электрическое притяжение распространяется на такие малые расстояния и без возбуждения трением» [43, с. 285].
Одним из первых химиков, обратившихся к проблеме химического сродства (1718), был француз Э. Жоффруа-старший, проводивший опыты по взаимодействию кислот и оснований. Получив соль действием едкой щелочи на кислоту, он затем действовал на эту соль другой кислотой. Если эта вторая кислота имела большее сродство к щелочи по сравнению с первой, она вытесняла из соли первую кислоту. Таким образом, Жоффруа получил ряд кислот, располагавшихся по степени убывания их сродства к щелочи.
«В химии наблюдаются,— писал Жоффруа,— между различными телами некоторые отношения, с помощью которых легко соединяются тела одни с другими. Эти отношения имеют свои... законы... среди множества смешанных веществ и тех, которые имеют расположение к соединению в совокупности. Замечено, что одно из этих веществ всегда соединяется постоянно с некоторым другим, предпочитая его всем» [60, с. 53].
82
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
Э. Жоффруа составил таблицы, которые представляют собой перрый опыт количественного определения сродства. Он опубликовал эти таблицы в то время, когда механистические картезианские взгляды еще разделялись физиками и химиками. Дж. Пар-тингтон отметил, что из-за отрицательного отношения Парижской Академии наук к учению Ньютона Жоффруа вместо - слова «сродство» употребил «отношение» [60, с. 54]. Однако среди ученых Франции идеи Ньютона получили некоторое распространение, и уже в 1723 г. Ж. Сенак рассматривает сродство как силу, подобную тяготению, однако действующую на весьма малых расстояниях [61].
В 1740-х годах во Франции окончательно победило ньютони-анство и в результате вновь повысился интерес к проблеме химического сродства [62]. Так, в 1749 г. П. Макер использовал таблицу Жоффруа и, внеся в нее поправки, разработал систему сродств, т. е. ввел новые виды сродства в зависимости от типа реакций.
В 1732 г. Г. Бургаве выступил с идеями о сродстве с позиций представлений Р. Бойля и И. Ньютона. Бургаве видел основное проявление сродства в растворении веществ. Согласно его взглядам, «частички растворителя полностью перемешиваются с частичками растворяемого вещества» [12, с. 669]. Понимая под растворителем вещество, способное изменить в химическом смысле другое (растворитель кислота изменяет металл), Бургаве указывает на сродство двух несходных веществ — кислот и металлов.
Химики и до Бургаве исследовали взаимодействие несходных веществ, в частности Жоффруа построил таблицу сродств на основе вытеснения различных по свойствам веществ. Но в теоретических воззрениях в основном преобладало представление, известное еще в древности (подобное растворяется в подобном), согласно которому два вещества по отношению друг к другу имеют тем большее сродство, чем они более сходны химически. Бургаве, наоборот, заметил, что чем более химически различны вещества, тем большее сродство они имеют. Это было новым в теоретических представлениях о сродстве.
Все изменения, которые тела претерпевают в химических процессах, Бургаве сводил к одному принципу — движению, которым обладают маленькие частички, входящие в состав тела. Когда происходит соприкосновение двух тел, маленькие частички, которые Бургаве называл «элементами», приводятся в активное движение и, соединяясь, образуют вещество, обладающее новым качеством, отличным от качеств соприкасающихся тел. О маленьких частичках — элементах — Бургаве писал, что они настолько тверды, что не подвергаются разрушению.
Взгляды о движении частиц при соприкосновении высказывал также М. В. Ломоносов. «Величина [частиц] — не единственная Причина сцепления, иначе твердые тела нс могли бы становить
ся
ГЛАВА ВТОРАЯ
ся жидкими, необходимо движение» [40, с. 259],—писал он. «Сцепление и притяжение — одно и то же» [40, с. 261],—выступал Ломоносов против картезианских представлений, однако, отдавая дань времени, русский ученый придерживался бойлев-ских механистических воззрений, согласно которым важнейшим считалось знание фигуры частиц.
«...Так как нельзя произвольно допустить притягательную силу или какое-нибудь другое скрытое качество, то необходимо, чтобы существовала некоторая материя, которая своим давлением толкала бы корпускулы в противоположных направлениях и которая была бы причиною их существования»,— объяснял Ломоносов взаимодействие веществ посредством законов механики с позиций материалистических представлений [40, с. 43].
Наблюдая прочное сцепление двух кусков мрамора, соприкасавшихся плоскими полированными сторонами поверхностей, Ломоносов предположил, что, когда «изгнан воздух из места соприкосновения», окружающий воздух, оказывая давление на соединенные куски мрамора, поддерживает сцепление. Но этому действию способствует противодействие некой невидимой жидкой материи, заключавшейся в промежутках тел. Но в каждом отдельном явлении сцепления, по мнению Ломоносова, следует учитывать, что поскольку корпускулы, например, «смешанного тела», т. е. химического соединения, различны по своей массе и фигуре, то и его составные части различаются «массою, числом, фигурою или способом соприкосновения» [40, с. 39]. То, что «мельчайшие корпускулы ртути жадно соединяются и срастаются с тончайши-ми молекулами золота» [40, с. 57], Ломоносов объясняет различием фигур или соприкосновением, обусловленными летучестью ртути и высокой тягучестью золота при слабом нагревании.
Явление кристаллизации солей, согласно Ломоносову, происходит так: «... соляные корпускулы, опускаясь под влиянием силы тяжести, ударяются в те, которые осели уже ранее на дно и своим ударом изгоняют из места соприкосновения водные корпускулы. Они приходят в непосредственное соприкосновение, прочнее сцепляются и, собравшись в большом числе, образуют кристаллы» [40, с. 63]. Полагая, что поверхность частиц может обладать вращательным тепловым движением, Ломоносов объяснял взаимодействие веществ свойством поверхности частиц приобретать форму, подобную зубчатым колесикам. Проникновение одних частиц в другие (сцепление устройств) содействует образованию однородного смешанного тела, например смеси спирта с водой. Вещества, не способные к химическому взаимодействию и растворению друг в друге, не обладают поверхностями, подходящими для проникновения одного в другое, например несмеши-вающиеся жидкости.
С середины XVIII в. учение о химическом сродстве привлекает к себе усиленное внимание химиков, прежде всего потому,
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
что практическая химия и металлургия нуждались в объяснениях химических процессов.
В 1750 г. в Германии X. Геллерт (1713-1785) пытался использовать представления о сродстве в металлургической практике и вскрыл различия между сродством веществ, проводя их • анализ мокрым и сухим путем. Геллерт применил идеи .о сродстве и к объяснению физических явлений. Заметив, что расплавленный свинец, подобно ртути, создает разрежение в капиллярной трубке, он справедливо объяснил это явление тем, что между частицами металлов действует большее притяжение, чем между частицами металлов и стеклянной стенкой капиллярной трубки [60, с. 710].
X. Геллерт видоизменил таблицу Жоффруа и составил таблицу из 28 столбцов, в которых размещены вещества, имеющие наименьшее сродство к веществам, расположенным в начале таблицы. Интересно, что он обозначал вещества символами, например кобальт —К, висмут —W, цинк —X, известь —С, а окись цинка — СХ.
Английский ученый В. Льюис, составивший таблицу сродства в 1750-х годах, сообщал, что «доктрина сродства» широко использовалась в фармации при изготовлении препаратов [63, с. 68—69].
Теоретические представления о сродстве носили, однако, в описываемое нами время в основном умозрительный характер.
П. Макер в 1760-х годах подразделял сродство на простое и сложное. Простое сродство действует между составными частями одного и того же вещества, вызывая агрегирование. Если же соединяются части различных тел и образуется вещество с отличными от двух первых свойствами, то в этом случае действует также простое сродство, называемое, однако, сродством соединения. Это сродство определяет, например, взаимодействие железа и серной кислоты с образованием «зеленого купороса».
Сложное сродство (affinite compliqee) проявляется при соединении более чем двух тел в одно. При взаимодействии трех тел с образованием одного, когда одно из трех тел может иметь сродство только к двум, действует промежуточное (intermediare) сродство. Сродство, действующее в случае соединения щелочи и соли металла с образованием щелочной соли и осадка, называлось взаимным (reciproque). И, наконец, выделялось двойное сродство, КогДа две соли обмениваются радикалами [39, с. 86].
О причине взаимодействия веществ в XVIII в. существовали определениые предположения. Так, Б. ле Саж считал, что основ-Ное влияние на соединение тел оказывают плотность и форма Молекул. Его соотечественник Ж. Лимбург полагал, что причина сродства лежит в подобии частиц, которая заставляет их приближаться достаточно большой поверхностью друг к другу и притягиваться [64, с. 6]. Можно сказать, что все объяснения причиц
85
ГЛАВА ВТОРАЯ
сродства до конца XVIII в. базировались на предположении о притяжении частиц за счет сил, действующих на малом расстоя-нии, однако в отдельных случаях учитывалось участие физических свойств частиц.
Немецкий химик и металлург И. Карстен подразделил определение сродства в XVIII в. на две категории: 1) сродство —это действие силы, присущей в природе неодинаковым веществам, благодаря которой они связываются друг с другом или соединяются глубочайшим образом; 2) сродством называется действие силы, свойственной неодинаковым веществам, с помощью которой они соединяются с различной степенью интенсивности. Первое определение предполагало существование между веществами слабого и сильного сродства. Во втором определении, которое, по мнению Карстена, более всеобъемлюще, чем первое, предполагалось, что «вещество одной силы „а“ охотнее соединяется с третьим веществом „с“, чем со вторым ,,Ь“, т. е. имеет место в природе избирательное сродство» [64, с. 1—2 |.
Новый этап в развитии учения о сродстве начался с работ Т. Бергмана, который в 1775 г. ввел понятие о нескольких видах избирательного сродства. Современники Бергмана высоко ценили его работы по исследованию химического сродства и прежде всего ставили ему в заслугу его счастливую идею о том, что «сродство как абсолютная сила представляло небылицу» [64, с. 44]. Бергман, как и некоторые его предшественники, причиной сродства признавал притяжение частиц вещества, от формы и положения которых зависят виды сродства. Он различал несколько видов притяжения, а именно притяжение между гомогенными частицами, называемое им притяжением сложения, и между гетерогенными, называемое притяжением соединения. Кроме того, для объяснения различия протекания реакций в растворах и расплавах он ввел понятие о притяжении растворения и притяжении сплавления.
В конце 1780-х годов Бергман составил более пятидесяти таблиц сродства для исследованных при высокой температуре (сухим путем) и в растворе при обычной температуре (мокрым путем) веществ. В таблицах для каждой кислоты дан ряд соединений, в котором щелочи, металлы и земли расположены в таком порядке, что каждая предшествующая щелочь, земля и металл вытесняет все следующие из их соединений с кислотами [65, с. 51—61]. Бергман ввел понятие «простого», «двойного» и «сложного» избирательного сродства.
По Бергману, величина притяжения у разных тел различна и определяет способность тел вступать в соединение. Изучая реакции вытеснения, Бергман сравнивал сродство двух тел по отношению к третьему. С последним соединяется нацело то из двух тел, которое имеет большую величину притяжения, т. е. большее сродство. Однако для каждого вещества оно является по
86
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
стоянной силой. Следовательно, по представлениям Бергмана, сродство вступивших во взаимодействие тел определяет направление химической реакции, которая при этом идет до конца. Бергман считал сродство независимым от изменения условий реакции.
Т. Бергман описал особые случаи реакций, когда имело место двойное, а также сложное избирательное сродство, которые он отличал от простого сродства. Явление двойного избирательного сродства проявляется тогда, когда вступают во взаимодействие два тела, АВ и CD, каждое из которых составлено из двух различных веществ, обменивающихся своими составными частями при соединении друг с другом, порождая два новых вещества АС и BD [64, с. 30].
Следует отметить, что на открытие двойного избирательного сродства претендовал английский ученый Дж. Блэк, считая, что все подобные наблюдения были им сделаны до Бергмана, а именно в 1756 или 1757 г., когда он представил «диаграммы» сродств.
Т. Бергман и его последователи, а также другие ученые при изучении химического сродства пользовались таблицами Блэка и результатами его наблюдений. Г. Копп обратил внимание, что изучение явления, названного впоследствии явлением двойного избирательного сродства, встречается еще у Р. Глаубера. Бергман впервые выступил с теоретическим обоснованием этого явления как явления химического сродства.
Учение Бергмана о химическом сродстве было прогрессивным шагом, поскольку он не только придавал значение количественной мере сродства, но и применил представления о сродстве конкретно к химическим реакциям.
Дж. Блэк, открывший скрытую теплоту плавления, размышляя над проблемами химического сродства, особенно подчеркнул «очень изобретательную мысль...» Жоффруа, «чья таблица была воспринята с большим одобрением и полезная для химиков, хотя вмешательство действий теплоты, летучести и количества недостаточно в ней присутствует» [66, с. 41].
Экспериментаторы обращали внимание на явление воздействия тепла на химическое сродство, поскольку наблюдали, что многие химические взаимодействия и разложения происходят при повышенных температурах. Г. Шталь, например, в конце 1730-х годов указывал, что каломель разлагается серебром на холоде, а роговое серебро — в присутствии ртути при нагревании. На эти факты также обращал внимание П. Макер. Во второй половине XVIII в. влияние температурных условий учитывалось при составлении таблиц, особенно при изучении взаимодействия веществ при растворении.
После работ Бергмана изучение сродства приняло еще более Широкий размах. Таблицы сродства были составлены И. Эрксле-беном (1775), Г. Руэлем (1777), К. Венцелем (1777), И. Вигле-
87
ГЛАВА ВТОРАЯ
бом (1780), И. Рихтером (1792) и др. Изучением причин химического сродства в 1780—1790-х годах занимались в Англии Р. Кирван, во Франции А. Фуркруа, Л. Гитон де Морво, в Германии Ф. Ахард, Г. Линк, А. Грен и многие другие.
Таблицы химического сродства отражали соотношения взаимодействия кислот и каких-либо веществ, установление точки насыщения, изменения температуры при взаимодействии веществ и т. п. Эти таблицы широко использовались для объяснения различных явлений, в том числе и физических. Дж. Блэк, объясняя процесс парообразования, писал, что «частица воды, становясь частицей пара, притягивает и соединяется с одной или большим числом атомов теплоты, являющейся причиной этого явления, если только поднимается необходимая температура, и удерживает их как части, или ингредиенты, своей парообразной формы, так что мы можем справедливо предположить, что когда частица пара становится водой, эти атомы теплоты отпускаются на свободу на основании установленных законов химического сродства» [66, с. 37].
Обращает на себя внимание тот факт, что Блэк, используя атомную теорию, в отличие от многих своих современников, представлявших сродство силой химической и действующей только между веществами разной природы, считал, что силы химической, а не физической природы действуют и при переходе вещества из одного агрегатного состояния в другое.
Дж. Блэк четко указал также четыре условия, которые содействуют химическому притяжению:
1) одно или два тела должны быть жидкими в момент смешивания;
2) растворению твердого тела в жидкое способствует механическое разделение;
3) свободное размешивание;
4) умеренная теплота [66, с. 41].
Изменение силы химического сродства отождествлялось с определением количества вещества, необходимого для насыщения определенного количества кислоты (или щелочи). В результате этих исследований появились работы К. Венцеля (1740—1793) по количественному определению соединительных (эквивалентных) весов. В основу их легло изучение количественных отношений между кислотами и основаниями в нейтральных солях. Венцель обнаружил, что при взаимодействии двух нейтральных солей получившиеся продукты нейтральны — явление, свидетельствующее о том, что химическое взаимодействие не влияет на нейтральность.
С целью измерения сродства Венцель определял скорость растворения металлов в кислотах различной концентрации и нашел обратную зависимость между сродством веществ к общему растворителю и временем растворения [67, с. 13]. Венцель сфор-
88
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
аудировал «первую систематическую идею о времени в химические реакциях» [68, с. 138].
В определении относительной меры сродства основную роль шрало представление о «насыщении» веществ друг другом. «Не было, быть может, предмета в области химии,— отмечал II. Карстен,—где царило больше путаницы и неопределенности, чем в понятиях насыщения и нейтральности» [64, с. 153]. Он приводит одно из определений: «Насыщение — это состояние веществ, при котором их взаимное стремление к соединению, их сродство или притяжение полностью бы удовлетворялось по отношению друг к другу» [там же]. Насыщение представлялось абсолютным или относительным (полным или неполным).
Наблюдаемое в многочисленных экспериментах явление нейтрализации, образования нейтральных растворов позволило немецкому химику И. Рихтеру исследовать стехиометрический состав химических соединений. Составив таблицы, в которых указывалось количество различных кислот, необходимое для нейтрализации 1000 частей одного и того же основания, и количество различных оснований, нейтрализующее 1000 частей одной и той же кислоты, Рихтер показал эквивалентность количеств этих веществ. Он указывал на условие нейтрализации, заключавшееся в том, что при взаимодействии, например, сернокислого натрия и хлористого бария количество бария, перешедшее от соляной кислоты к серной, должно быть химически эквивалентно количеству натрия, перешедшего от серной кислоты к соляной. Его таблицы «пропорциональных чисел» давали возможность рассчитать состав солей, которые еще не были разложены. По существу, это были химические эквиваленты составных частей кислот, оснований и солей, означавшие, что при образовании сложных веществ элементы соединяются в строго определенных количествах, сохраняясь при переходе от одного химического соединения к другому. Система обозначений Рихтера давала возможность изображать химические реакции и их относительные количественные соотношения [69, с. 268].
У Рихтера были непосредственные предшественники, занимавшиеся вопросами эквивалентности и нейтральности. Кроме К. Венцеля, определению количеств различных кислот, требующихся для нейтрализации одного и того же количества основания, немало времени в 1770-х годах посвятил Т. Бергман. К более определенным выводам о количественных отношениях он пришел позже, при изучении процессов осаждения серебра из азотнокислых солей [70, с. 46]. К 1790-м годам относятся исследования Р- Кирвана, который занимался определением «частей истинных кислот» и «их нейтральных солей» [71, с. 266]. Приверженность Боргмана и Кирвана к флогистонной теории мешала им сделать правильные выводы, весовую «эквивалентность» они связывали с одинаковым количеством флогистона, содержащегося в веществах.
ГЛАВА ВТОРАЯ
Стехиометрические исследования Рихтера не были в полной мере поняты его современниками по причине использования им одновременно флогистонной и антифлогистониой терминологий [22, с. 96] и потому, что они вообще еще не могли быть объяснимы до возникновения атомистики Дальтона, их значение было оценено позже. Они содержали важную закономерность, объединенную в общий химический закон — закон эквивалентов.
К концу XVIII в. учение о химическом сродстве завершилось новой теорией сродства К. Бертолле. Излагая свои соображения о принципах химического действия, Бертолле писал, что все силы, производящие химические явления, представляют собой производные взаимного притяжения молекул тел, которому дали название «химическое сродство» [72, с. 1]. Однако, по его мнению, математическое выражение эффектов сродства показывает определенную зависимость сил сродства от количеств участвующих во взаимодействии веществ.
К. Бертолле высказал взгляды, отличные от представлений Бергмана о сродстве, господствовавших в химии в последней четверти XVIII в. Для Бертолле сродство — понятие, изменяющееся в зависимости от условий эксперимента — количества, растворимости, температуры, давления и агрегатного состояния. Величина сродства, по его мнению, зависит от количества реагирующих тел и не может быть величиной постоянной для каждого индивидуального вещества, как указывал Бергман. Следовательно, направление химической реакции должно зависеть, по теории Бертолле, от сродства и массы реагирующих тел. При реакции двух тел с третьим последнее распределяется между двумя в зависимости от сродства и масс, и при этом наступает равновесие, при котором существуют не только бинарные соединения первого с третьим и второго с третьим, но и свободные первое и второе. Реакция лишь в тех случаях идет до конца, когда продукт «полностью удаляется из сферы действия» [73].
К. Бертолле ввел понятие «химическая масса» — произведение силы сродства на количество вещества, проявляющее эту силу сродства. «Если два вещества конкурируют при соединении с третьим, то каждое из них испытывает степень насыщения в соответствии с их массой»,— писал Бертолле [72, с. 69]. Взаимодействие веществ в растворе, когда из сферы действия не удаляются продукты реакции, приводит к состоянию равновесия *.
* В истории учения о химическом сродстве и химическом равновесии XVIII век знаменателен тем, что именно в это время механика подарила химии столь фундаментальное понятие, как понятие о равновесии. В химии уже был для этого подготовлен материал — в таблицах химического сродства. Но горделивый замысел Бергмана, Кирвана и других ученых конца XVIII в.— авторов таблиц химического сродства — создать основу для учения о химическом равновесии был обречен на неудачу. Химики еще слишком мало знали о действии масс в химических реакциях, о влия-
90
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
Изучение другой стороны учения о химическом сродстве, касающейся природы химического сродства веществ в дореакцион-дОм состоянии, привело в конце XVIII в. к признанию единой химической природы сродства. Но словам Бертолле, «отличие, устанавливаемое некоторыми физиками между химическим срод-стВом и физическим сцеплением, необоснованно» [73, с. 142]. Он настаивал также на том, что в органических соединениях действуют обычные силы химического сродства, а не какие-то особые силы, как утверждалось ранее.
Несмотря на то что сущность химического сродства тогда еще не могла быть установлена, в развитии представлений о нем следует отметить ряд новых результатов. Учение о химическом сродстве явилось источником новых исследований, в частности первых исследований К. Венцеля зависимости скорости реакции от времени растворения и концентрации растворителя, т. е. зарождения химической кинетики. Развитие учения о химическом сродстве связано с открытием стехиометрических законов, которые сыграли большую роль в развитии химии.
ПНЕВМАТИЧЕСКАЯ ХИМИЯ
Важную роль в возникновении и развитии химии газообразных веществ играли технические потребности. Так, методы обработки металлов были связаны с изучением процессов окисления и восстановления сложных веществ, в состав которых, как выяснилось впоследствии, входили газообразные продукты.
В период теории флогистона пневматическая химия, являясь частью аналитического и технического направлений, вызывала особый интерес у химиков.
В истории химии принято считать, что впервые химическим путем получал газы И. Ван Гельмонт, обнаруживший в 1620 г. лесной газ (gas silvestre), т. е. углекислый, газ, образующийся при сгорании угля. Но, полагая, что газы невозможно собрать в сосуд, он оставил их изучение.
И. Ван Гельмонт впервые ввел слово «газ» [13, с. 154—155] и утверждал, что существуют газы, отличные от воздуха. Однако и после его исследований примерно до середины XVIII в. воздух продолжал считаться единственным газом в природе, принимав-щим различные формы из-за попавших в него примесей.
В большой степени исследованиям химического состава воздуха способствовало изучение его физических свойств, которым занимались ученые XVII в.—Г. Галилей, Е. Торричелли, Б. Паскаль, О. Герике, Р. Бойль, Р. Тоуилей, Э. Мариотт и др. Вопреки
нии на химический процесс различных факторов (температура, природа растворителя, присутствие других компонентов).— Прим. отв. ред.
91
Оборудование пробирной лаборатории Лемана
1, 4, 5, 6 — лабораторная печь со сменными блоками 4 и 6 для различных пробирных работ; 2 — печь для нагревания одной реторты; 3 — керамическая печь для отгонки из реторт (получение цинка, фосфора и др.). На блоке 6 топочное отверстие ошибочно отсутствует в оригинале XVIII в.
Пробирные весы Лемана
1 — весы в весовом шкафчике; 2 — разновесы; 3 — весовые чашечки; 4 — ареометр («соляные весы»)
ГЛАВА ВТОР АЙ
учению Аристотеля, представлявшему воздух как одну из четы-рех стихий-качеств, существующих неизменно в природе, в XVII в. появились новые представления о том, что воздух это не пер-вичная, а вторичная субстанция, возникающая путем истечения из жидких и твердых сред, в частности из воды, земли и других тел (О. Герике, X. Гюйгенс, Ф. Сильвий). Воздух представлялся в то время веществом гомогенным, но сложным [74, с. 17].
Р. Бойль также рассматривал воздух как конгломерат различных истечений, например, из солей, ио считал, что эти истечения зависят от времени года. Важнейшая заслуга Бойля в применении первых аппаратов для собирания воздуха, что способствовало исследованию других газов.
В 1660 г. Р. Бойль описал способ собирания газов в длинно-горлую колбу, в которой находились разбавленная серная кислота и железные гвозди. Колбу ставили вверх дном в плоскую чаш ку с той же жидкостью. Выделявшийся при действии кислоты на железо газ — водород — вытеснял кислоту из колбы. Заменив серную кислоту азотной, Бойль получил другой газ — окись азота.
С помощью воздушного насоса Бойль и его ассистент Р. Гук определили вес и упругость воздуха. У Бойля имеются описания методов и аппаратов для перегонки под пониженным давлением. Он же наблюдал, что воздух необходим для горения и дыхания. Бойль разработал план исследований воздуха — настолько эта проблема казалась ему важной [22, с. 52].
В 1665 г. английский ученый Р. Гук (1638—1703) писал об особом веществе, сходном с тем, которое находится в селитре в «сжатом» (связанном) состоянии [75]. По мнению Гука, это вещество при достаточно высокой температуре способно растворять все горючие тела. Тело полностью сгорает при постоянном притоке растворителя, т. е. воздуха. На основании опытов в замкнутом сосуде Гук установил, что горение прекращается, как только горючее тело насыщается особым веществом, т. е. растворителем.
Через четыре года, в 1669 г., английский ученый Дж. Мейоу (1641—1679), изучив составные части селитры, развил представления Гука об особом веществе, содержавшемся в воздухе, назвав его «воздушный спирт селитры» (spiritus nitro-aereus). Этот спирт, поддерживающий горение, по мнению Мейоу, «есть главный источник жизни и дыхания» [76].
Кальцинацию металлов Дж. Мейоу считал процессом горения, обусловленным соединением «сернистых частиц» с воздушно-селитряным спиртом. Обнаружив увеличение в весе при обжигании сурьмы, Мейоу заключил: «Я отлично знаю, что, по общепринятому взгляду, кальцинация сурьмы заключается в удалении содержащейся в ней серы. Несмотря на это, я склонен думать, что взгляд этот неверен» [77, с. 187].
Несмотря на то, что Мейоу высказал справедливое предполо-
92
развитие химии в конце xvii—xviii ВИ.
?кение об участии частиц вещества, содержащегося в воздухе, в горении и дыхании, его представления о составе воздуха были далеки от истины.
Значительный успех развитию пневматической химии принесло изобретение английским ботаником С. Гейлсом (1677—1761) пневматической ванны (1723). Нагревая различные сложные вещества в изогнутом ружейном стволе, Гейле собирал выделяющиеся газы в стеклянный сосуд, наполненный водой и погруженный горлышком в воду. Этот опыт позволил установить, что «воздух» входит в состав большинства тел. Гейле проводил количественные измерения воздуха, выделявшегося из химических продуктов при нагревании. В 1727 г. в Предисловии к изданному им труду «Растительная статика» Гейле останавливается на необходимости количественного подхода в исследованиях, считая, что «наиболее вероятным путем проникновения в природу тех частей мира, которые попадают в сферу нашего наблюдения, должны быть число, вес и мера» [78, с. 31]. Гейлса можно считать родоначальником метода измерения объемов газов. Однако он ие пытался определить качественный состав воздуха.
Изучение Гейлсом превращения упругого (свободного) возду ха в фиксированное (связанное внутри вещества) состояние, ко торое, по его мнению, происходило под воздействием притягивающей силы серных частиц, представляло основу для последующих исследований [74, с. 35].
Для Г. Бургаве, так же как и для Гейлса, воздух — всеобщий растворитель. В 1732 г. он писал, что воздух представляет собой «...хаос всех существ, перемешанных и соединенных вместе: в нем плавают вверх и вниз разжиженные частицы всех тел» [12, с. 314]. Но, в отличие от Гейлса, Бургаве все же особое внимание обратил на газ (gas silvestre), полученный Ван Гельмонтом и, как писал Бургаве, «обширно существующий» и образующийся при горении, ферментации, гниении и перегонке [там же]. Он проводил также опыты над животными, замечая, что они гибнут в безвоздушном пространстве.
«В нынешние времена довольно известно, что животные без иоздуха умирают»,—писал М. В. Ломоносов в 1746 г. в переведенной им на русский язык «Волфианской экспериментальной физике» [40, с. 518]. Для Ломоносова воздух, как огонь и вода, представлялся одним из растворителей всякого рода мельчайших частиц.
С 1750-х годов начался новый этап в развитии пневматической химии.
В диссертации Дж. Блэка, опубликованной в 1754 г. на латинском, а в 1756 г. на английском языке, описан обнаруженный Ученым новый вид воздуха, названный им «связанный воздух».
Дж. Блэк изучал свойства открытой в 1722 г. Ф. Гофманом Магнезии (основного карбоната магния) [66, с. 48]. Когда он
93
ГЛАВА ВТОРАЯ
a i x
нагревал магнезию в тигле, то обнаружил, что она уменьшилась в весе, и уже кальцинированная магнезия (MgO.—3. Ш.) составила 712 от ее первоначального веса. «Я начал подозревать,— писал Блэк,— что выделение пузырьков из магнезии происходило от воздуха, который она содержит, в то время как рожденная (кальцинированная магнезия.— 3. Ш.) не выделяла пузырьков газа...» [там же].
Дж. Блэк описал один из опытов следующим образом: «В разбавленную кислоту, заключенную в баллоне, положил мел или магнезию и... сделал отверстие в пробке, в которое вставил согнутую трубку. Другой ее конец ввел (способ д-ра Гейлса) в перевернутую банку, наполненную водой и помещенную в кадку с водой. Можно было наблюдать выделение пузырьков газа, которые обильно поднимались кверху, где собирались и двигались через воду в банку» [66, с. 49].
Таким образом, Блэк получил углекислый газ, названный им «связанным воздухом», и показал, что это новое химически индивидуальное вещество, т. е. окончательно установил существование газа, отличного от воздуха. Такое открытие стало возможным благодаря применению количественного метода исследования.
В 1764 г. английский ученый, работавший в Дублине, Дж. Макбрайд (1726—1778) повторил опыты Блэка, а также получил «связанный воздух» пз гниющих животных веществ. Он указал, что холодная вода способна поглощать некоторое количество полученного им газа и приобретает при этом способность осаждать известь. Результаты этих исследований уже тогда оказались полезными в практике. Макбрайд предложил их для использования в медицине [49, с. 29].
В 1765 г. английский химик У. Браунриг с помощью нового пневматического прибора обнаружил газ, открытый Блэком, в минеральной воде одного из предместий и установил связь исследований Венцеля и Блэка [там же].
В 1766 и 1767 гг. Г. Кавендиш с помощью усовершенствованной аппаратуры изучил «связанный воздух», определив его растворимость в воде количественным методом, а также плотность. Он указал, что при сгорании угля также образуется «связанный воздух».
Г. Кавендиш открыл еще одно «воздухообразное» вещество, отличное от воздуха, выделив горючий газ (водород.— 3. Ш.), и определил его свойства, но ошибочно думал, что это флогистон.
Область исследований «воздухообразных» веществ была значительно расширена работами Дж. Пристли и Дж. Резерфорда в Англии, К. Шееле и Т. Бергмана в Швеции.
Дж. Пристли начал свои пневматические исследования примерно в одно и то же время с Г. Кавендишем. Первые результаты исследований газов были опубликованы Пристли в 1767 г. в работе, посвященной изучению электричества. Пристли иссле
94
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
довал «мефитический воздух», который позднее был назван азотов и который получался пропусканием воздуха через раскаленный уголь, а образующийся при этом углекислый газ («связанный воздух») поглощался щелочью. Пристли заметил, что в этом «воздухе» гаснет свеча.
Пневматические эксперименты Пристли были связаны с его исследованиями по электричеству и привели к выводам, касавшимся проводимости, которой обладал «мефитический воздух», но не обладал воздух, лишенный влаги [79, с. 21].
Различие между «связанным воздухом» и «мефитическим», который оставался после горения серы и фосфора, а также кальцинации металлов в воздухе, окончательно было установлено в 1772 г. английским ученым Дж. Резерфордом.
В 1772 г. Дж. Пристли опубликовал большую статью «Наблюдения над различными видами воздуха», где описывались многочисленные новые открытия в области химии газов [80]. Открытию и получению новых газов способствовало усовершенствование Пристли пневматической ванны Гейлса, состоявшее в замене в ее приемнике воды ртутью. Гем самым появилась возможность собирать газы, растворимые в воде. В названной статье Пристли описал получение «селитряного воздуха» (окись азота) действием азотной кислоты на металлы, и «солянокислого воздуха» (хлористый водород), получение чистого азота, который он назвал «фло-гистированным воздухом». Пристли открыл также «летучий щелочной воздух» (аммиак), дефлогистированный селитряный воздух (закись азота), получавшийся восстановлением окиси азота железом и серой в присутствии влаги.
Дж. Пристли заметил, что свойства «селитряного воздуха» изменялись в присутствии железа и серы, а именно в нем уже не гасла зажженная свеча, не наблюдалось покраснения от смешения с атмосферным воздухом (речь идет о превращении на воздухе окиси азота в двуокись). Пристли получил также сероводород и четырехфтористый кремний. Изучая свойства атмосферного воздуха, Пристли показал, что воздух участвует в процессах горения, дыхания, гниения и жизненных процессах растений.
Исследователи творчества Пристли отмечают, что самым плодотворным периодом его научной деятельности были 1773— 1780 гг., когда ему удалось выделить и изолировать, помимо Указанных газов, двуокись серы и «дефлогистированный воздух» (кислород) [81,с. 139].
Открытие кислорода (1774)—одно из крупнейших открытий в науке. Пристли получил его нагреванием двух веществ — окиси Ртути (HgO) и сурика (РЬ3О4), направив на каждое из них пучок солнечных лучей при помощи сильной линзы. При этом он обнаружил, что получается воздух, в котором свеча горит так же ярко, как в измененном «селитряном воздухе». Пристли назвал Газ «дефлогистированным воздухом», т. е. лишенным флогистона.
95
ГЛАВА ВТОРАЯ
Замечательные открытия в описываемой области принадлежат К. Шееле. Его работы, проводимые с 1768 по 1775 г., появились в печати лишь в 1777 г. [82]. Однако в 1775 г. Т. Бергман опубликовал статью о получении Шееле «огненного воздуха» (кислорода) [83, с. 125], в которой сообщал об открытии Шееле кислорода, по крайней мере, на три месяца раньше открытия Пристли [84].
Кислород был получен Шееле несколькими способами, а именно разложением селитры или нитрата магния при нагревании а также перегонкой смеси селитры с серной кислотой. «Огненный воздух», по мнению Шееле,— тот, посредством которого поддерживается циркуляция крови и соков у животных и растений.
Взаимодействием черной магнезии (МпО2) и соляной кислоты Шееле в 1774 г. получил хлор, который назвал «дефлогистиро-ванной соляной кислотой», а в 1775 г.—мышьяковистый водород.
Анализы воздуха, проведенные Шееле для доказательства его состава, привели ученого к выводу о присутствии в воздухе «упругих флюидов» [85, с. 43—45], однако сущность этих флюидов правильно была истолкована только А. Лавуазье.
Почти одновременно с Шееле и Пристли кислород получил П. Байей (1725—1797) во Франции (1774). Байен выделил кислород нагреванием «ртутной извести» и, назвав его «упругие флюидом», высказал справедливую точку зрения, что этот газ является причиной увеличения веса металлов при кальцинации. Он наблюдал превращение ртути в красную окись при действии на нее этого флюида [13, с. 318, 344].
К. Шееле, Дж. Пристли и П. Байен, будучи убежденными сторонниками флогистонной теории, пытались увязать с ней экспериментальные факты. Шееле считал, что «огненный воздух» состоит из воды, земли и флогистона. Сложным телом считал кислород также и Пристли, который постоянно колебался в объяснениях и допускал возможность превращения одних газов в другие. Они не смогли понять значения открытия кислорода даже в то время, когда Лавуазье шаг за шагом показывал несостоятельность теории флогистона.
В 1776 г. К. Бертолле опубликовал свои наблюдения над воздухом, среди которых имеются интересные заключения о том, что органическое вещество — уксусная кислота,— разлагаясь в большом количестве, выделяет «горючий» (водород) и «связанный воздух» (углекислый газ). Кроме того, он подтвердил, что в азотной кислоте и металлических известях содержится «жизненньш воздух» (кислород).
Исследования в области пневматической химии в последней четверти XVIII в. значительно расширились в связи с усовершенствованием техники эксперимента. С помощью нового сконструированного эвдиометра Г. Кавендиш в 1783 г. провел серию анализов воздуха с целью определения «доброкачественности»
96
Достижения экспериментальной химии были бы невозможны без постоянного совершенствования химической аппаратуры. Важнейшую, хотя и не единственную ее часть составляла аппаратура для проведения химических анализов. До середины XVIИ в. она сравнительно проста, предназначена преимущественно Оля разделения перегонкой и для пробирного анализа плавлением и купеляцией. Приборы эти в основном металлические и керамические; использование стекла огра ни чивается рето р там и.
Начиная со второй половины XT ILL в. экспериментальная техника быстро усложняется. Появляются чувствительные и достаточно точные весы. Л. Лавуазье начинает использовать сложную стеклянную аппаратуру с кранами и многими соединениями, изготовление которых требовало специальных уплотнений и замазок.
В начале XIX в. особой изобретательностью отличались Я. Берцелиус и Ж.-Л. Гей-Люссак, создавшие приборы, которые использовались в препаративной и аналитической химии более 100 последующих лет.
На вклейках помещены изображения приборов, имевших широкое применение или сыгравших важную роль в истории химии. В их числе лаборатория М. В. Ломоносова, оборудование типовой пробирной лаборатории XVI—XVIII вв., паяльная трубка Т. Бергмана, весы и приборы Р. Бойля, А. Лавуазье, Я. Берцелиуса и некоторые другие.
Химическая лаборатория М. В. Ломоносова. Конструкция
Химическая лаборатория V. В. Ломоносова. Оборудование
'иг. 22 — пробирные печи: А—D — для перегонки из реторт; BDE — плавильная печь. Фиг. 23 — большая плавильная печь. Фиг. 24—инструменты: F ~~ муфель; g — пробирные иглц; II — щипцы; I — форма для отливки; 9 — Щипцы; р — разновесы; t — тигель; V — форма для капеллен; h — стеклянная посуда; 8 —реторта; т — каиелль; п — подставка; /—форма для Набивки капеллей; z — набойник. 1»иг. 25 — весы
и
I ШИ ШИ I
"tn • Ж if
I Ы.ьйиГ •Мнчи И мм
чта^пнгиини ГЯЦВД8Л|ПЦГ w литии umwnnuwejmf. jt-ппч Чниицпч .
~ МЛ »Ш инна».. ММШММЦ. •млмитп ии '
8л*|1НГИШЦ. ‘Н1ЙШЛ М ЛНИПИЙИ1 •чНнИНП 1«яЛ5ли,на#-
ЧШ ri4 HU I йМ«| hLHteltlMH I ,М null» ЫШ
>"aW4Hir... _
a dWafljfflOi > гч»:^.№
ВИЧ чЧ, ЬяМ-
И||Г Й^иЯй*!- ЭД MVi ПГ 1
IHW мини I №м №№№. .HfHW Ut !Ш!1И
»tti Г.’l*4Ub Ч'' '•ч*
Лабораторные
^~ПРИ^°Р для разложения веществ образных продуктов;
ртути., 6 — баллон, для
7 -- Олл для vw.w-nivxiA
С
•J — прибор для проведения реакций
VI 1ГЗВ<‘ III VI 14
поглощением газо-разложения окиси в токе газа;
приборы Лавуазье
ных продуктов; 9 — поджигание при помощи лупы; ю - реторта г
газооГраз ны х 11 р< >д у кт<>в
фосфора пузырем
под колоколом для собирания
Перегонный куб (Лавуазье)
Прибор Лавуазье для элементного анализа органических веществ
iiiiiiiiiiluuliiiluqiiiiiiiiuiiiiiiiiiiiiiimlinniiiiiiiiiiiiiiiiiiiiiiiiiiwiiiiiNnHinnniuimmiHiHiiniiiiiiiiiiiiiiiiiii
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ.
воздуха в зависимости от загрязненности флогистоном [74, с. 67-70].
Развитие пневматической химии способствовало открытию и
и3учению почти двадцати газов, обладавших характерными для каждого особыми свойствами и весом. Но среди них не было вещества, идентичного флогистону. Количественный анализ позво
лил установить различия между отдельными газами — углекис
лым газом и азотом, а также водородом и окисью углерода, неспособными поддерживать жизнь.
С середины XVII до конца XVIII в. пневматическая химия
прошла, по существу, три стадии развития:
1) до 1750 г., когда все виды газов представлялись разновидностями атмосферного воздуха, основной задачей исследователей было изучение физических свойств воздуха, выделение «воздуха» из сложных веществ и улучшение техники исследований;
2) 1750—1770 гг.—открытие и изучение газов, отличных от
воздуха;
3) с 1770-х годов — установление и изучение состава атмосферного воздуха, разработка кислородной теории горения.
Пневматическая химия предоставила обширный материал для создания новых теоретических представлений.
5 Всеобщая история химии
ГЛАВА ТРЕТЬЯ
НОВАЯ СИСТЕМА ХИМИЧЕСКИХ ЗНАНИЙ
В середине XVIII в., в период становления капиталистических отношений, рост производства требовал совершенствования технологических процессов на основе научных знаний. Социаль-но-экономические условия, сложившиеся в то время во Франции, обусловили причины, которые объясняют, почему революция в химии началась именно в этой стране [1—3].
А. Фуркруа так охарактеризовал состояние химии, способствовавшее началу химической революции: «...Множество открытий, новых фактов, интересных экспериментов следовали друг за другом с удивительной быстротой. Академических трудов, периодических изданий, частных диссертаций едва хватало, чтобы опубликовать все новинки. Наука заняла все умы, но, обогащаясь бесчисленными фактами, ее теория... казалась потерянной... среди этих обширных приобретений. Каждый химик имел свою частную теорию» [4, с. 35—36].
ИЗУЧЕНИЕ А. ЛАВУАЗЬЕ ЯВЛЕНИЙ ОКИСЛЕНИЯ И ВОССТАНОВЛЕНИЯ
Началом исследований процессов горения можно считать работу А. Лавуазье* * 1772 г. по сожжению алмаза с помощью за-
Примечание. Литературу см. стр. 401—402.
* Антуан Лоран Лавуазье родился 26 августа 1743 г. в Париже в семье богатого прокурора. Под влиянием отца сын поступил на юридический факультет Парижского университета, который окончил в 1764 г. Но адвокатом Аптуан Лавуазье пе стал. Он решил посвятить себя естественным наукам. Уже в 1766 г. Парижская Академия паук наградила 23-летпего Лавуазье золотой медалью за предложенный проект освещения улиц большого города. Проходит еще два года. Молодой ученый избирается (18 мая 1768 г.) адъюнктом Парижской Академии наук. Окрыленный поддержкой Лавуазье намечает большую программу физико-химических исследований, в результате которых он смог ниспровергнуть учение Шталя. В 1778 г. Лавуазье избирается действительным членом, а в 1785 — директором Парижской Академии наук. Жизнь и активная научная деятельность Лавуазье трагически оборвалась 8 мая 1794 г. Он был казнен как бывший генеральный откупщик, наживший огромное состояние (1 200 000 ливров). Узпав о казпи Лавуазье, знаменитый математик Ж. Лагранж сказал своему коллеге Ж. Даламберу: «Понадобилось лишь
98
НОВАЯ СИСТЕМА ХИМИЧЕСКИХ ЗНАНИЙ
мигательного стекла. Подобные исследования уже проводились в конце XVII в. во Флоренции. Лавуазье провел ряд экспериментов над алмазами и впервые правильно объяснил результаты этих опытов, показав, что при сгорании алмаза образуется «связанный воздух» (углекислый газ) [5, с. 76—79].
Предусмотренные программой (имевшейся в дневниковых запи- . сях Лавуазье 1772 г.) исследования «упругого флюида»., выделявшегося при брожении и вообще при различных химических реакциях, а также воздуха, поглощавшегося при горении веществ, имели целью познать природу этих, по его мнению, различных видов воздуха, опираясь па работы предшественников: «Я поставил перед собой задачу все повторить с новыми предосторожностями, дабы объединить все то, что мы знаем о том воздухе, который связывается или выделяется из тел, с другими добытыми познаниями и создать теорию» (цит. по [6, с. 73—74]). Эти опыты были изложены в «Мемуаре о кислоте фосфора и ее соединениях с различными солеобразными, землистыми и металлическими субстанциями» [6, с. 75], в котором процесс горения фосфора Лавуазье объяснял еще в духе теории флогистона, но показал, что при горении необходим доступ воздуха,— факт, вскрывавший противоречие в господствовавшей тогда теории.
В заметке Лавуазье, переданной им в запечатанном пакете в Парижскую Академию наук 1 ноября 1772 г., отсутствуют объяснения явлений горения на основе теории флогистона [7, с. 103]. Уже из этой записки, вскрытой 5 мая 1773 г., стало ясно, что взгляды Лавуазье отражали «ход эволюции его химической мысли» [8, с. 152]. В ней приводились объяснения причины увеличения веса тел при горении. Речь шла об увеличении веса серы и фосфора при горении и аналогии этого явления с процессом сгорания металлов. В подтверждение своих взглядов Лавуазье привел также опыт по восстановлению свинцового глета в замкнутых сосудах, в котором он наблюдал, что «в момент перехода земли в металл выделяется значительное количество воздуха» (цит. по [6, с. 78]).
В 1773 г. Лавуазье продолжил опыты по кальцинации свинца, олова, цинка, восстановлению сурика и окиси ртути углем. Установив, что «воздух» при горении фосфора, серы и других веществ присоединяется к ним, он с 5 февраля по 5 марта 1774 г. повторил опыты, из которых ранее Р. Бойль сделал вывод, что при кальцинации металлов в закрытых сосудах вес металла увеличивается за счет проникавшей через стенки сосуда одно мгновение, чтобы отрубить эту голову, но, быть может, и столетия будет мало, чтобы создать подобную ей».
О жизни и деятельности А. Лавуазье см.: Grimaux Е. Lavoisier (1743— 1794) d’apres sa correspondance, ses manuscrits, ses papiers de famille et d’antres documents inedits. P., 1888; Дорфман Я. Г. Лавуазье. 2-е изд. М.: Изд-во АН СССР, 1962.— Прим. отв. ред.
99
5*
ГЛАВА ТРЕТЕЙ
огненной материи. Этот опыт Бойля повторил в 1756 г. М. В. Ло-моносов и нашел, что мнение Бойля неверно. Опыты по кальцинации олова привели Лавуазье к выводу, что «...порция воздуха, соединившегося с металлами, несколько тяжелее, чем воздух атмосферы, а часть воздуха, оставшаяся после обжига, наоборот, несколько легче его» [9, с. 119]. В работе, посвященной кальцинации олова, Лавуазье уже представлял себе состав атмосферного воздуха [9, с. 120].
Предвидя, что его взгляды встретят упорное сопротивление сторонников теории флогистона, Лавуазье последовательно разъяснял свои теоретические представления, подкрепляя их экспериментами.
В январе 1774 г. Лавуазье опубликовал книгу, посвященную физическим и химическим исследованиям роли газов в химических реакциях [10]. Книга состоит из двух частей. В первой автор представил краткий исторический очерк о работах, проводившихся химиками в этой области. Во второй содержится описание его собственных опытов, касавшихся кальцинации фосфора, серы, а также олова и свинца в герметически закрытых сосудах. Лавуазье изложил свои новые представления о том, что при обжиге к сжигаемым веществам присоединяется часть атмосферного воздуха. Здесь же он высказал предположение, что воздух не может быть элементарным веществом, как было принято. Об этой книге опубликован рапорт, представленный в декабре 1775 г. комиссару Академии наук в Париже Ж. Трюдену де Монтиньи, президенту Академии в то время физику Ж. Ле Руа, химикам П. Макеру и Л. Кадэ. Как выяснилось впоследствии [11, с. 5—6], этот рапорт был написан не Лавуазье, как считалось в истории химии, а Макером, сторонником учения о флогистоне, и характеризовал интерес современников Лавуазье к его методу и экспериментам, объединившим физику и химию и представлявшим «реальный прогресс науки» [11, с. 8].
ОТКРЫТИЕ КИСЛОРОДА
Как отмечалось в главе второй, в большинстве историко-химических работ правильное объяснение открытия кислорода принадлежит только Лавуазье. Объяснения его современников и в определенном смысле предшественников весьма контрастируют с представлениями Лавуазье. Пристли, например, полагал, что воздух, годный для дыхания и способствующий более яркому горению свечи, состоит из земли и азотной кислоты. Лавуазье же описал его как новое элементарное вещество.
Опыты, проводившиеся в марте — апреле 1774 г., были продолжены затем с ртутной известью в ноябре 1774 г. Тогда Лавуазье указал, что воздух, содержащийся в ртутной извести,
100
НОВАЯ СИСТЕМА ХИМИЧЕСКИХ ЗНАНИЙ
является «наиболее чистой частью того самого воздуха, который пас окружает». К экспериментам с ртутной известью Лавуазье обратился, по-видимому, услышав об этом опыте от Пристли, удивленного свойством «воздуха», в котором свеча горела ярче, нежели в атмосферном. То, что Пристли называл случайностью, а именно получение флюида при разложении окиси ртути, ,у Лавуазье было запрограммированным поиском, приведшим в результате удачных экспериментов к подтверждению его предположений
В марте — апреле 1775 г. Лавуазье сообщал и об опытах по восстановлению окиси ртути без добавления посторонних веществ, а также при непосредственном контакте ее с углем либо с другим веществом, «содержавшим флогистон» (Лавуазье тогда еще пользовался терминологией теории флогистона).
Эксперименты Лавуазье с кислородом, проводимые им в феврале и марте 1775 г., были сообщены Академии наук в конце апреля 1775 г. и изложены в работе «Мемуар о природе вещества, соединяющегося с металлами при прокаливании и увеличивающего их вес» [12]. В этой статье Лавуазье доказал, что «...начало, соединяющееся с металлами во время их прокаливания, которое увеличивает их вес и превращает их в землю (окись.— 3. Ш.), есть не что иное, как наиболее чистая часть воздуха, таким образом, если воздух, находившийся ранее в соединении с металлом, из него выделяется, то в таком виде, в котором очень легко вдыхается, и легче, чем атмосферный воздух, способен поддерживать воспламенение и горение тел» [12, с. 123].
Для выяснения природы «воздуха», соединявшегося с металлами при кальцинации, Лавуазье предпринял восстановление известей. Он утверждал, что восстановление возможно лишь при контакте извести с углеобразной материей и с какой-либо субстанцией, содержащей то, что называется «флогистоном». Поскольку уголь, применяемый для восстановления, полностью исчезал в этом процессе, Лавуазье заключил, что «воздух, выделяющийся при востановлении металлов посредством угля, не является простым веществом, что он в известной мере есть результат соединения упругого флюида, выделенного металлом, и другого флюида, выделенного углем» [там же]. Речь здесь идет об углекислом газе, который, по мнению Лавуазье, не мог содержаться в металлической извести до ее соединения с углем.
Лавуазье, пытаясь попять природу флюида, содержавшегося в «известях», разлагал известь с помощью большого зажигательного стекла. Такие опыты он проводил вначале над железными известями, однако указывал на невозможность изучить флюид, Отделявшийся из них, поскольку он смешивался с атмосферным воздухом. Тогда Лавуазье обратился к опытам по восстановлению окиси ртути с добавлением угля и без добавления посторонних ВеЩеств. Лавуазье описал получение в первом случае углекисло-г° газа, во втором — «воздуха», который «не обладает пи одним
101
ГЛАВА ТРЕТЬЯ
из свойств „связанного воздуха41: совершенно нс умерщвляя, подобно последнему, животных, он, казалось, напротив, скорее способен поддерживать их дыхание; свечи и другие зажженные предметы не только в нем не гасли, а, напротив, пламя в нем заметно расширялось, давая значительно больше света и блеска, чем в обыкновенном воздухе; уголь горел в нем с яркостью, почти равной яркости фосфора, и вообще, все горючие вещества сгорали в нем с поразительной быстротой» [13, с. 40—41].
А. Лавуазье объяснил, что «если он получается в состоянии „связанного воздуха44 при всех случаях восстановления металлов с применением угля, то это явление обязано своим происхождением соединению последнего с чистой частью воздуха, и весьма вероятно, что все металлические земли дали бы лишь, подобно ртутной, воздух, в высшей степени пригодный для дыхания, если бы удалось их восстановить до конца без посторонних веществ, как восстанавливается ртуть, осажденная per se (в момент получения, тотчас же.—3. 77/.)» [12, с. 127]. «Удобовдыхательный» воздух, по замечанию Лавуазье, получается также из селитры при взрыве и является частью селитряной кислоты [12, с. 128].
На основании этих исследований Лавуазье впоследствии подробнейшим образом описал состав атмосферного воздуха. «...Можно двумя способами,— писал он,— определить свойства составных частей атмосферного воздуха: путем разложения и путем синтеза.
Прокаливание ртути дало нам пример того и другого способов, так как после отнятия у годной для дыхания части ее основы при помощи ртути мы снова ввели ее, чтобы получить воздух, совершенно схожий с атмосферным. Но этот синтез воздуха можно также произвести, взяв из различных областей составляющие его вещества. Из дальнейшего будет видно, что, когда растворяют вещества животного происхождения в селитряной кислоте (acide nitreux), выделяется большое количество воздуха, который гасит огонь, вреден для животных и во всем схож с частью воздуха, негодного для дыхания. Если к 73 частям этой упругой материи прибавить 27 частей легко поддерживаемого дыхание воздуха, извлеченного посредством прокаливания из ртути, превращенной ранее в красную ртутную землю, то образуется смесь, совершенно схожая с атмосферным воздухом и обладающая всеми его свойствами» [13, с. 49].
РАЗРАБОТКА НОВОЙ ТЕОРИИ КИСЛОТ
В 1776 г. А. Лавуазье проводил опыты по разложению и синтезу азотной кислоты и установил, что «содержавшийся в ней „эластичный флюид44 может быть получен из атмосферного воздуха».
102
НОВАЯ СИСТЕМА ХИМИЧЕСКИХ ЗНАНИЙ
Д. Лавуазье указал на ошибочные выводы Пристли: «Очевидно что не воздух составлен из азотной кислоты, как утверждал Пристли, а, напротив, азотная кислота является составленной из воздуха, и это одно замечание дает ключ к большому числу экспериментов, содержащихся в III, IV и V разделах 2-го тома Пристли» [14, с. 138]. Лавуазье показал, что растворение металлов в кислотах можно рассматривать как один из методов ана лиза кислот, способствующий выделению различных «эластичных флюидов».
Проводя аналогию с получением фосфорной и серной кислот из фосфора и серы, которые образуют кислоты при поглощении одной пятой части объема воздуха в процессе горения, Лавуазье сделал обобщение, что «кислоты отличаются одна от другой лишь основанием, соединенным с воздухом», обусловливавшим кислые свойства этого класса соединений.
А. Лавуазье выступил против существовавшего в эпоху флогистона представления о кислотах как веществах, которые содержат одну и ту же первичную кислоту, изменяющую кислые свойства химических соединений вследствие взаимодействия с другими веществами. Однако Лавуазье полностью не отказался от представлений о кислоте как носителе некоего первичного принципа. Он назвал основание чистого воздуха «кислым принципом» (oxigine) и высказал окончательно мнение, что все свойства кислот обусловлены присутствием в них этого принципа.
Представления Лавуазье о природе кислот были опубликованы в 1778 г. [15]. Он обосновал существование двух типов кислот, содержавших азот, но с различными пропорциями его в этих кислотах [16]. В это время Лавуазье рассматривал воздух «состоявшим из дыхательного воздуха и азота» (воздуха, лишавшего живые организмы жизни).
СОЗДАНИЕ КИСЛОРОДНОЙ ТЕОРИИ ГОРЕНИЯ
1777 год был, по выражению А. Фуркруа, «годом истинной славы Лавуазье в создании новой пневматической доктрины» [4, с. 38]. В этом году в нескольких статьях Лавуазье обосновал свои взгляды многочисленными новыми фактами, полученными в точных экспериментах. Изучая явление горения фосфора, Лавуазье вновь обратил внимание на количественный состав атмосферного воздуха, который заключает четверть «чистого воздуха», и три четверти «удушливого воздуха».
В статье, посвященной процессу дыхания, Лавуазье указывал на тождество явления горения фосфора с процессом горения рту-ти‘ «...Ртуть, окаливаясь поглотила лучшую, легко вдыхаемую часть воздуха, так что оставила лишь часть мефитическую, иди негодную для дыхания» [17, с. 176].
103
ГЛАВА ТРЕТЬЯ
Сравнив процесс горения с функцией дыхания, Лавуазье прп^ шел к выводу, что «...воздух, служивший известное время для поддерживания жизненной функции, имеет большое сходство с воздухом, в котором прокаливались металлы» [17, с. 177]. «Более глубокое исследование,— продолжает далее Лавуазье,— заставило меня заметить два весьма примечательных различия между обоими этими видами воздуха — я имею в виду воздух, который служил при прокаливании ртути, и воздух, которым дышал живой воробей: во-первых, уменьшение объема было значительно меньше у второго, чем у первого; во-вторых, воздух, оставшийся после дыхания, давал осадок с известковой водой, между тем как воздух, оставшийся после прокаливания, пе вызывал в нем никаких изменений» [17, с. 178].
А. Лавуазье назвал углекислый газ, действовавший па известковую воду, «меловой кислотой». При поглощении меловой кислоты известью или едкой щелочью Лавуазье обнаружил «ме-фитический воздух» (азот), оставшийся в опытах по прокаливанию ртути.
В процессе дыхания, согласно Лавуазье, «содержащаяся в атмосферном воздухе часть воздуха, безусловно, доброкачественная для дыхания, пройдя через легкие, превращается в воздухообразную меловую кислоту» [17, с. 180]. Эти представления легли в основу учения о физиологических процессах [18].
В 1777 г. Лавуазье опубликовал восемь статей-мемуаров, в которых рассмотрены различные проблемы горения и пневматической химии: поглощение объемов вдыхаемого воздуха фосфором, доказательство, что сернистая кислота представляет собой купоросную кислоту, лишенную части своего воздуха [19], соединение квасцов с веществами, содержащими уголь, сопровождающееся превращением «чистого» воздуха в меловую кислоту (СО2) [20], взаимодействие серной кислоты и пирита [21], а также первые обобщения о природе тепла, о состояниях тепла — испарении и охлаждении [22]. И, наконец, в 1777 г. Лавуазье опубликовал мемуар «О горении вообще» *, в котором представлена теория горения, полностью отбрасывавшая теорию флогистона [23].
«При горении тел постоянно наблюдаются четыре явления, что, по-впдпмому, должно считать законом, от которого природа никогда пе уклоняется»,— писал Лавуазье [23, с. 226]. Эти наблюдения показывают, что 1) при горении происходит выделение света или огня, 2) горение может происходить только в «чистом воздухе» (т. е. кислороде), 3) вес сгоревшего вещества увеличивается точно на количество поглощенного воздуха, 4) при горении неметаллических веществ в результате соединения с кислородом образуются кислоты, а при обжиге металлов — металлические извести (т. е. окислы) [23, с. 226—227].
1 См. Приложение VI.
104
ЙОВАЯ СИСТЕМА ХИМИЧЕСКИХ ЗНАНЙЙ — ....... ................................. *
Таким образом, в описываемый период уже сложилась кислородная теория горения веществ. Лавуазье проводил исследования процесса горения одновременно с другими учеными (Пристли, Ц1ееле, Байен и др.) и во многом использовал их опыт, ио только ему удалось на основании своих наблюдений представить . ясную картину процесса горения.
ЗАКОН СОХРАНЕНИЯ МАТЕРИИ *
Применение физических методов исследования к решению химических проблем оказалось чрезвычайно плодотворным в творчестве Лавуазье. «Пришло время поставить па место химию более строгим и обоснованным методом... указать до некоторой степени предел, которого достигли химические знания, и то, чему мы можем следовать с этого момента, и перейти с уверенностью к продвижению науки»,— писал Лавуазье [24].
Количественный метод Лавуазье базировался на аксиоме сохранения материи — фундаментальном принципе, который высказывался еще в древности в виде идеи о неуничтожаемости материи. Своими исследованиями Лавуазье экспериментально обосновал этот принцип.
В работах по истории науки отмечалось, что принцип сохранения материи безоговорочно принимался в естествознании. Французский ученый Э. Мариотт еще в 1678 г. отмечал, что «природа не делает ничего из ничего и материя вовсе не теряется (цит. по [25, с. 377]). М. В. Ломоносов сформулировал это положение в 1748 г. в письме к Л. Эйлеру, придав ему форму закона природы: «...Все встречающиеся в природе изменения происходят так, что если к чему-либо нечто прибавилось, то это отнимется у чего-то другого. Так, сколько материи прибавляется какому-либо телу, столько же теряется у другого... Так как это всеобщий закон природы, то он распространяется и на правила движения» [26, с. 183-185].
А. Лавуазье, впервые сознательно применив в химии положение И. Ньютона о том, что количество (масса) материи «определяется по весу тела, ибо оно пропорционально весу» (1686), считал взвешивание «лучшим средством для определения количества веществ, употребляемых в химических операциях и получаемых в результате опытов» [7, т. 2, с. 5]. По его мнению, «определение веса исходных веществ и продуктов до и после опытов — основа всего полезного и точного, что может быть сделано в химии» [7, т. 2, с. 11]. Поэтому он особенно заботился ° точности взвешивания. По его заказу знаменитый парижский мастер Фортэн изготовил весы неслыханной до того времени чув-
* Данный раздел написан С. А. Погодиным.
105
ГЛАВА ГРЕГЬЯ
i. g—i ТЛ« п . 1,1. -.5 . .. . —i I 14 Г i«m> 4.1—j ствительности. Вот некоторые данные об этих весах. Большие весы Лавуазье при нагрузке около 10 кг допускали взвешивание с точностью до 20 мг, средние при нагрузке 600 г были чувстви-тельны до 5 мг и малые при нагрузке 4 г позволяли определять вес до 0,1 мг, Лавуазье подчеркивал необходимость проверки разновесов, двойного взвешивания, охлаждения взвешиваемых тел до комнатной температуры. Он ввел в практику принятый и ныне обычай держать весы в отдельной весовой комнате, вдали от разъедающих газов и паров [7, т. 2, с. 11].
Таким образом, А. Лавуазье не только сделал весы главным инструментом химика, но и поднял технику взвешивания на небывалую до него высоту. Не ограничиваясь определением веса твердых тел и жидкостей, он впервые стал систематически измерять и вес газов. Измеряя газы по объему, он приводил этот объем к условиям, принятым им за нормальные, т. е. к давлению 28 дюймов и температуре 10° Реомюра [7, т. 2, с. 48—56], что имело следствием введение в обиход химика барометра и термометра. Он определил вес кубической единицы важнейших газов [15, т. 2, с. 250]. С современной точки зрения ошибки определений довольно велики; их относительная погрешность лежит в пределах от 3 (состав углекислого газа) до 20% (состав воздуха). Однако благодаря своей гениальной интуиции он сумел за этими погрешностями, обусловленными несовершенством измерительной техники, увидеть весовые законы химических превращений.
Весьма поучительно проследить, как постепенно и осторожно подходит Лавуазье в своем «Учебнике» к утверждению закона сохранения материи. Описывая сжигание фосфора в кислороде, он говорит, что устройство прибора не позволило собрать и взвесить белые хлопья продукта горения, и добавляет: «его вес можно найти только вычислением, предполагая, что он равен сумме весов кислорода и фосфора; ио как бы очевидно ни было это заключение, в физике и химии никогда не позволительно предполагать то, что можно определить прямыми опытами» [27, т. 1, с. 61]. Сделав опыт в другом приборе, Лавуазье путем взвешивания находит, что «вес нового вещества равен сумме весов сожженного фосфора и кислорода, который был им поглощен, что, впрочем, было легко предвидеть a priori» [27, т. 1, с. 63]. Лавуазье неоднократно возвращается к закону сохранения материи и постоянно подчеркивает его важность. Например, в главе о замазках он настаивает на необходимости герметически соединять сосуды, что имеет особенное значение с того времени, «когда опыты признаются удовлетворительными лишь постольку, поскольку вес полученных продуктов оказывается равным весу материалов, взятых для опыта» [7, т. 2, с. 146]. Наиболее законченную формулировку закона сохранения материи Лавуазье дает при описании процессов винного брожения: «Ничто не тво
106
НОВАЯ СИСТЕМА ХИМИЧЕСКИХ ЗНАНИЙ
рится ни в действиях искусства, ни в действиях природы, и можно положить в виде принципа, что во всякой операции количество материи одинаково до и после операции, что качество и количество начал (т. е. элементов) остаются теми же самыми, что происходят только превращения, изменения. На этом принципе основано все искусство делать опыты в химии: необходимо предполагать существование равенства или уравнения между началами (элементами) исследуемых тел и получаемыми из последних посредством анализа» [27, т. 1, с. 140—141].
В этих словах Лавуазье не только выражает закон сохранения материи, как это отмечают все историки химии, но высказывает и закон сохранения элементов при химических реакциях, иа что обычно не обращается внимания. Оба эти закона являются, как он пишет, основой для составления химических уравнений, т. е. материальных балансов всех химических превращений. Нет надобности разъяснять, какое первостепенное значение имеет знание материального баланса химических реакций для всех естественных наук, а также для химических и металлургических производств.
Третий основной закон химии — закон постоянства состава — Лавуазье, насколько мы могли установить, нигде не формулировал. Но его многочисленные определения количественного состава химических индивидов как путем анализа, так и путем синтеза, использование им постоянства состава углекислого газа и воды для анализа органических веществ были бы совершенно непонятны, если бы он не был убежден в постоянстве состава химических соединений.
ПРОБЛЕМА ХИМИЧЕСКОГО СРОДСТВА
Проблема химического сродства, связанная с образованием химических соединений, стала одной из основных в работах Лавуазье после установления теории горения. На основе этих работ Лавуазье утверждал, что существуют два типа растворения.
Следует отметить, что ранее Ломоносов также подчеркивал существование двух типов растворения. «Эти явления обусловлены противоположными причинами, и мы подозреваем, что металлы в кислых спиртах2 растворяются иначе, чем соли в воде»,— писал он в «Диссертации о действии химических растворителей вообще», опубликованной в 1750 г. в «Новых комментариях Петербургской Академии наук» на латинском языке и, следовательно, доступной европейскому читателю того времени [28, с. 349].
Мнение Ломоносова противоречило общепринятым в XVIII в. представлениям о растворении как едином процессе.
2 Кислыми спиртами в то время назывались кислоты.
107
ГЛАВА ТРЕТЬЯ
Позже П. Макер и А. Боме также различали растворение, при котором, согласно Макеру, происходит соединение цельных частей одного тела с цельными частями другого (dissolution) и разведение, а именно растворение солей в воде (solution).
В 1782 г. в работе «Общие соображения о растворении металлов в кислотах» Лавуазье, отметив выделение азотистого ангидрида при растворении металлов в азотной кислоте, подчеркнул, что «растворение металлов не есть простая операция, как полагали до сих пор» [29]. В этом же году Лавуазье опубликовал еще несколько мемуаров, посвященных обсуждению проблемы химического сродства и, в частности, сродства кислорода.
В «Мемуаре о сродстве кислородного начала к различным веществам, с которыми он способен соединяться» [30] Лавуазье составил таблицу сродства кислорода по отношению к различным веществам, отражавшую последовательность замещения одних веществ другими в процессе соединения их с кислородом, и высказал предположение о возможности количественного измерения сродства. Такое же предположение возникло при изучении природы теплоты совместно с II. Лапласом (1749—1827). Эта работа была вызвана практическим интересом к процессу окисления различных веществ, в частности железа, который описан в «Мемуаре о соединении кислородного начала с железом» [31]. Пред-метом исследования послужило изучение различных степеней окисления железа, из которого следовало, что сродство одного и того же вещества зависит от условий реакции и содействует образованию ряда химических соединений.
В «Начальном курсе химии» Лавуазье изложил свое представление о различных типах растворения: «При растворении солей в воде молекулы соли просто удаляются одна от другой, но ни соль, ни вода не испытывают разложения, и их можно найти в растворе в том самом количестве, что и до процесса. Можно то же самое сказать о растворении смол в спирте и спиртовых растворителях. При растворении металлов, напротив, всегда происходит или разложение кислоты, или разложение воды; металл окисляется, он переходит в окись; выделяется газообразное вещество, таким образом, каждое вещество после растворения (dissolution) не сохраняется в том состоянии, каким было ранее» [27, т. 2, с. 423-424].
Таким образом, в результате процесса dissolution, рассматриваемого как химическая реакция, могут быть получены новые химические индивиды, в отличие от процесса solution, тоже химического по своей природе, но не сопровождающегося образованием новых химических индивидов.
Практические задачи, связанные с изучением теплового эффекта при сжигании различных видов топлива, положили начало калориметрическим и термохимическим исследованиям Лавуазье. Совместно с Лапласом Лавуазье провел количественные опреде-
108
НОВАЯ СИСТЕМА ХИМИЧЕСКИХ ЗНАНИЙ
ления тепла, содержащегося в ряде веществ. Был применен калориметрический метод для измерения тепла, выделявшегося при горении фосфора, углерода, при образовании азотной кислоты, при дыхании [32].
Проблема передачи теплоты «в системе неравномерно нагре-. тЬ1Х тел, в особенности когда их смешение приводит к их разложению и к образованию новых соединений» [6, с. 189—190], интересовала ученых XVIII в. (М. В. Ломоносова, Г. В. Рихма-па, Дж- Блэка и др.).
А. Лавуазье и П. Лаплас отмечали, что «физики расходятся в представлениях о природе теплоты... Многие придерживаются теории теплового флюида... Другие... полагают, что теплота есть... результат незаметных движений молекул материи...» [32, с. 285]. Проблема теплоты изучалась Лавуазье и Лапласом в разных аспектах. Ученым принадлежит определение теплоемкостей многих неорганических и органических веществ с помощью изобретенного ими калориметра, теплотворности различных видов топлива, изучение проблемы теплового расширения тел и т. д. С этими исследованиями связано возникновение термохимии. Однако новаторские идеи в области теплоты сочетаются в этих работах с представлениями о невесомой материи. Лавуазье и Лаплас пришли к заключению, что изменения агрегатного состояния тел и калориметрические явления происходят под действием тонкой отталкивательной материи теплорода (calorique).
С исследованиями о теплоте Лавуазье и Лаплас связывали проблему химического сродства, предполагая в будущем возможность проводить количественные измерения сил сродства при измерении теплот реакций.
А. Лавуазье и П. Лаплас высказали важное для химии положение о том, что «теплота реакции — величина, характерная для образования каждого данного соединения» [6, с. 200].
В учение о сродстве Лавуазье ввел важное уточнение: изучение реакции осаждения требовало одних и тех же температурных условий. Лавуазье повторил опыты Т. Бергмана, определившего количества металлов, осаждавших одно и то же количество серебра из ее соли, и заключил, что по таким количественным соотношениям возможно определение количества кислорода, соединяющегося с равными количествами металлов [33, с. 46].
УСТАНОВЛЕНИЕ СОСТАВА ВОДЫ
В тот же период, когда Лавуазье изучал природу теплоты, °н провел исследования по разложению и синтезу воды.
Современники Лавуазье Г. Кавендиш, Дж. Пристли, Дж. Уатт наблюдали образование воды при сжигании «горючего воздуха» (водорода). Лавуазье в 1774—1775 гг. сжигал «горючий воздух»,
109
ГЛАВА ТРЕТЬЯ
названный Кавендишем флогистоном, но тогда он не заметил результатов опыта, ошибочно предположив, что должна в этом случае образоваться кислота.
В 1777 г. Уолтайр и Дж. Пристли, пропуская искру в сосуде, содержавшем водород и воздух, наблюдали оседание пара на стейках сосуда [33, с. 160]. Такое же наблюдение имеется у II. Макера (1778).
Г. Кавендиш пытался выяснить природу жидкости, образующейся на стенках сосудов при сжигании «горючего воздуха», и пришел к выводу, что «...вытекает безусловное основание считать, что дифлогистированный воздух (кислород.— 3. Ш.) представляет собой воду, лишенную своего флогистона, и что горючий воздух является... либо флогистированной водой, либо даже чистым флогистоном, по всей вероятности, первым» (цит. по [6, с. 163]).
В апреле 1783 г. Дж. Уатт высказал мысль, что вода состоит из «дефлогистированного воздуха» (кислорода) и флогистона.
Парижская Академия наук поручила Лавуазье повторить уже известные опыты в большом масштабе [6, с. 165—167]. Из этих опытов Лавуазье сделал вывод, что «вода отнюдь не является простым веществом, а она полным своим весом состоит из горючего газа и живительного воздуха» (цит. по [6, с. 170—171]). Но, помимо констатации этого факта, в работе Лавуазье установлено и весовое соотношение объемов реагирующих газов, которое примерно составляло 1 :2. Лавуазье тщательно изучал химические, физические и органолептические свойства полученной воды [34, с. 126].
После синтеза воды Лавуазье приступил к ее разложению, действуя на воду железными опилками. Затем совместно с Ж. Менье (1754—1793) Лавуазье провел термическое разложение воды в присутствии железа [35].
Опытами по разложению и синтезу воды Лавуазье завершил полемику по поводу ее состава, начатую Кавендишем в 1781 г. Эти исследования имели тогда не только теоретическое, но и практическое значение.
Сочетая теорию с практикой, А. Лавуазье окончательно доказал сложность состава воды путем анализа и в то же время дал дешевый технический способ получения водорода, необходимый для развития только что зародившегося воздухоплавания и со хранивший значение до наших дней.
Установление сложности состава воды, неопровержимо доказанное опытами Лавуазье, нанесло, как говорит Бертло, «последний удар теории флогистона, уже столь сильно потрясенной его предыдущими работами» [35а, с. 135]. Последние сомнения были рассеяны, и Лавуазье смог в 1783 г. выступить во всеоружии опытных данных с блестящими по форме и по содержанию «Размышлениями о Ллогистоне» [36, т, 2, с, 623]. В них он дает
ПО
ЙОЁАЯ СИСТЕМА ХИМИЧЕСКИХ ЗНАНИЙ
уничтожающую критику как первоначальной теории Шталя, так п ее последующих видоизменений и излагает свои собственные взгляды. Лавуазье пишет: «Моя задача была развить в этом ме-муаре теорию горения, опубликованную в 1777 г.; показать, что флогистон Шталя — воображаемое существо, которое он без всяких оснований принял составной частью металлов, серы, фосфора и всех горючих тел; что все явления горения и обжигания объясняются гораздо проще и легче без флогистона, чем при его помощи. Я не жду, что мои взгляды будут сразу приняты; человеческий ум привыкает видеть вещи определенным образом, и те, кто в течение части своего поприща рассматривали природу с известной точки зрения, обращаются лишь с трудом к новым представлениям; итак, дело времени подтвердить или опровергнуть выставленные мною мнения» [36а, с. 341].
РАЗРАБОТКА ПОВОЙ
ХИМИЧЕСКОЙ НОМЕНКЛАТУРЫ *
Необходимо сказать о коренном преобразовании химического языка, которое также явилось одним из важнейших следствий работ Лавуазье.
Химики флогистического периода получили от своих предшественников — алхимиков и иатрохимиков — не только богатый запас фактических сведений о различных веществах, но и великое множество названий для их обозначения. Эти названия были длинны, трудны для запоминания и неудобны для произношения; одно и то же вещество имело по нескольку названий. Флогистики также немало способствовали этому номенклатурному хаосу. Па-пример, во времена Лавуазье для сульфата меди существовало четыре названия, для карбоната магния — десять и для углекислого газа — двенадцать. Никакой системы в химической номенклатуре не было. Разобраться во всей этой путанице становилось все более и более трудным. Единичные попытки улучшить положение не давали ощутимых результатов.
В 1782 г. Луи Бернар Гитон де Морво (1737—1816), юрист по профессии, много занимавшийся химией, составил по поручению редакции «Методической энциклопедии» проект реформы химической номенклатуры. В то время он еще был сторонником теории флогистона, и поэтому предложенная им номенклатура оказалась не соответствующей развитию науки. Вскоре к воззрениям Лавуазье стали присоединяться видные французские математики: Пьер Симон Лаплас (1749—1827), Жак Антуан Кузен (1739—1800), Гаспар Монж (1746—1818), Шарль Огюст Вандермонд (1735—1796). Их примеру последовали и химики. Первым был Клод Луи Бертолле (1748—1822), признавший в 1785 г.
* Данный раздел написан С. А. Погодиным.
111
ГЛАВА ТРЕТЬЯ
флогистон «ненужной гипотезой» [37], в следующем году к Лавуазье примкнули Гитон де Морво и Антуан Франсуа Фуркруа (1755—1809). Эти трое ученых совместно с Лавуазье приступили летом 1786 г. к разработке новой номенклатуры. 18 апреля и 2 мая 1787 г. они доложили свои предложения Академии наук, которая назначила специальную комиссию для их рассмотрения. Все четыре члена комиссии: Антуан Боме, Луи Клод Кадэ, Жан Дарсэ и Бальтазар Жорж Саж были флогистиками. Поэтому, не одобряя, но и не порицая прямо проект новой номенклатуры, они предложили опубликовать ее, но так, «чтобы нельзя было из этого заключить, принимается или опровергается Академией наук новая теория; беспристрастие, которое всегда было основой поведения Академии, требует выждать испытания времени и суда физиков» [38, с. 259]. Такое уклонение от прямого высказывания весьма типично для характеристики отношения современников к взглядам Лавуазье.
Основные принципы, которыми руководствовался Лавуазье с сотрудниками, изложены им с большим мастерством в докладе «О необходимости преобразовать и усовершенствовать химическую номенклатуру» [38, с. 1—26] и кратко повторены в «Предварительном рассуждении» [27, т. 1]. Следуя взглядам очень близкого к материализму философа-сенсуалиста Этьенна Бонно де Кондильяка (1715—1780), Лавуазье считает, что «языки —это настоящие аналитические методы, при помощи которых мы продвигаемся от известного к неизвестному, до некоторой степени подобно математикам» [38, с. 6]. «Но если языки — настоящие орудия, которые люди создали себе для облегчения действий своего ума, то важно, чтобы эти орудия были возможно лучшими, и заниматься их усовершенствованием значит работать для успеха наук» [38, с. 8]. «Хорошо построенный язык... повлечет за собою необходимую революцию в способе преподавания; он не позволит преподающим химию уклоняться от пути, по которому идет природа; придется или отбросить номенклатуру, или без сопротивления следовать по начертанной ей дороге. Так логика наук существенно зависит от их языка» [38, с. 15]. «Настало время освободить химию от всякого рода препятствий, замедляющих ее успехи, внести в нее истинный дух анализа, и мы достаточно убедительно показали, «что эта реформа должна быть произведена усовершенствованием языка» [38, с. 17].
Новая химическая номенклатура прежде всего делит вещества па простые и сложные. В первой редакции (1787) в число простых веществ были включены радикалы (этот термин предложен Лавуазье) известных тогда 12 органических кислот, а также едкое кали, едкий натр и аммиак. Для сложных веществ, состоящих из двух простых, устанавливаются названия из двух слов: одно из них обозначает принадлежность данного «индивида» [38, с. 20] к определенному классу или роду, другое — к определенно
го
НОВАЯ СИСТЕМА ХИМИЧЕСКИХ ЗЙАЙЙЙ
му виду. Например, все кислоты, по представлениям Лавуазье, состоят из кислорода — начала кислотности — и «основания» или «радикала» кислоты — азота, серы, фосфора, углерода и др. Поэтому названия кислот составляются из существительного «кислота» и прилагательного, образованного от названия радикала с • окончанием «ique» (по-русски «ная», например, серная кислота). Если известны для одного и того же «радикала» две кислоты, то видовое название «менее насыщенной» кислородом получает окончание «еих» (по-русски «истая», например, сернистая кислота). Не будем останавливаться на номенклатуре окислов, сернистых, фосфористых и тому подобных двойных соединений и перейдем к названиям солей. Каждая средняя или нейтральная соль (это понятие ввел еще Руэль в 1744 г.) также имеет название, указывающее и родовую, и видовую принадлежность соли. Родовое название имеет окончание «ат» или «ит». Например, соли серной кислоты имеют родовое название «сульфаты», сернистой кислоты «сульфиты». Видовые названия производятся от названия основания соли: сульфат магнезии, сульфат барита, сульфит извести и т. д.
Не вдаваясь в дальнейшие подробности, отметим, что основная мысль новой номенклатуры, заключающаяся в том, чтобы каждый химический индивид имел одно определенное название, характеризующее его химическую функцию и состав, оказалась весьма плодотворной. Она внесла порядок и стройность в обозначения веществ, позволила систематизировать громадный фактический материал, чрезвычайно облегчила изучение химии. Несмотря на все видоизменения, которым, начиная с Берцелиуса (1811), химическая номенклатура подвергалась на протяжении свыше полутораста лет, ее основные принципы сохранились до наших дней.
ЗАВЕРШЕНИЕ ХИМИЧЕСКОЙ РЕВОЛЮЦИИ.
УТВЕРЖДЕНИЕ АНТИФЛОГИСТИЧЕСКОЙ ХИМИИ
Изложение антифлогистической системы в целом содержится в изданном в 1789 г. «Начальном курсе химии» А. Лавуазье *.
* Несмотря на напряженную политическую обстановку, эта книга была уже в 1789 г. выпущена вторым изданием. В нем, в отличие от первого, каждый том имеет отдельную пагинацию. Кроме того, к нему в виде третьего тома приложена система химической номенклатуры. В 1794 г. вышло второе издание, являющееся перепечаткой первого, а в 1801 г.— третье, дополненное некоторыми мемуарами Лавуазье. «Начальный учебник химии» Лавуазье неоднократно издавался и на иностранных языках: голландском (1789, 1792, 1795, 1801), английском (1790, 1793), итальянском (1791) п немецком (1792 и 1803). Это свидетельствует о распространении новых взглядов в других западноевропейских странах. Конечно, как и во Франции, учение Лавуазье было принято не сразу и не без борьбы. — Прим. С. А. Погодина.
113
ГЛАВА ТРЕТЬЯ
el£mentaire
D Е С Н I М I Е,
PRESENTE DANS UN ORDRE NOUVEAU
ET D APRES LES DECOUVERTES M0DERNE1,;
Avec Figures :
Par M. La rc> t s r r ft , de t'Acadcmie des Sciences, de la Svcleic Rayale de Medecuie, des Soeietes d' Agriculture de Paris & d’Orleans , de la Saciete Roy ale de Lvndres , de I'inflituc de Bologne , de la Societe Helvetians de В a fie , de celles de Philadelphie, Harlem , Manc/iejler , Padoue , &c.
TOME PREMIER.
A PARIS,
Chez Си Chet, Libraire, rue A hotel Scrpente.
M. D C G L X X X Г X.
Svus Ic Privilege de Г Academic des Sciences & de la Socieie Royals de Mcdecinc,
Титульный лист книги А. Лавуазье ((Начальный курс химии» (1789)
Содержание двух томов учеб, пика Лавуазье распределено в трех частях, из которых две части представлены в первом томе.
В первой части Лавуазье, основываясь на новых физических и химических представлениях, дал обоснование антифло-гистической химии. Новизна его взглядов проявилась прежде всего в представлении о материи как субстанции, способной существовать в различных агрегатных состояниях. Эти взгляды опровергали ранее используемые представления о существовании постоянных твердых тел, жидкостей и газов.
А. Лавуазье писал, что в твердом состоянии молекулы удерживаются друг около друга силами притяжения, превышающими силы отталкивания. В жидком состоянии молекулы
удалены друг от друга настолько, что силы притяжения становятся равными силам отталкивания, а превращению жидкости в газ мешает атмосферное давление. В газообразном состоянии «упругий флюид теплород» способствует тому, что молекулами управляют в основном силы отталкивания. Отсюда следует, что в основе воззрений Лавуазье лежали ньютоновские корпускулярные представления о структуре материи.
Лавуазье отметил, что «отсутствие в «Начальном курсе химии» главы о составных и элементарных частях тел неминуемо вызовет удивление, но я позволю себе заметить, что стремление считать все тела природы состоящими лишь из трех или четырех элементов происходит от предрассудка, перешедшего к нам от греческих философов». Представления об аристотелевских элементах имели место в учебниках видных флогистиков эпохи Лавуазье П. Макера и А. Боме. «Курс химии» Лавуазье был первым, в котором не было отведено места флогистону.
В «Курсе химии» Лавуазье дал определение элемента. Он отождествлял понятия «элемент» и «простое вещество» # скептически относился к мнениям «о числе и природе элементов», «если названием элементов обозначить простые и неделимые молекулы, составляющие тела... Если же, напротив,— продолжал
114
НОВАЯ СИСТЕМА ХИМИЧЕСКИХ ЗНАНИЙ
Лавуазье — мы свяжем с названием элементов или начал тел лредставление о последнем пределе, достигаемом анализом, то все вещества, которые мы еще не смогли никаким образом разложить, являются для пас элементами» [27, т. 1, с. XVI—XVIj].
А. Лавуазье не применял корпускулярные представления к оп- , ределению понятия «элемент», и в этом вопросе пошел по пути определения элемента как вещества, обладавшего определенным характерным признаком — неразложимостью его существовавшими тогда аналитическими методами исследования. Понятие «элемент», данное Лавуазье, оказалось широким, так как включало не только реальные химические элементы, но и не разложенные тогда химические соединения — многие извести металлов (окислы), различные земли. Лишь едкие щелочи не были включены в таблицу элементов. Лавуазье предполагал, что они сложны по составу [39, с. 21].
Не будучи уверенным в фактах, касавшихся «избирательных притяжений», т. е. химического сродства, Лавуазье не включил в свой «Курс» этих вопросов, составлявших содержание той части химии, которая, по словам Лавуазье, «наиболее способна со временем стать точной наукой».
Изложив общие представления об агрегатных состояниях тел, Лавуазье переходит в первой части учебника к вопросам теплоемкости, рассматриваемой как «способность тел содержать в себе материю тепла». Здесь Лавуазье обращается к форме молекул, их размерам и расстоянию между ними, которое определяется «соотношением между их взаимными силами притяжения и силой отталкивания, вызванной теплородом». Переходя затем к вопросу образования атмосферы посредством испарения различных жидкостей, Лавуазье детально излагает опыты по анализу и синтезу воздуха атмосферы, употребляя слово «газ» вместо «воздухообразный флюид».
Одна из глав первой части «Курса» посвящена «разложению» кислородного газа серой, фосфором и углем с образованием кислот. Речь в ней идет о горении этих простых веществ, при котором образуются ангидриды кислот. Лавуазье ошибочно предполагал, что кислородный газ — сложное вещество, состоящее из «кислородного начала», соединенного с теплородом. В результате горения элемент (фосфор, сера и т. п.) соединяется с кислородным началом, освобождая теплород [6, с. 221]. По аналогии с процессом образования кислот Лавуазье объясняет «разложение кислородного газа» металлами с образованием их окислов. Затем Лавуазье излагает опыты по разложению и синтезу воды.
Описание термохимических измерений связано с рассмотрением процессов горения и окисления.
В дальнейшем Лавуазье рассматривает взаимодействие разных веществ, отличавшихся различным сродством к кислороду. Он посвящает также ряд глав анализу органических соединений,
115
ГЛАВА ТРЕТЬЯ
на основе которого описывает природу явлений брожения, фер. ментации, гниения.
От рассмотрения химического взаимодействия и образования химических соединений Лавуазье переходит к щелочам, еще не разложенным в то время, и веществам, способным образовывать соли. И, наконец, Лавуазье составляет таблицу простых тел, в которую включены 16 металлов, три неметаллических простых вещества (сера, фосфор, углерод, к ним Лавуазье отнес муриевый, плавиковый, борный радикалы, содержавшие хлор, фтор, бор), солеобразующие (землистые) вещества — известь, магнезия, барит, глинозем и кремнезем.
В литературе отмечено, что первая таблица простых веществ и первая их классификация, данные Лавуазье в его «Учебнике химии», представляют «документ огромной исторической важности» [36а, с. 342].
А. Лавуазье делит простые вещества на четыре группы: 1) простые вещества, принадлежащие к трем царствам природы, которые можно считать элементами тел: свет, теплотвор, кислород, азот, водород; 2) простые неметаллические вещества, окисляющиеся и дающие кислоты: сера, фосфор, углерод и радикалы — муриевый, плавиковый, борный; 3) простые металлические вещества, окисляющиеся и дающие кислоты: сурьма, серебро, мышьяк, висмут, кобальт, медь, железо, марганец, ртуть, молибден, никель, золото, платина, свинец, вольфрам, цинк; 4) солеобразующие землистые простые вещества: известь, магнезия, барит, глинозем, кремнезем.
Необходимо отметить, что в таблицу простых веществ Лавуазье включил и так называемые «невесомые начала» — свет и теплотвор, к которым прибегали физики вплоть до середины прошлого века для объяснения световых и тепловых явлений. Радикалами Лавуазье называл вещества, дающие при соединении с кислородом кислоты; он ошибочно считал, что соляная (мурис-вая) и плавиковая кислоты содержат кислород. В число простых веществ не были включены «постоянные щелочи», т. е. едкие кали и натр, так как Лавуазье предвидел их сложность (цит. по [36а, с. 343]). Он предполагал, что и «земли» являются не простыми веществами, а окислами, но, верный своему принципу: «не смешивать того, что я даю как опытные истины, с тем, что еще гипотетично» [там же], он поместил «земли» в список простых веществ. Лавуазье писал: «Можно ожидать, что эти земли вскоре перестанут причисляться к числу простых веществ. Они — единственные из всего данного класса веществ, которые не имеют охоты соединяться с кислородом, и я весьма склонен думать, что эта индифферентность по отношению к кислороду... происходит оттого, что они уже сами по себе насыщены кислородом. Земли, рассматриваемые с такой точки зрения, могли бы оказаться окислами, т. е. простыми телами, уже окисленными до известной
116
НОВАЯ СИСТЕМА ХИМИЧЕСКИХ ЗНАНИЙ
TABLEAU DES SUBSTANCES SIMPLES.
Norns nouveaux.
Noms anc. correspond.
Lumiere
Substances simples quiap-
aux trois re~ gnes, et qu’on
Oxygene
commc les ele-
Azote
Substances mples non
Hydrogene
Soufre....
Phosphore.
Carbone. .
oxidablcs et acidifiablcs.
Radical boracique.
Antimoine........
Arsenic. .. Bismuth... Cobolt.... Cuivre.... Etain....
Fer......
liques oxida- Manganese bits et acidi-fiables.
Substances simples vnital-
Mercure.. Molybdene Nickel....
Platine Plomb.
Substances
Chaux... Magn^sie
fables Мии- jAlumine ses, Ш
Silice
I Lumiere.
< Chaleur.
к Principe de la chaleur.
jFluide igne.
\ Feu.
/Matiere du feu et de la
к chaleur.
f Air dephlogistique.
I Base de Fair vital, Gaz phlogistiqu£.
« Mofete.
Base de la mofete.
< Gaz inflammable.
I Base du gaz inflammable Soufre.
Phosphore.
Charbon pur.
Inconnu.
Inconnu.
Inconnu.
Antimoine.
Arsenic.
Bismuth.
Cobolt.
Cuivre.
Etain.
Fer.
Manganese.
Mercure.
Molybdene.
Nickel.
Platine.
Plomb.
Zinc.
Terre calcaire, chaux.
t Mkgnesie,base dusel d’Ep-1 som.
| Barote, terre pesante.
5 Argile , terre de Falun, c base de Falun.
r Terre siliceuse, terre vi-
Таблица простых тел Лавуазье
ГЛАВА ТРЕТЬЯ
степени». Это предположение позже, как известно, подтвердилось [6, с. 225].
Дав совершенно ясное определение понятия «простое веще, ство», Лавуазье, однако, обошел молчанием определение такого важного понятия, как «химическое соединение». В отличие от своих современников, он для обозначения сложных веществ, со-стоящих из двух или нескольких простых, постоянно пользуется термином «соединение» (combinaison) вместо термина «смешанное тело» (corpo mixtum), под которыми подразумевались и химические соединения, и механические смеси, и растворы [36а с. 343].
Во второй части «Курса» автор предложил новую номенклатуру химических соединений. Название веществ, состоявших из двух простых, должно указывать, с одной стороны, на принадлежность данного индивидуального вещества к определенному классу или роду, с другой — на принадлежность к элементу или радикалу, образовавшему это соединение (серная кислота, азотная кислота и т. п.).
А. Лавуазье привел названия веществ в зависимости от степени окисления простого тела, соединявшегося с кислородом. Название окись серы (oxide de soufre) отражает первую степень окисления серы, сернистая кислота (acide sulfurique) — вторую степень, серная кислота (acide sulfureux) — третью степень и окисленная серная кислота (acide sulfurique oxygene) — четвертую степень окисления серы. Введенные Л. Гитоном де Морво, А. Фуркруа, А. Лавуазье названия — окись (oxide), для солей — нитрат, оксалат, сульфат и многие другие сохранены до сих пор; «мефитический воздух» получил новое название — азот.
Во второй части книги, в разделах об «Окислении простых веществ», Лавуазье охарактеризовал природу горения, опираясь на свои работы по исследованию теплоты [40, с. 143—145]. Эта часть труда особенно интересна тем, что она содержала первые идеи о кинетике химических реакций [6, с. 226—227].
Третья часть «Курса» (второй том) представляет собой руководство по практике экспериментирования в химии. Оно дает «подробное описание всех относящихся к современной химии приемов» и приборов, которые должны отличаться, по мнению Лавуазье, исключительной тщательностью и точностью [6, с. 228] • «Начальный курс химии» Лавуазье был переиздан в 1791 и 1801 гг. и, переведенный на другие европейские языки, способствовал распространению новых химических знаний.
В письме 2 февраля 1790 г., написанном Лавуазье знаменитому физику В. Франклину, охарактеризовано положение в химик после выхода «Начального курса химии» в свет: «Мне кажется, что химия, представленная в этом виде, гораздо проще, чем ста' рая химия, и молодые люди, головы которых не заняты еШе никакой системой, с большой жадностью схватывают эту новУ10
118
Новая сйстёма химических зйаний
доктрину. По им опа кажется простою, между тем как старые хцмики ее отвергают и испытывают даже больше трудностей при восприятии этой повой доктрины, чем все те, кто вообще не изучал химию.
французские ученые разделены в этот момент между старой и новой доктринами. На моей стороне г. Де Морво, г. Бертолле, г. Де ФуРкРУа, г- Де ла Плас, г. Монж и вообще физики в Академии. Ученые Лондона и Англии также незаметно покидают доктрину Шталя, но немецкие химики прочно держатся за нее.
Вот такова революция, которая совершилась в химии*...» (цит. по [6, с. 269-270]).
Как мы уже отмечали, в 1785 и 1786 гг. к взглядам Лавуазье примкнули Бертолле, Гитон де Морво и Фуркруа; последний (в начале 1787 г.) впервые начал с большим успехом преподавать химию по новой системе [36а, с. 351]. Они показали пример другим французским химикам. Назовем из них только Жана Антуана Клода Шапталя (1756—1832), выдающегося химика-технолога и государственного деятеля, владельца химических заводов, автора очень распространенного учебника химии. В 1791 г. Лавуазье писал Шапталю: «Только пожилые люди, которые уже не имеют мужества переучиваться или не могут заставить свое воображение подчиниться новому порядку вещей, придерживаются учения о флогистоне. Вся молодежь приняла новую теорию, и из этого я заключаю, что революция в химии завершена» (цит. по [36а, с. 351]).
Результат исследований, приведший к коренному изменению в химических представлениях, возможно подытожить следующим образом: ниспровержение теории флогистона на основе кислородной теории горения; обоснование сущности процессов горения, дыхания, окисления и их тождества; обоснование принципа сохранения материи с помощью химических уравнений; создание новой химической номенклатуры.
К концу XVIII столетия воззрения Лавуазье стали общепризнанными.
Антифлогистическая химия, сформулировавшая понятия о простых веществах и химических соединениях как химически индивидуальных веществах, подготовила почву для возникновения химической атомистики.
* Открытие и изучение углекислого газа и кислорода оказало огромное влияние на развитие естествознания. Решение таких кардинальных проблем, как проблемы дыхания, окисления, фотосинтеза, пищеварения, обмена веществ, стало возможным только после фундаментальных открытий Лавуазье. Последующие страницы истории изучения этих проблем богаты примерами широкого обмена идеями и объектами исследования в тРУДах химиков и биологов [40].— Прим. отв. ред.
119
ГЛАЁА ЧЕТВЁРТАЯ
РАЗВИТИЕ МЕТОДОВ
ХИМИЧЕСКОГО АНАЛИЗА И АНАЛИТИЧЕСКОЙ ХИМИИ* 1
ВВЕДЕНИЕ
Аналитические определения в том смысле, который охаракте-ризован ниже, проводились уже в глубокой древности. Некоторые описания мы можем найти у средневековых авторов, сочинения которых рассмотрены в вышедшем ранее томе «Всеобщей истории химии» — «Возникновение и развитие химии с древнейших времен до XVII века». С XII в. н. э. все большее значение в разработке методов анализа начинает играть горное право, стимулировавшее государственный контроль за частной добычей полезных ископаемых.
Слово анализ по-гречески означает разложение, расчленение. В научной литературе этот термин встречается впервые у Аристотеля, но не в применении к химии, а скорее к логике. Р. Декарт применил термин «аналитический» к своей новой геометрии. В применении к химии слово анализ в современном смысле впервые использовал Р. Бойль: «The Chemical Analysis of Seed Pearles» — «Химический анализ мелкого жемчуга» — такой заголовок встречаем мы в одной из его работ [6]. В XVIII в. термин «химический анализ» постепенно входит в научную литературу, а в 1801 г. во Фрейберге вышло в свет «Руководство по химическому анализу минеральных тел», написанное профессором местной Горной академии В. А. Лампадиусом [7]. В этом сочинении, кстати сказать, впервые появляются и термин аналитическая химия, и ее определение как деятельности по разложению минеральных, в данном случае, тел с целью анализа, т. е. определения составных частей.
Примечание. Литературу см. стр. 403—406.
1 Истории аналитической химии посвящено немало публикаций в мировой и отечественной литературе. В данной главе па основании обобщающих [1—5] и ряда оригинальных химико-аналитических работ [8—21] рассматривается история развития методов и принципов химического анализа в том плане, в каком оно способствовало формированию нового понятия об элементах и формулированию в XIX в. основных законов химий.
120
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
Так в истории химии утвердились термины химический анализ и аналитическая химия, однако сами по себе химико-аналитические исследования начались в глубокой древности, и почти сразу же сформировались три основные химико-аналитические задачи:
— идентификация или отождествление испытуемого вещества с уже известным;
— полное или частичное качественное определение состава испытуемого вещества;
— полное или частичное количественное определение содержания составных частей в исследуемом веществе.
В сущности те же задачи стоят и перед современным химическим анализом.
Итак, химический анализ в общем смысле — это изучение химического состава исследуемых веществ. Такое определение представляется, однако, неполным, так как надо еще определить, что понимается под словом состав.
Уже древние мыслители различали несколько уровней состава. Наиболее отчетливую форму они имеют у Аристотеля, у которого в иерархии состава мы усматриваем уровень качеств (сухость, влажность, тепло, холод), уровень элементов (огонь, воздух, вода, земля), уровень «химически однородных» тел— гомеомерий,— образующихся в результате химического соединения—миксиса, и, наконец, уровень ангомеомерий — сложных тел, состоящих из ряда гомеомерных образований так, как состоит тело животных из мяса, костей, сухожилий, кожи и т. п.
Аристотель отличал «химическое соединение» — миксис от смеси — синтезис.
В миксисе составные части вступили во взаимодействие и существуют лишь потенциально, т. е. в том смысле, что могут быть выявлены путем разложения миксиса — путем анализа. В синтезисе составные части лишь смешаны, а не связаны. Отдельные частички этой смеси могут обладать различными свойствами, и составные части смеси существуют, таким образом, не потенциально, а актуально [8, с. 135—165].
В наших современных представлениях нетрудно найти аналогии аристотелевской иерархии. Это естественно, поскольку в основе умозрительных представлений Аристотеля лежали пытливые наблюдения над объектами природы.. Переходя к кругу современных понятий, мы усмотрели бы дохимический уровень организации материи — до элементарных частиц включительно; Уровень элементов, т. е. атомов, уровень химических соединений н Уровень систем, состоящих из различных химических соединений или частиц, разделенных поверхностями раздела, т. е. уровень фаз. В соответствии с этим мы различаем сегодпя следующие уровни химического состава: элементный (и в его рамках
ГЛАВА ЧЕТВЕРТАЯ
изотопный), вещественный (или компонентный) и, наконец, фазовый. С углублением представлений о природе химических веществ еще в позднем средневековье начал выявляться дополнительный уровень состава, который мы в настоящее время называем структурно-групповым [9].
Как читатель увидит далее, на любых этапах истории химии никогда не отдавалось заметного предпочтения выявлению какого-либо одного из этих видов состава. Так, при исследовании рудных пород с древнейших времен всегда изучали фазовый состав руды, т. е. присутствие в монолитной массе рудной породы определенных рудных минералов и их спутников. При исследовании золы растений и ее пригодности для изготовления стекла с давних времен интересовались компонентным составом золы, т. е. содержанием вымываемого водой компонента — поташа. Компонентный состав — разнообразие кристаллизующихся солей — изучали и при анализе минеральных вод, и при извлечении растительных масел, и во множестве других случаев. Понятие о групповом составе определенно сформулировалось после открытия минеральных кислот. С тех пор стали аналитически выявлять, например, в солях начало серной кислоты — «серный спирт» (spi-ritus vitrioli), начало азотной кислоты — «селитряный спирт», начало соляной кислоты — «соляной спирт» (spiritus salis) и т. д.
Таким образом, отнюдь не только с XIX в., но и во всей истории химии перед химическим анализом стояли задачи выявления фазового, компонентного, группового и, разумеется, элементного состава, столь существенного сначала для алхимии, а затем и теоретической химии.
Разумеется, любые представления о химическом составе в каждом историческом периоде основывались на господствующих теоретических представлениях. В эпоху алхимии несомненную роль играли ртутно-серная, а впоследствии ртутно-серно-соляная концепции.
Затем наступила эпоха флогистона, когда, например, металлы считались сложными веществами, а их кислородные соединения — «земли» или «основания металлов» — простыми. Соответственно и задачу анализа сводили к определению «земель» (см., например, [10, рус. пер., с. 115—119]).
Алхимики, пытаясь выделить свои «элементы» и добиться желаемой трансмутации, разработали различные способы разложения вещества и последовательность операций разложения и превращения. Их операции включали и прокаливание, и обжиг веществ, их растворение и выделение из растворов (подробнее см. [8, ч. II, в частности с. 240, с. 347]). Все эти процедуры подхватывали и применяли ремесленники, но с другой целью: им надо было выделить золото, серебро, медь, железо, а не эфемерные элементы. Тем не менее очень долго, в некотором роде вплоть до Лавуазье, считалось, что компоненты и фракции, вьь
122
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
еденные из исходных веществ путем разложения, как бы просуществуют в разлагаемом теле и являются его истинными составными частями. II лишь после формирования нового понятия об элементах, открытия кислорода и появления учений о стехиометрических отношениях, об атомах, а затем и о молекулах, у аналитической химии появляется надежная теоретическая основа.
Только постоянно имея в виду историческое изменение основных представлений о химических веществах и их составе, мы можем обратиться к собственной истории химического анализа, памятуя также, что анализ всегда служил познанию состава веществ и развитие химического анализа как ветви химии заключается в развитии аналитических методов.
РАЗВИТИЕ ХИМИЧЕСКОГО АНАЛИЗА В XVIII ВЕКЕ
В XVI—XVII вв. наиболее активные аналитические исследования велись в традиционных горнорудных областях центральной Европы и некоторых других пунктах с развитой промышленностью и торговлей. В XVIII в. крупная школа химиков-аналитиков возникает в Швеции. Эстафету европейских аналитических исследований передал туда немецкий ученый И. Кункель.
Горнохимическая служба была в Швеции и ранее. Так, в 1637 г. решением тогдашнего регента и крупного политического деятеля Швеции Акселя Оксеншерна были основаны Берг-коллегия и химическая лаборатория. По мнению историков химии [22, с. 195; 23, с. 57—62], это была первая химическая лаборатория в Северной Европе. Деятельность ее никакими достижениями отмечена не была до тех пор, пока в 1683 г. руководителем не стал выдающийся шведский ученый-энциклопедист, врач и химик Урбан Иерне (1641—1724). С этого момента ее деятельность оживилась, были проведены многочисленные исследования почв, минералов, руд и минеральных вод. Исходя из представлений о сродстве веществ Иерне высказал и в своей практике придерживался точки зрения, что если вещество растворяется в щелочи, то оно должно высаживаться кислотой, и наоборот; вода противоположна жирам, и вещества, растворимые в жирах, осаждаются от прибавления воды и т. д. В 1712 г. Иерне издал первый шведский учебник химии.
Иоганн Кункель (1613—1703), сын алхимика при дворе герцога Голштинского, был придворным аптекарем и химиком в Разных немецких княжествах, руководил стекольным производном. В 1688 г. его пригласили в Швецию на должность горного советника. В 1679 г. при участии Кункеля вышло в свет Руководство по стеклоделию, в котором Кункель, описывая па
123
ГЛАВА ЧЕТВЕРТАЯ
яльную трубку, указал на полезность применения этого ипстру, мента при пробирных анализах металлов и рудных минералов. Он подчеркивал эту полезность и в своих химических сочинениях.
Возникновение способа химического анализа с помощью паяльной трубки
Вообще говоря, паяльная трубка известна с древних времен. Мы видим ее изображение и в эллинистическом Египте, и па фресках Помпеи [14, с. 127]. Тогда и впоследствии ее применя-ли в ювелирном деле. В практике алхимиков опа тоже была, по крайней мере в XVI в. Так, ее применение для запаивания ампул можно видеть на гравюрах 10 и 13 «Немой книги», издать ной до руководства Кункеля и отражающей опыт алхимиков более давнего времени [8, с. 256—257].
К. Ф. Платтнер [54, с. 1] указывал, что еще датский ученый XVII в. Расмус (Эразм) Бартолин (1620—1698) пламенем от паяльной трубки накаливал исландский пшат (СаСО3), превратив его в негашеную известь. Наконец, в самом металлургическом производстве дутье мехами было в ходу с древних времен. Тем не менее предложение Кункеля использовать паяльную трубку для химических реакций в малом масштабе сыграло в истории аналитической химии выдающуюся роль.
Считается, что впервые совету Кункеля последовал Г. Шталь, который в 1703 г. опубликовал сообщение о получении металлической сурьмы и свинца из их «известей», т. е. окислов, нагреванием с помощью паяльной трубки на небольшом кусочке древесного угля. 40 лет спустя к этому методу обратились и другие исследователи, в том числе А. Маргграф, но наибольшую активность в разработке метода и его применении проявили шведские ученые, получившие паяльную трубку от самого Кункеля.
А. Ф. Кропштедт последовательно применял паяльную трубку для практического анализа минералов [27]. В 1770-х годах Т. Бергман и И. Г. Ган разработали аппаратуру и методику анализа с помощью паяльной трубки уже не только для минералов, но и для выделяемых в лаборатории химических соединений [34]. Эти методы были проверены, усовершенствованы п подробно описаны в 1812 и 1818 гг. Я. Берцелиусом [ 24, 25], в середине XIX в. Э. Гаркортом [52, 53] и К. Ф. Платтнером [54]. Во второй половине XIX в. анализ с помощью паяльной трубки постепенно уходит из практики химико-аналитических лабораторий, но не вследствие появления горелки Бунзена, как считают некоторые историки химии, а потому, что анализ «мокрым путем» оказался более чувствительным и точным. ПаяльпаЯ трубка остается, одпако, в практике анализа руд и минералов и в XX в.
124
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
ф. Шабадвари [2, с. 67] полагает, что претензии шведских исследователей считать паяльную трубку чисто «шведским методом» преувеличены. Иоганн Андреас Крамер (1710—1777) в £739 г., А. Маргграф в 1740 г., ученик Г. Шталя И. Г. Потт (1692—1777) в 1746 г. и последующих годах также употребляли паяльные трубки, хотя и не систематически. Впрочем, Крамер и следовавший за ним К. Ф. Циммерман применяли не устное, а принудительное дутье, т. е. род малых мехов. Г. Копп [3, с. 45] считал, что, несмотря па наличие предшественников, применение паяльной трубки для собственно химических опытов принадлежит все-таки шведам. Среди шведских же исследователей XVIII в. следует упомянуть еще и Свена Ринмана (1720—1792), использовавшего в 1746 г. паяльную трубку для определения цинка.
Пробирный анализ руд и возникновение химической минералогии
Быстрое развитие в XVIII в. горнорудного дела и торговых связей потребовало расширения пробирной службы во всех европейских странах. Естественно, потребовались и специальные руководства по минералогическому и пробирному анализу. Они появлялись в течение всего столетия, причем некоторые переводились на многие европейские языки.
Так, во Фрейберге в 1725 г. вышла книга И. Генкеля «Pyrito-logie oder Kies-Historie», которая в русском переводе 1775 г. названа «Руководство по химическому рудословию» [26]. Книга эта выдержала несколько немецких изданий и, кроме русского, была переведена на французский и английский языки.
Иоганн Фридрих Генкель (1678—1744), медик по образованию, с 1712 г. работал врачом во Фрейберге, а затем стал горным советником саксонского курфюрста. Хотя Фрейбергская горная академия — первое в истории высшее учебное заведение такого рода —обрела этот статус только в 1765 г., во Фрейберг, где издавна существовала для обслуживания серебряных рудников и заводов, а при Генкеле расширилась Саксонская государственная химическая лаборатория, постоянно приезжали практиканты из других княжеств и стран. Так, с осени 1739 г. и до весны ^740 г. у Генкеля проходили обучение три русских студента: Гейзер, Виноградов и Ломоносов, которые, правда, не сошлись Характерами с педантичным и весьма расчетливым руководителем.
Книга Генкеля дает ясное представление о теоретическом и Методическом уровне аналитической химии начала XVIII в., ее Целенаправленности и установках. Мы цитируем ее здесь по несвершенному русскому переводу 1775 г., поскольку архаичный мзык хорошо подчеркивает характер знаний того времени.
125
ГЛАВА ЧЕТВЕРТАЯ
В «Предуведомлении» Генкель пишет: «Конец химии есть тот, чтобы во внутренний состав тел проницать, разделять их па существенные час ти, купно наки соединять; тела в их целости и порознь исследовать ц таким образом всячески домогаться, чтобы внутреннее оных содержание откровенным учинить» [26, с. 2].
Специальные области химии: а) наука о квашении и закисании (Zi-motechia), b) наука об огне (Pyrotechnia), с) наука о солях (Halotechnia) [26, с. 5].
«Польза химии есть следующая:
а. Познание вообще естества и того и другого тела порозно.
Ь. Искусство полученное тело основательно испытать и благоразумно исследовать, каковое называется химиею умозрительною (Chimiapliysica).
с. Лекарства для целения тела человеческого составлять, чему учит нас химия врачебная (Chimia pharmaceutica).
d. Химия, горному производству служащая, называется химией металлургической, которая вмещает " '
ства (Ars docimastica et flusoria).
e. Знать, составлять к пользе например, изрядно делать краски, и проч.
f. Высшее познание в химии,
в себя пробирное и плавильное искус-и увеселению общего жития служащее, варить мыло, делать стекла, гнать вино именно: небогатые металлы превращать в дорогие, называется алхимиею» [26, с. 6].
Генкель посвящает свою книгу только химии минеральной или металлургической. «Способ преподавать оную можно взять синтетический или аналитический, или средний способ предметом нашим быть имеет, яко наилучший и удобнейший» [26, с. 71.
«Добродетели, которые любитель химии иметь должен, суть главнейшие: а) охота к делу; Ь) терпение и постоянство; с) рачительное наблюдение; d) исправная всему записка; е) предмет — не богатства, а благоразумия; f) предосторожность для здравия своего, опасность огня и денежной траты» [26, с. 8].
Пожалуй, эти заповеди вполне современны. Привлекает внимание противопоставление алхимическим поискам золота: стремиться надо не к богатству, а к знанию.
И. Генкель обладает немалым уже знанием об основных химических веществах. Так, он знает, что в купоросах, квасцах, глауберовой соли содержится «купоросная кислота», что уксусную кислоту для концентрирования следует вымораживать. Принципиальная сторона пробирного анализа металлов у него не отличается по существу от прописей Агриколы, однако подробнее говорится о флюсах, в частности для качественных проб путем получения окрашенных стекол. «Металл,— пишет он,— должно брать в виде земли или извести. Для получения стекла берут кварцевый песок и щелочь». «Медный пепел дает изрядную изумрудную зелень». «Медная известь с раствором свинцовой соли... дает прекрасную яхонтовую лазурь, которая, однако, склоняется вообще к зелени». «Оловянный пепел дает стеклу желтоватый цвет, но делает его мутным, наподобие опалфлюса». «ВизмутоваЯ руда и коболд дают высокого цвета голубое стекло». «Марганец, или гвоздишный камень, дает фиолетовое голубое стекло».
Марганец и, по-видимому, кобальт, о которых идет здесь речь, в 1725 г. пока еще не известны как свободные металлы; Генкель оперирует только с окислами.
126
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
В отличие от предшествующих пробирных руководств, Ген-кель активнее вводит методы анализа «мокрым путем». В числе* реактивов, которыми он оперирует, мы, кроме традиционных царской водки и азотной кислоты, видим серную, соляную и уксус-ную кислоты, нашатырь и аммиак, а также органические реактивы, такие, как кровяная соль, фиалковый сок, дубильный, экстракт.
Ассортимент определяемых металлов у Генкеля близок к традиционному: серебро, золото, медь, сурьма, свинец, висмут, железо, ртуть. Но в числе предметов аналитического поиска появляются сера, купоросы, квасцы и «синяя краска сафлор, или ко-болд»,— окись или сульфид кобальта, используемые для получения синего стекла и синих глазурей.
Знаменательнее всего то, что Генкель начинает характеризовать рудные минералы не просто как природные тела определенных физических свойств и облика, но как химические вещества, закладывая тем самым основу химической минералогии.
Вышедшая 33 года спустя (1758) книга шведского ученого А. Ф. Кропштедта (1722—1765) уже представляет собой в сущности химическую минералогию [27]. Здесь все минеральные тела разделены на 4 класса: земли, соляные породы, земляные смолы, металлы. Кронштедт — чистый эмпирик, прекрасно знающий изучаемый материал. Каждый минеральный объект, о котором он пишет, рассмотрен им всесторонне и хорошо охарактеризован различными химическими реакциями, которые можно использовать как аналитические.
Некоторые приводимые Кропштедтом сведения имеют давнее происхождение, иные являются прямым результатом поисков алхимиков. Дело, однако, в том, что во все времена, вплоть до сегодняшнего дня, но особенно на заре научной химии, психологически труден был переход от познания химических превращений веществ к использованию их в качестве аналитических характеристик или аналитических приемов. Знания, изложенные Кропштедтом, носят, пожалуй, впервые в минералогической литературе, характер химико-аналитических признаков.
Становление химического анализа твердых минеральных тел «мокрым путем»
Для дальнейшего развития аналитической химии громадную Роль сыграли исследования Маргграфа.
Андреас Сигизмунд Маргграф (1709—1782), фармацевт по первоначальному образованию, изучал затем пробирное искусство во Фрейберге у Генкеля, а с 1738 г. и до конца жизни работал й химической лаборатории Берлинской Академии наук.
ГЛАВА ЧЕТВЕРТАЯ
Он не написал специальной книги по аналитической химии но его статьи, появлявшиеся в трудах Академии, и выпуски со~ брания сочинений быстро становились достоянием тогдашнего научного сообщества. Его сразу же начинали цитировать совре-мепники и продолжали последующие поколения ученых.
Важнейшее значение для аналитической химии имели систе-матические исследования Маргграфом химического поведения металлов и их соединений. Он впервые заметил, что осадки соединений золота и серебра, образующиеся под действием щелочи и не растворяющиеся в ее избытке, хорошо растворяются в присутствии аммиака. Соединения цинка и висмута растворяются и в щелочах, и в аммиаке, тогда как осадок соединений ртути вовсе нерастворим. Полученные результаты сразу же приобрели практическое приложение при разделении соединений металлов.
А. Маргграф исследовал также реакции желтой кровяной соли с разными металлами и использовал открытое в 1709 г. образование «берлинской лазури» как аналитическую реакцию на железо. В 1740-х годах он впервые получил синтетическим путем натриевую селитру, установил цвет калиевого и натриевого пламени и последовательно сравнил формы и свойства целого ряда солей этих металлов, а также получил растворимое натриевое стекло. Забегая вперед, заметим, что заключительное слово о различении натрия и калия сказано было венгерским ученым Яковом Винтерлем (1732—1809), предложившим в конце 1770-х годов винную кислоту для осаждения калия. Ф. Шабадвари, правда, замечает, что и до Маргграфа на различие солей калия и натрия обращали внимание в 1703 г. Г. Шталь и в 1736 г. парижский академик Дюамель де Монсо (1700—1780), но считает Марггра-(Ьа одним из величайших исследователей во всей истории аналитической химии [2, с. 73]. И это связано не только с установлением Маргграфом природы и свойств каких-то определенных веществ, но в первую очередь с драгоценнейшими для аналитической химии результатами — выработанными им общей системой и методикой исследования. Так, Маргграф отметил, что при дробной перекристаллизации целесообразно отбрасывать несущие с собой примеси первую и последнюю порции кристаллов. Он ввел в широкое употребление микроскоп для изучения кристаллов, унифицировал процедуры кислотного растворения и т. д.
Об уровне аналитических возможностей середины XVIII в., после работ Маргграфа, можно судить по имевшим тогда хождение аналитическим руководствам. Типична в этом отношении вышедшая в 1761 г. книга «Пробирное искусство» Иоганна Готлоба Лемана (1719—1767), медика по образованию, затем прусского королевского горного советника, а с 1761 г. до конца жизни " профессора химии и академика в Петербурге.
Исходная позиция Лемана такова: для того, чтобы удобнейшим способом разыскивать ценные минералы, необходимо на
128
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
учиться познавать их химические составные части. Именно:
твердую огнепостоянную стекловатую землю; 2) составные части солей; 3) загорающееся существо (флогистон); 4) содержание металлов.
Флогистон, отождествляемый с горючим началом, со способностью гореть, определяли, присыпая измельченное исследуемое вещество к расплавленной селитре. Если происходит вспышка, значит, есть флогистон. Проба, разумеется, качественная.
«Под именем огнепостоянной земли разумеется все то, что может сдержать самый наикрепчайший жар» и не сплавляется в металлоподобное тело ни с чем, а только прокаливается до белизны и сплавляется само или с добавками в стекло.
Для обращения твердых земель в стекло применяли зара
D. ЗФ*пи (Gottlob SfbtnannS
<Preug. $crgra$£, SXitgliebtf bfr S'pniglitfjen 2!tabf>niie bfr 9Ci|Jcnjcbaff«iz b?r ^burmeyufeiidu’U nu|li d)cr ber Snglifd'cu Socirtit jnr 2fuftiahme
isrr 9J?antifactnrctt unb bcr Jpatibhing, unb bfr
2lf<ib«nic ju €>t. (p«ftsburg crbcntlid)<n SJJitgficbs,
Я5 e r I i n z be») $tnolb SSJc&cr, pritil.
1761.
i
Титульный лист книги И. Г. Лемана «Пробирное искусство» (1761)
нее заготовленное дисперсное стекло, легко затем сплавляющееся, и буру. Другой способ заключался в том, что чистый песок и Щелочь (соду, поташ) сплавляли в процессе исследования пробы, добавляя глауберову соль, гипс, сурик, белый мышьяк или некоторые другие компоненты. В присутствии металлов будут, по Леману, образовываться цветные стекла и шлаки.
При изучении составных частей солей «купоросную кислоту» можно изгонять сплавлением с песком. Можно также с помощью «флогистона», т. е. нагревая с сажей, маслами, древесным углем и Другими веществами, «богатыми флогистоном». При этом образуется род «серной печени», откуда сера обнаруживается потом Действием кислоты.
При анализе руд следует «рудную глыбу истолочь, просеять, й^ремешать и взять из кучи со всех четырех углов и из середи-ны чистою ложкою». Можно рудную пробу сначала промыть н лотке.
Общий подход к анализу таков:
У вещества изучают внешний вид, вкус, запах, изменение его воздухе, на свету, в тепле, в горячей печи, наблюдая и измеряя также потерю или увеличение веса.
6
Всеобщая история химии
129
ГЛАВА ЧЕТВЕРТАЯ
Твердую пробу обрабатывают «гоненой водой», изучая и ее действие, и изменение ее цвета, запаха и вкуса. Затем жидкую фазу отделяют и, испарив, изучают остаток.
Имея индивидуальное по виду вещество, подвергают его част^ ным реакциям, описанным для отдельных минералов, солей и «земель».
Если же исследованию подвергается «земля», то пробу ее «отмучивают в чистой двоеной, дважды перегнанной, воде в стеклянной посуде. Фильтруют через чистое, в гоненой воде вьь стиранное, полотенце».
Тяжелые части откладывают, отделяя от песка. Муть с поло-тенца надлежит еще раз размешать и через непроклеенную бумагу отцедить. Отдельно исследовать вещество на фильтре и фильтрат [10, рус. пер., с. 230].
В России к этому времени, в 1739 и 1767—1768 гг., было издано два руководства Ивана Андреевича Шлаттера (1708—1768). работавшего директором Петербургского монетного двора [28, 29]. Раздел о пробирном искусстве имеется также в книге М. В. Ломоносова «Первые основания металлургии или рудных дел», вышедшей в 1763 г. [30]. Позднейшие авторы отмечали, что пробирные методы, приводимые Ломоносовым, отличаются от тех, которые использовались 100 лет спустя, только несколько меньшей точностью исполнения, оставаясь по существу теми же [1, с. 146].
Таково было состояние химического анализа к концу второй трети XVIII в., к тому периоду, когда в печати начали появляться работы еще одного замечательного представителя шведской школы химиков Т. Бергмана.
Торберн Бергман (1735—1784) прожил жизнь, недолгую даже по не очень щедрым нормам XVIII в. Он получил в Упсальском университете юридическое, медицинское и даже математическое образование, но основные его интересы сосредоточились на химии. Для характеристики Бергмана как ученого большой интерес представляет его международная переписка с крупнейшими учеными того времени [31].
Среди многочисленных научных результатов Бергмана особо выделяются его химико-аналитические достижения. Сегодня, спустя 200 лет, оглядываясь на путь, пройденный химическим анализом, мы можем сказать, что именно с Бергмана начинается подлинно систематический и не эмпирический, а подлинно научный подход к химическому анализу.
Основные методические результаты Бергмана в этой области содержатся в четырех ого сочинениях: «De analyse aquaruin» («Анализ вод»), изданном в 1778 г. [32], «De minerarum doci-masia humida» («Пробирный анализ минералов мокрым путем»), напечатанном в 1780 г. [20], «De praecipitatis metallics» (осаждение металлов) [33], «De tubo ferruminatorio» [34] — руковод-
ил
Развитие методов химического анализа
по использованию паяльной трубки, которое уже упоминалось выше.
Для анализа минеральных вод Бергман предложил следующие реактивы. Для кислот — лакмус. Для щелочей — вытяжку из фернамбукового дерева. Для железа — вытяжку из чернильных ореш- . ков. Желтую кровяную соль он рекомендовал для железа, (синий осадок), меди (коричневый осадок) и марганца (белый осадок); для меди рекомендован также аммиак, углубляющий синюю окраску ее солей. Для барита (металлический барий тогда еще получен не был, но были известны его соли п окись) — серная кислота; ее же рекомендовалось употреблять для выделения углекислоты. Для сероводорода — белый мышьяк. Для извести — мочевая кислота и щавелевая (ее тогда называли «сахарная»). Для углекислого газа — известковая вода. Для серной кислоты и ее солей — хлористый барий. Для соляной кислоты и ее солей --азотнокислое серебро. Для осаждения металлов и «земель» — «летучая щелочь» (аммиак), карбонат аммония, поташ, сода и едкое кали. Для разделения некоторых солей предлагался винный спирт. Кроме того, для «магнезии» рекомендовался натрий-аммонийфосфат.
В арсенале Бергмана были также ацетат свинца и нитрат ртути. В других своих сочинениях он упоминает еще цианистый калий и хлористый аммоний.
Во многих случаях Бергман указывает чувствительность аналитических реакций. Например, по его данным, 1 гран серной кислоты может окрасить в красный цвет 172 300 гран синего лакмуса.
Аналитические реактивы и реакции, найденные эмпирическим путем, использовались и ранее. Бергман впервые ввел понятие реагент, которое определял следующим образом: реагентами такие вещества
раствору, немедленно или через короткое время изменяют его окраску или прозрачность и тем свидетельствуют о присутствии в растворе какого-то другого вещества» [32].
Мы видим также, что Бергман, по существу, ввел групповые реактивы, которые стали использоваться во всех последующих методах анализа.
При анализе минеральных вод Бергман сначала определяет газы, собирая их над ртутью. Затем он исследует их по запаху (сероводород) и по осаждению известковой воды (углекислый газ). Остальное считается воздухом. Саму минеральную воду Упаривают, собирая и взвешивая последовательно выпадающие Фракции. Из осадка отдельные компоненты экстрагируют сначала спиртом (хлориды кальция и магния), потом последовательно восьмикратным количеством холодной воды, кипячением с четырехкратным количеством чистой воды, разбавленной уксусной кислотой (карбонаты кальция и магния), горячей концентриро
«Я называю которые, будучи прибавлены к
131
6*
fЛАВА ЧЕТВЕРТАЯ
ванной уксусной кислотой (окислы кальция и магния), соляной кислотой (окись железа, глинозем). В остатке — кремниевая кислота, которую идентифицируют паяльной трубкой. Все вытяжки исследуются групповыми реактивами и специфическими реакциями.
Любопытно, что Бергман исследовал способность сероводорода осаждать многие металлы, но не использовал это явление в аналитических целях.
Наиболее интересны методические положения, высказанные Бергманом в книге об анализе минералов мокрым путем [20]. Во-первых, Бергман замечает, что при традиционном исследовании сухим путем из породы выплавляется, как правило, не весь металл. Во-вторых, выплавляемый королек несколько теряет в весе при его получении в силу несовершенства метода. В-третьих, эти потери тем более искажают результат, чем меньше содержание металла во взятой пробе.
В противовес этому, при реакциях мокрым путем вес осадков всегда больше веса содержащегося в пробе металла, что снижает относительную ошибку определения.
И еще одно обстоятельство подчеркивает Бергман: «Весьма часто случается, что не только нужно знать и определять разнородные металлические части, коих иногда 3—4 и более в выплавляемом корольке находится, но также нередко и сокрытую земляную породу, чего, однако же, до сего времени совершенно не-можно было сделать сухим путем» [20, с. 8].
Процедура анализа такова. Руду измельчают и растворяют в серной, азотной или соляной кислотах, а затем из полученных растворов осаждают известные металлы.
«Осаждение должно производить с величайшей осторожностью в прозрачнейших стеклянных сосудах, дабы от недостатка осаждающего средства не осталось чего в растворе или от излишества оного вновь не растворилось. Когда осаждение совершенно кончится и осадок на дне сосуда уляжется, то прозрачный жидкий раствор тихонько слить и вместо оного налить новое количество горячей воды, взболтать и потом дать вновь устояться. После произошедшего осаждения жидкость опять слить и на остаток налить свежей воды, и еще повторять до тех пор, пока наливаемая вода не будет более извлекать вкуса и цвета... Окончив сию работу, осадок, на цедилке находящийся [на фильтре], должно собрать на белую и не содержащую квасцовых частей бумагу; в умеренной теплоте высушить и наконец в закрытом стеклянном сосуде через 5 минут держать в такой степени жара, в которой может кипеть вода. Потом, по охлаждении сосуда, взвесить вместе с цедилкою, которой вес, при начале замеченный, после сего вычесть. Осадок гораздо безопаснее прополаскивать в стеклянном сосуде, поелику цедилку, однажды напитавшуюся соляным раствором, весьма трудно лишить сего качества, наипаче когда уже прошло несколько часов.
...Все осадки должно тщательно обмывать; все кислоты и при осаждении самые растворы разводить перегнанною водой, а для отделения от посторонних металлических частей вообще процеживать сквозь белую пропускную бумагу; осадки и остатки на цедилке рачительно обмывать, высушивать, а потом взвешивать» [20, с. 10—12].
132
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА 1.яа„д.. =j... j-.. _ш... ---'! 1
Для каждого из известных ему металлов Бергман указывает т0 что мы называем сейчас аналитической формой, и фактор пересчета ее веса на вес чистого металла. Так, для серебра — это хлорид, а фактор 100: 129 (правильно 100 : 133, т. е. относительная ошибка 3%). Для свинца — карбопат, фактор 100:132 (правильно 100: 129, т. е. относительная ошибка 2,35%), а-также сульфат, фактор 100 : 143 (правильно 100 : 146,5, т. е. ошибка 2,3%)- Для сурьмы — полупятиокись, фактор 100:138 (правильно 100:132,8, т. е. ошибка 3,87%). Для цинка — карбонат, фактор 100:193 (правильно 100:191,8, т. е. ошибка 0,63%). Указанные «ошибки» рассчитаны, разумеется, от теоретических значений, но обычный эмпирический аналитический фактор совокупно включает и систематические погрешности метода, так что истинные правильности анализов Бергмапа могли бы быть и выше.
Впрочем, анализы солей легких металлов у Бергмана шли даже с большими погрешностями. Так, для кристаллической соды (в процентах):
У Бергмана Правильно Погрешность
абс. % отн. %
Основание (Na2O) 20 21,66 1,66 7,66
Кислота (СО2) 16 15,38 0,62 4,03
Вода (Н2О) 64 62,96 1,04 1,65
Как мы увидим далее, у некоторых современников Бергмана анализы были более правильными.
Очень интересны некоторые частные методы Бергмана, приобретшие впоследствии весьма общее значение. Так, для растворения киновари (HgS) он предлагает концентрированную соляную кислоту с добавлениями 0,1 от ее количества пиролюзита (МпО2), т. е. окисление сульфидов хлором, элементарная природа которого еще ие была ясна.
Для случая серебра Бергман, по-видимому, впервые в истории химии ввел косвенное определение, впоследствии столь важный и общий прием. Серебро в виде сульфата он предлагает определять, осаждая, отделяя и взвешивая сульфат бария. Правда, Фактор пересчета не очень понятен. Бергман считал, что серебра в сульфате содержится 68,75% (в действительности 69,19%, т- е. ошибка 0,64 отн. %).
133
ГЛАВА ЧЕТВЕРТАЯ
Повышение точности аналитических результатов
Более правильными, чем у Бергмана, были анализы, выполненные в тот же период времени К. Ф. Венцелем.
Карл Фридрих Венцель (1740—1793) занимал руководящие посты на Фрейбергском горно-металлургическом производстве и как любитель занимался различными, преимущественно аналитическими, исследованиями, в основном следуя работам Бергмана. Довольно многочисленные труды Венцеля остались неоцененными, и лишь много лет спустя, уже в XIX в., Берцелиус обратил внимание на исключительную точность полученных Венцелем результатов. К количественному сравнению их с результатами других исследователей мы еще вернемся, но заметим, что достигнутая Венцелем правильность связана не столько с преимуществом его аналитических приемов, повторявших приемы Бергмана, сколько с подготовкой веществ для анализа. Так, соли для анализа Венцель получал растворением чистых металлов или карбонатов в чистых кислотах с последующей тщательной перекристаллизацией.
Выше уже шла речь о шведской школе химиков XVIII в. Совершенствовавшееся в ней искусство химического анализа привело в XVIII в. к открытию ряда новых элементов. Так, были открыты: в 1735 г.—кобальт (Георг Брандт); в 1751 г.—никель (А. Кропштедт); в 1774 г.—марганец (К. Шееле и И. Ган); хлор (К. Шееле); в 1783 г.— вольфрам (ученик Бергмана X. Д. Эль-гуяр); в 1790 г.—молибден (П. Гьельм). Некоторые историки приписывают Шееле и Гану открытие в 1774 г. еще и бария, Шееле — в 1778 г. молибдена и в 1781 г. вольфрама, а Гадолину в 1794 г.—иттрия. Наконец, к той же шведской школе принадлежит великий Берцелиус.
Нет ничего удивительного в том, что развитие аналитической химии и открытие новых элементов проходили, как взаимно ускоряющие процессы. Чем больше появлялось предметов для анализа, тем большее значение приобретала в нем система, но чем развитее оказывался анализ, тем больше предоставлял он возможностей для новых открытий.
Немалое значение для развития аналитической химии во второй половине XVIII в. имели работы французских ученых.
Парижский профессор пробирного искусства и академик Бальтазар Жорж Саж (1740—1824) проанализировал множество минеральных, растительных и животных объектов.
А. X. Баталин [1, с. 111], анализируя работы Сажа, отмечает заслуги последнего в изучении действия сероводорода на растворы металлов. Так, Саж заметил, что с помощью сероводорода можно отделить железо, марганец, кобальт и висмут от цинка, олова, свинца и других металлов.
134
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
результаты анализов Саж выражал в скрытой процентной форме, указывая число фунтов, унций и гран на сто фунтов вещества, взятого для анализа (1 фунт=16 унциям). Целочисленность приводимых им значений дает представление о метрологических характеристиках полученных результатов и применявшихся методов. В качестве типичного примера приведем один из анализов мышьяковой руды [35, т. 3, с. 322] :
Фунты Унции
Мышьяк 75 —
Медь 11 0
Железо 8 0
Кобальт 3 7
Золото — 9
Серебро — 3
Кварц 1 13
100
Определение 3 унций серебра на 100 фунтов руды, что составляет 0,19%, свидетельствует не о правильности, а лишь о чувствительности методов анализа, данные которого, несомненно, подогнаны под сумму 100%.
Заметим, что у Сажа, не разделявшего воззрений А. Лавуазье, были с ним и с А. Фуркруа общие аналитические исследования. Важнейшие из них в методическом смысле — это уже упомянутое применение сероводорода для осаждения металлов.
Стремление к систематизации химико-аналитических знаний нашло в руководстве Сажа отражение в виде ряда таблиц. Одна из них содержит интересное перечисление металлов, цвета их «земель» (окислов), цвета стекол, полученных непосредственно из этих окислов, и окраски, которую эти окислы, взятые в малом количестве, придают «бесцветному стеклу» [35, т. 2, с. 319].
Представляет интерес принятый Сажем способ выражения состава анализированных им биологических объектов. В качестве типичного примера приведем результаты сухой перегонки 4 унций ячменного крахмала (1 унция=8 гроссов=72 грана) [36, с. 64] :
Унции Гроссы Граны
Кислота 1 6 —
Тяжелое масло — 3 —
Легкое масло — — 14
Летучая щелочь — -— 10
Уголь 1 1 60
Итого 3 3 12
Минеральный остаток — 4 60
Всего 4
135
ГЛАВА ЧЕТВЕРТАЯ
Такой общий подход еще раз свидетельствует о том, что од, на из методологических проблем анализа заключается в опреде, ленпп понятия состава. Тем большее методологическое значение имела для химического анализа кислородная теория Лавуазье Она не только ответила на вопрос, что же считать подлинно элементарным веществом, металл или его «землю» — окисел. Она тем самым в значительной степени определила химический смысл аналитических превращений. С появлением кислородной теории и возникновением новой системы химических веществ, идущей от Лавуазье, для химии вообще и для химического анализа в частности, весьма актуальным стал вопрос о том, какие мате-риальные физические отношения скрываются за данными химического анализа.
Ответами на этот вопрос послужили освещаемые в других главах этой книги работы И. В. Рихтера по стехиометрии и вытекающие из них более общий закон эквивалентов (1791—1802), закон постоянства состава Ж. Л. Пруста (1794—1806), химическая атомистика и закон кратных отношений Дж. Дальтона (1803—1808) и закон объемных отношений Ж.-Л. Гей-Люссака (1805-1809).
Вообще говоря, идея постоянства состава в невысказанной форме жила в сознании химиков. В этом убеждало их ежедневно наблюдаемое постоянство внешних признаков и свойств, например, минеральных солей, добытых из различных источников или получающихся при лабораторной кристаллизации. Были и более определенные высказывания. Так, итальянец, натурализовавшийся в Германии, врач и химик XVI—XVII в. Анджело Сала (1576—1637) утверждал, что в природе имеется только од-на-единственная вода, одинаковая во всех реках и источниках, а наблюдаемые конкретные различия связаны только с примесями-веществами, растворенными в воде [37].
Полтораста лет спустя, в 1777 г., К. Ф. Венцель уже убежденно заявлял: «То, что каждое соединение веществ должно иметь определенную и неизменно сохраняющуюся меру (аналитическое соотношение частей.—Л. /(.)...уже очевидно» [38].
Приводившиеся Бергманом (см. выше) факторы пересчета, отвечающие содержанию металлов в их соединениях, также основываются на убеждении, что в любом хлористом серебре или сульфате свинца всегда присутствует одна и та же постоянная доля металла. Эта идея постоянства состава, высказанная Прустом в форме закона в 1799 г., основывалась, таким образом, на многовековом опыте человечества и на точных химических анализах, но далее сама служила основанием для суждений об индивидуальности вещества по данным его анализа.
Идея эквивалентности тоже была естественной для химиков-аналитиков. Примером и подтверждением тому может служить рассмотренный ранее предложенный Бергманом косвенный ено-
186
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
сОб определения количества серебра в его сульфате путем осаждения и взвешивания сульфата бария. Сформулированная Рихтером в форме закона в последнем десятилетии XVIII в. и развитая затем Волластоном и Берцелиусом в начале XIX в., эта идея, рожденная химическим анализом, послужила затем его верным • помощником и надежным критерием. Правда, чрезмерное почитание установленных соотношений имело, как и всякая чрезмерность, свою отрицательную сторону, и тот же Берцелиус в 1827 г. писал: «С тех пор как начали руководствоваться химическими пропорциями для проверки количественных разложений, наибольшее число химиков обратили внимание свое на усовершенствование аналитических процессов, менее же опытные выводили совершенно ложные заключения, подтверждая мнимую верность своих результатов отношением и согласованием, которые они представляли с законами химических пропорций; сие заблуждение препятствовало им сомневаться в своих грубых погрешностях.
Невозможно полагаться слишком много на результаты несовершенных разложений, верных единственно потому, что они соответствуют химическим пропорциям...» [39, с. 40].
Эти слова великого химика и сегодня звучат актуальным предостережением против необоснованных спекуляций, возможности которых отнюдь не уменьшились с развитием современных аналитических методов.
Ведущие ученые всех времен понимали, что критерием истины были и будут факты, а не умозрительные построения. Именно поэтому на всем протяжении развития новой химии особое значение придавалось и придается получению достоверных аналитических результатов. И хотя проблема «точности анализа» четко сформулирована была лишь в первом десятилетии XIX в. Берцелиусом, она, пусть и в неявной форме, стала особой заботой химиков-аналитиков уже с конца XVIII в. К тому времени в распоряжении химиков-аналитиков имелись достаточно чувствительные и точные весы. Например, большие «опытовые» весы Ломоносова, хранящиеся в его музее, при испытаниях в новейшее время под нагрузкой 1 кг отклонялись уже при перегрузках °0 мг, т. е. чувствительность их составляла 5-10-5. Эти весы были изготовлены на Сестрорецком заводе в 1747 г. [40, с. 176— 180].
В распоряжении Лавуазье имелись различные весы. Большие, изготовленные в 1787 г., при нагрузке 10 кг имели чувствительность 20 мг, т. е. 2-10“6; средние весы при нагрузке 600 г отклонялись от перегрузки 7 мг; малые, наиболее чувствительные, при нагрузке 4 г отклонялись уже при перегрузке 0,1 мг, что соответствует чувствительности рядовых аналитических весов се-Рсдины XX в.
Дж. Блэк (1728—1799) в своих экспериментах использовал собственной копструкпии весы с градуированным коромыслом и
137
ГЛАВА ЧЕТВЕРТАЯ
рейтером. Он утверждал, что с помощью этих весов мог различать массы, отличающиеся на 71гоо грана, т. е. около 0,05 мг, что при. ближается к метрологическим характеристикам микроаналитиче-ских весов первой трети XX в.
Как сообщает историк химии А. Айд [41], механик Рамсден в 1789 г. изготовил для Лондонского Королевского общества весы чувствительностью 0,5 мг. Такие же весы Рамсдена имелись у известного аналитика XVIII—XIX в. М. Г. Клапрота. Аналитические навески того времени составляли около 5 г. Таким образом, погрешность взятия единичной навески составляла сотые доли процента. Между тем, отклонения дошедших до нас реальных анализов того времени от теоретически вычисленных оказываются на два порядка выше. Следовательно, эти расхождения обусловлены не столько погрешностями измерительных приборов, сколько чистотой исследуемых веществ и самим способом работы, т. е. методическим уровнем исследований.
Чтобы оценить методический уровень работы аналитиков конца XVIII в., рассмотрим результаты трех из них, а именно тех, чьи исследования сыграли важную роль в развитии химии вообще и химического анализа —в частности: Р. Кирвана, М. Клапрота и Л. Воклена.
Ричард Кирван (1733—1812), ирландец по рождению, член Английского Королевского общества, получил юридическое образование и сначала был адвокатом, но большую часть жизни провел в свободных самостоятельных научных исследованиях, преимущественно аналитических. Его опубликованные работы по анализу минеральных солей и вод имеют немалый объем. Их появление произвело впечатление не только на английскую научную общественность, и вскоре Лоренц Крёлль стал переводить Кирвана на немецкий язык [42]. Из немногочисленных методических нововведений Кирвана историки химии отмечают применение борной кислоты для обнаружения свинца и систематизацию фактов «несовместимости» некоторых солей. Так, в растворе, указывал Кирван, не могут одновременно существовать карбонаты щелочных металлов и соли щелочных земель и др. Из этой систематизации у Кирвана вытекал групповой подход к определению минеральных компонентов природных вод. Кирвану же принадлежит идея использования раствора желтой кровяной соли для титриметрического определения железа. В 1784 г. он подробно описал процедуру анализа. Конец титрования определяли по прекращению образования осадка при дальнейшем прибавлений реактива. Титр реактива устанавливали по раствору точно отмеренного 1 грана железа в серной кислоте; Кирван отмечал его на этикетке сосуда, содержащего раствор реактива. Заметим, однако, что к тому времени титриметрические методы уже получили некоторое развитие (см. ниже). И все-таки основное значение работ Кирвана состоит в большом числе проделанных им анали
138
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
зов, которые послужили современникам для сравнений и выводов.
Мартин Генрих Клапрот (1743—1817) начал свою деятельность как аптекарский помощник, в частности, с 1771 г. работал У берлинского аптекаря Валентина Розе. Последний был учеником Маргграфа, сам проводил химические исследования (химикам известен составленный им легкоплавкий сплав); его • сын и, особенно, внук стали в свое время крупными химиками-аналитиками.
Приобретя собственную аптеку, Клапрот получил возможность шире проводить интересовавшие его аналитические исследования минералов. В 1788 г. за научные заслуги Клапрота избрали членом Берлинской Академии наук. В 1809 г. он стал также первым профессором химии во вновь открытом Берлинском университете [43].
Необычайно искусный аналитик, Клапрот открыл три новых элемента: уран (1789), цирконий (1789), титан (1795), а четвертый, церий (1803),—в виде окиси.
М. Клапрот опубликовал по анализу минералов более 200 работ, которые затем были изданы в шести томах под общим названием «Вклад в химическое познание минеральных тел» (1795—1815) [44]. В этих публикациях, ценных и своими конкретными результатами, особенно важно было то, что Клапрот сообщал не только конечные результаты, что делали химики и до него, но подробно, шаг за шагом, описывал весь ход анализа, величины навесок и масс выпадающих осадков, количество растворителей и прочие подробности анализа. Они настолько точны, что позволяют легко воспроизвести все описанные процедуры. Собственно на это и рассчитывал Клапрот, говоря, что его результаты не только понятны, но и воспроизводимы, и что он подробно описал свои повседневные приемы и применявшиеся химические процедуры для каждого, кто имел бы достаточно терпения и желания пойти по его стопам.
М. Клапрот работал столь точно, что сколь-нибудь заметное отклонение суммы компонентов от 100% заставляло его искать причины таких отклонений. На этом пути он и открыл упомянутые новые элементы и некоторые неожиданные для современников составные части. Особое значение имело обнаружение им калия не только в золе растений, как это было давно известно, но и в минеральных объектах, первоначально в алюмосиликате калия—лейците [44, т. 1, с. 48].
Для последующих аналитиков очень важно было то, как Клап-Рот подтверждал свои качественные выводы многочисленными частными реакциями, а количественные заключения базировал на вычисляемых им самим химических пропорциях и встречных экспериментах. Клапрот разработал или усовершенствовал методы анализа силикатов, минеральных вод, сульфидных руд, в частности на содержание серы, и др.
139
ГЛАВА ЧЕТВЕРТАЯ
Правильность полученных им результатов удивительна. Так в 46 гранах AgCl он определил 34,5 грана серебра (по нынешним данным 34,62), т. е. абсолютная погрешность составляет 0,26% а относительная к содержанию серебра 0,35%. Железо в Fe2O3 Клапрот определял с относительной, по железу, погрешностью 7,23%, a Na2SO4 по осаждению BaSO4 с 1,59 абс.% и 2,61 отн. %.
М. Клапрот открыл немало новых качественных реакций и способов количественного осаждения. Шабадвари [2, с. 140] считает, что именно Клапрот первым наблюдал, во всяком случае первым описал, образование молибденовой сини. Он сумел отделить железо в присутствии марганца и алюминия прибавлением тартрата натрия (осаждается гидроокись железа).
Фосфорную и мышьяковую кислоты Клапрот осаждал в виде свинцовых солей, хромовую кислоту определял весовым путем в виде серебряной соли или осаждал в виде соли ртути, удаляя затем ртуть. Возможно, что этот способ Клапрот заимствовал у Воклепа [2, с. 140]. Как ни удивительно, Клапрот нигде не описал применение сероводорода, который как реактив уже был в ходу. Единственный раз — для определения сурьмы — использует он и сульфид аммония.
При всем аналитическом искусстве Клапрот иногда впадал в серьезные ошибки. Наиболее неожиданней в этом плане результат анализа минерала вавеллита: (А1ОН)3(РО4)2-5Н2О. В этом веществе Клапрот не идентифицировал фосфат алюминия, приняв его за обычную гидроокись.
Третий крупнейший химик-аналитик конца XVIII в. Луи Николя Воклен (1763—1829) происходил из простой крестьянской семьи, был учеником аптекаря, затем работал в Париже у Фуркруа, сначала лаборантом, затем сотрудником. Написал много раз переиздававшееся пробирное руководство. В 1791 г. избран членом Парижской Академии наук, позже стал профессором в Коллеж де Франс и Парижском университете. Одновременно он был совладельцем фабрики, выпускавшей химические реактивы высокого качества, в том числе аналитической чистоты.
Аналитические методы Воклена и Клапрота в основном совпадали, хотя Клапрот, как считают историки химии [2, с. 141], внес в анализ много больше нового, чем Воклен. Существенно новое Воклен внес в аналитическую химию открытого им хрома, изучив условия переходов хром (III)хром (VI) и способы осаждения хроматов.
Разработка Вокленом аналитической химии хрома — показательный пример того, как развивалась аналитическая химия в то время. Исходным материалом послужил хромат свинца — оранжево-красный минерал крокоит, доставленный во Францию из России [45, с. 142]. Наличие свинца в крокоите было известно давно, по только Воклену, и то не сразу, удалось, высадив свинец в виде карбоната кипячением с поташом, из оставшегося окра-
740
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
ценного раствора выделить бихромат калия. Из последнего была чатем получена хромовая кислота, а из нее металлический хром (1797). Любопытно, что Клапрот также исследовал крокоит и тОже, хотя и позже Воклена, получил металлический хром, правда иным путем — не через СгО3, а через Сг2О3.
Пути этих двух замечательных аналитиков пересекались не однажды. Воклену еще раз удалось «опередить» Клапрота в открытии бериллия (1798).
К тому времени химики знали несколько особого типа бесцветных «земель»: кремнезем, глинозем, горькозем (Bittererde — Talkerde — MgO). И Клапрот, и Воклен исследовали химический состав минерала берилла, но именно Воклен усмотрел, что «белая земля», которую удается выделить, разлагая берилл, например, содой, неоднородна: наряду с глиноземом {alumina) в ней содержалась другая, очень похожая по свойствам, «земля», которую Воклен за ее вкус назвал «сладкой» {glycina — от греч. слова «гликос» — сладкий).
Хорошо известно, что гидроокиси алюминия и бериллия весьма близки по химическим свойствам. Отличие, замеченное Вок-леном и, как ни удивительно, не замеченное Клапротом, состоит в том, что гидроокись бериллия частично растворяется в избытке карбоната натрия, а еще лучше — карбоната аммония. Химикам известны трудности аналитических операций со смесью алюминия и бериллия, и открытие Вокленом бериллия хорошо иллюстрирует высокий уже уровень аналитического эксперимента, достигнутый к концу XVIII в.
Завершая обзор развития приемов и методов химического анализа в XVIII в., отметим два важных достижения французских ученых. Первое из них — элементный анализ органических веществ, второе — основание анализа мерой.
Подход к определению элементного состава органических веществ
То, что при сгорании органических веществ выделяется вода, Документально зафиксировал еще И. Ban Гельмонт, полагавший, что вода составляет основную часть древесины. То, что при сгорании органических веществ образуется именно углекислый газ, было установлено почти сразу же после того, как Дж. Блэк описал свойства этого вещества (1756). Однако лишь А. Лавуазье показал, что, во-первых, при сгорании всех органических веществ кьщеляется углекислый газ, а во-вторых, он не находится в скрытом состоянии в органическом веществе, не просуществует в нем, к лишь образуется при горении, т. е. при соединении органического вещества с кислородом. Это заключение, основанное па Многочисленных опытах, произведенных Лавуазье в 1770-х годах,
141
ГЛАВА ЧЕТВЕРТАЯ
само легло в основу органического анализа. Из него следовало что по количеству образовавшегося углекислого газа можно судить о количестве углерода, содержащегося в органическом веществе. Проведя свои анализы, Лавуазье, кстати, показал, что все исследованные им органические вещества содержат углерод и водород [46].
В наиболее совершенном аппарате Лавуазье сожжение анализируемых органических веществ, в первую очередь различных масел, производилось в специальной лампе в токе воздуха или кислорода так, что образующиеся газообразные продукты поднимались по вертикальной трубке вверх, проходили через хлоркальциевую трубку для поглощения образующейся воды, а затем через два последовательных сосуда с раствором щелочи. И хотя Лавуазье не опубликовал полученные таким путем результаты, а в более простом оформлении (сожжение под колоколом) они были недостаточно правильны, именно его работы и его прибор легли в основу дальнейших разработок по элементному анализу органических веществ.
Историческое значение всех аналитических исследований Лавуазье многократно повышается конечно же тем, что он последовательно проводил их на основе сформулированного им принципа сохранения массы вещества, ранее принимавшегося лишь интуитивно *.
Второе важное достижение французских аналитиков этого периода заключалось в разработке принципов анализа измерением не конечного продукта, а расходуемого для реакции вещества, т. е. принципов, из которых вырос объемный анализ.
Основание анализа мерой* 2
Хотя упоминание о титровании имеется еще у Глаубера в 1658 г. [2, с. 215 сл.], но оно носит не аналитический, а препаративный характер. Первое же аналитическое использование измерения количества реагирующих веществ принадлежит, по-видимому, Клоду Жозефу Жоффруа (1683—1752), известному под именем Жоффруа-младший. В 1729 г. в трудах Парижской Академии появилась его публикация об определении крепости уксуса. Методика была такова: к точно взвешенному, всегда одинаковому количеству уксуса (2 драхмы) в колбе постепенно, при взбалтывании, прибавляли тонкоизмельченный, хорошо вы-
* В XVIII в. принцип сохранения массы вещества при химических реакциях широко применялся в практике химического количественного анализа, хотя как один из основных законов природы этот принцип был осмыслен значительно позднее.— Прим. отв. ред.
2 Более подробно см. книгу датского историка химии Е. Ранке Мадсс-па [4].
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
сущенный порошок поташа до прекращения бурного «кипения», расход поташа соответствует крепости уксуса. И хотя Жоффруа сравнивал не объемы, а массы реагирующих веществ, мы видим Б технике эксперимента все элементы титриметрического определения. Отметим, что Жоффруа ссылается как на предшественников на В. Гомберга и Г. Шталя.
В 1747 г. Л. Ж. Ле Монье, придворный врач Людовика XV, изучал содержание в природных минеральных водах «щелочных земель» — в сущности карбонатов,— обрабатывая одно и то же количество сухого остатка серной кислотой, прибавляя ее по каплям до прекращения «вскипания». Необходимое число капель характеризовало состав используемой пробы. Эта публикация появилась лишь в 1752 г.
В 1756 г. эдинбургский профессор Френсис Хьюм описал способ определения качества коммерческого поташа по объему стандартного раствора азотной кислоты, используемого для нейтрализации пробы. Конец титрования определяли опять же по прекращению «кипения». И только в 1767 г. англичанин Льюис использовал для определения конца титрования поташа кислотой растительные индикаторы в реакционном сосуде или на индикаторной бумаге. Для определения абсолютных количеств поташа в пробе Льюис предварительно титровал свою кислоту по точно отмеренному количеству (Vs унции) заведомо чистого поташа.
Спустя 10 лет Лавуазье пишет о нейтрализации кислотой как о рутинной процедуре, известной каждому химику.
Ф. Хьюм дозировал раствор кислоты чайной ложкой (3—5 лм), отсчитывая объем с точностью до 7г ложки. Чтобы установить метрологические характеристики, Е. Ранке Мадсен воспроизвел методику Хьюма, сравнив результаты с параллельно проводимым титрованием тех же проб той же кислотой (271/) по метиловому оранжевому. Как ни удивительно, при пересчете полученных результатов на содержание карбоната средние значения из 10 определений по Хьюму и одним из методов XX в. совпали, хотя вычисленное значение стандартного отклонения для метода Хьюма оказалось в 5 раз больше, чем у метода XX в. [4, с. 38].
Важное значение для дальнейшего развития метода сыграли исследования сподвижника Лавуазье Л. Б. Гитона де Морво. В своей публикации 1782 г. он приводит принципы и методы определения кислотности растворов путем титрования их раствором, содержащим точно отмеренное (взвешенное) количество поташа или соды. Индикаторами служили куркумовая или фернамбуковая бумажка. Расход раствора карбоната устанавливали по Уменьшению его веса.
Понимая, что при титровании соляной и серной кислот надо использовать разные факторы пересчета, Гитон де Морво прибегает к осаждению и даже титрованию соляной кислоты взвешенным раствором нитрата свинца; конец титрования оп уста
U3
ГЛАВА ЧЕТВЕРТАЯ
навливал по прекращению образования мути при дальнейшем прикапывании раствора свинцовой соли. Для вычисления использовались соотношения, установленные Венцелем: то количество соляной кислоты, которое связывает 11 частей карбоната калия реагирует с 16 частями свинца. Испытуемый объем составлял около 50 мл; каждую пробу следовало производить дважды.
Чтобы оценить точность метода, Е. Ранке Мадсен по экспериментальным данным Гитона де Морво (1784) и по табличным данным из курса «Объемный анализ» И. Кольтгофа и В. Стенгера (1942) вычислил константы равновесия АГ=[Са2+] [НСОз ]/[СО2]. Оказалось: по данным ученых XVIII в. А=4,25-10“5, а по данным ученых XX в. А?=3,7 • 1СГ5 [4, с. 100].
Титриметрические методы развивались в первую очередь под влиянием практических задач. Бурно развивавшаяся во второй половине XVIII в. текстильная промышленность требовала большого количества качественного сырья и эффективного крашения пряжи и тканей. Поташ употребляли для отбеливания льняного волокна, серную кислоту —для растворения красильного индиго. Гитон де Морво разрабатывал свои методы для анализа щелоков селитряного производства, которым он руководил с 1778 г. А селитра шла в первую очередь для приготовления пороха.
Следующий важный шаг был сделан также для решения конкретной практической задачи.
Беление льна даже с помощью щелочной обработки — процесс чересчур длительный. Поэтому вскоре после открытия хлора К. Бертолле, изучавший его свойства, предложил использовать хлор для беления льняных тканей, в частности в виде щелочных растворов (1785), получивших впоследствии (1792) название «жавелевая вода» (Eau de Javelle) — по месту первого ее производства. Однако на текстильном производстве в Руане первая же попытка беления хлором привела к резкому ослаблению волокон, и ткань расползлась. В сущности, к промышленному применению метода нельзя было и приступить.
Задача заключалась в том, чтобы правильно дозировать хлор, т. е. в том, чтобы научиться точно определять химическую активность белильных растворов. Эту задачу поставил и решил Ф. Де-круазиль.
Франсуа Антуан Анри Декруазиль (1751—1825) родился в семье потомственных аптекарей в г. Дьепп (Франция) и сам был аптекарем, но проявлял разнообразные интересы. Впоследствии он состоял на государственной службе, руководя, в частности, в 1793—1795 гг. организацией производства селитры и ружейного пороха. Первая широкая информация о методе Декруазиля принадлежит не ему, а Бертолле, который в общих чертах описал методику в своей статье о белении тканей в 1789 г. Эта публикация имела почти немедленный резонанс в других странах. В 1791 г. в Англии и в 1792 г. в Германии публикуются аналогичные сооб
144
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
щения. Одним из первых новый метод оценил Кирван, который сам занимался титриметрическими методами.
В 1792 г. лондонский врач Джордж Фордайс, весьма вероятно, что под влиянием метода Декруазиля, описал алкали-ацидиметри-ческое титрование по фиалковому соку, применяя, по-видимому впервые, растворы не карбонатов, а едких щелочей.
Принципиальное методическое значение для титриметрических методов имел разработанный Дж. Блэком прием обратного титрования. Блэк применил его для определения небольшой природной щелочности воды, к которой приливал точно отмеренное заведомо избыточное количество кислоты и затем оттитровывал его стандартным раствором щелочи по лакмусу.
Сам Декруазиль описал методику и аппаратуру лишь в 1795 г. Сущность метода заключалась в том, что в градуированный цилиндр, нулевая метка которого несколько возвышалась над дном, вводили объем испытуемой хлорной воды до нулевой метки. Затем с помощью шаровой пипетки осторожно, при взбалтывании, вводили заранее приготовленный раствор индиго (1 вес. ч. индиго : 8 вес. ч. конц. серной кислоты: 1000 вес. ч. дистиллированной воды). Концом титрования считался момент, когда прибавляемый синий раствор перестанет окрашиваться в красный цвет и общая окраска раствора станет оливковой. Декруазиль предлагал тот же принцип для определения самого индиго и даже двуокиси марганца. По-видимому, эта методика — первый в истории химии пример окислительно-восстановительного титрования.
Ф. Декруазиль назвал свою мензурку «бертоллиметр», щелочной раствор хлора — «бертоллетовый щелок», беление этим раствором— «бертоллирование». Возможно, что это было данью уважения основателю метода, возможно — лестью высокой персоне, но названия эти не привились. Впрочем, от тех же времен идет укоренившееся в химии название «бертоллетова соль», полученная этим ученым при пропускании хлора в нагретый раствор едкого кали.
Говоря о Бертолле, нельзя не упомянуть о его важном вкладе в аналитическое изучение сульфидов. В русле ранее упоминавшегося интереса французских химиков к этой проблеме, Бертолле в 1798 г. опубликовал важный, систематизированный в таблицу, материал об осаждении сульфидов металлов из растворов. Он доказал солеобразную природу сульфидов, отметил некоторые металлы, осаждаемые сероводородом в кислой среде, и возможность °саждения сернистым аммонием. Он предложил также вариант склянки для получения сероводорода из сернистого железа по ме-Т°ДУ Шееле (1774), и его можно считать одним из основоположников сероводородных методов анализа.
145
ГЛАВА ЧЕТВЕРТАЯ
Химико-аналитические знания к концу XVIII в.
Завершая обзор научных и методических достижений химического анализа к концу XVIII в., мы можем констатировать, что к этому времени химики-аналитики имели в своем арсенале научные принципы сохранения общего веса веществ при их химических превращениях, постоянства состава веществ и эквивалентности одних и тех же компонентов в различных химических соединениях, откуда следовал принцип химических пропорций. С ниспровержением флогистонной теории аналитики получили более ясное представление о иерархии сложности химических веществ, цели аналитических определений и способе их интерпретации.
В методическом отношении аналитики обогатились различными методами как качественных, так и количественных определений прямыми и косвенными путями, взвешиванием конечных продуктов и измерением израсходованных количеств веществ. Усовершенствованы были процедуры осаждения, отделения, подготовки аналитической формы вещества и набор самих аналитических форм, в то время практически только весовых.
Основные аналитические достижения к началу XIX в. были обобщены в уже упоминавшемся учебнике В. А. Лампадиуса [7а], вышедшем в свет в 1801 г.
Книга Лампадиуса была не первым в истории химии специальным руководством по химическому анализу,— им, если не считать пробирных книжек, пожалуй, была книга Бергмана [20]. До Лампадиуса издавались и учебники по химическому анализу. Копп и Шабадвари называют первым вышедшую в 1790 г. книгу йенского профессора И. Ф. А. Геттлинга «Полный химический пробирный кабинет», а вторым — книгу Воклена «Руководство для исследователя» (1799). Тем не менее все историки химии особенно выделяют книгу Лампадиуса, подчеркивая, что она «уже подобна обстоятельным аналитическим руководствам наших дней» [2, с. 171].
Знаменательно, что автор этой книги — Вильгельм Август Лам-падиус (1772—1842) был профессором Фрейбергской горной академии: в истории металлургии и аналитической химии с областью Рудных гор и, в особенности, с Фрейбергом связано много выдающихся имен и научно-технических достижений [7,б].
Сам Лампадиус не был сколько-нибудь видным или оригинальным аналитиком. Достаточно сказать, что, получив сероуглерод, он не смог правильно определить его состав и полагал, что имеет дело с неким серным спиртом. Но, видимо, он был хорошим педагогом.
В. А. Лампадиус начинает свою книгу описанием основный аналитических приборов и приемов, перечисляет и характеризуя1.
146
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
необходимые реактивы и способы проверки их чистоты, признаки н характерные химические реакции неорганических веществ, руд и минералов, а затем излагает собственно способы разложения минералов и количественного определения составных частей, следуя прописям Бергмана, Воклена и Клапрота.
«Каждая наука,— напоминает Шабадвари,— время от времени нуждается в авторах, умеющих хорошо ее описать, но они редко оказываются также выдающимися исследователями. В истории аналитической химии мы встречаемся с этим не один раз» [2, Св 174]. И действительно, с начала XIX в. роль некоторых систематизирующих руководств оказывается для развития химического анализа отнюдь не меньшей, чем роль многих новых открытий и оригинальных разработок. Историк химии может сделать отсюда вывод, что в аналитической химии энтропия информации в XIX в. возрастала быстрее, чем в других отраслях химической науки, реже испытывавших аналогичные состояния.
РАЗВИТИЕ ХИМИЧЕСКОГО АНАЛИЗА
В ПЕРВОЙ ПОЛОВИНЕ XIX В.
Подобно другим областям химической науки, аналитическая химия развивалась как благодаря научным результатам, накопленным в результате общего развития химии, так и в связи с конкретными прикладными задачами, которые ставили перед аналитической химией развивающиеся промышленность и химическая наука.
В начале XIX в. бурное развитие различных производств в связи с промышленной революцией резко стимулировало поиски новых источников природного, в том числе и горнорудного, сырья, а также способов определения состава и качества как сырья, так и промышленной продукции. Этим поискам не помешали бурные политические события и войны, потрясавшие Европу. Более того, они заставили искать замену некоторым материалам, временно недоступным вследствие войны и блокады. Проводившиеся с этой практической целью изучение и освоение новых объектов давали обширный материал для чисто научных исследований.
Не меньшее значение для развития науки имел и общий подъем, порожденный, в конечном счете, революционными преобразованиями в производстве и обществе.
Все эти процессы удивительно ярко отражаются на динамике открытия химических элементов. Если до середины XVIII в. было известно 16 элементов, то за последующие 25 лет их было откры-т° 8, с 1775 по 1800 г.— 10, а в первую четверть XIX в.— 18 новых элементов.
Аналитическое изучение новых веществ и новых элементов Девало громадную новую химическую информацию о свойствах веществ и их способности к превращениям. Эти характеристики
147
ГЛАВА ЧЕТВЕРТАЯ
можно было бы использовать затем для химического анализа однако многие объекты имели близкие свойства, затруднявшие д идентификацию, и количественное определение их в совместном присутствии.
Научная и производственная практика вынуждали разрабатывать систему методов химического разделения смесей и систему анализа сложных смесей. С другой стороны, необходимо было иметь способы быстрого и достаточно легкого качественного и полуколичественного определения рудных минералов и содержания в них ценных металлов. Нужен был такой способ, которых! могли бы использовать поисковики. Между тем, производство требовало оперативных и достаточно точных методов анализа про-мышленно-потребляемых или промышленно-выпускаемых химических веществ.
Наконец, сама развивающаяся химия нуждалась в надежных аналитических методах, которые позволили бы не просто познавать состав веществ, но и постигать процесс и характер их превращений.
Химия не могла развиваться без анализа, не говоря уже о том, что химические исследования первой четверти XIX в. продолжали оставаться в основном аналитическими.
Перечисленные выше основные потребности аналитическая химия минимально удовлетворила в течение первой трети XIX в.
Для полевых экспресс-анализов и полуколпчественных исследований руд были надежно отработаны методы исследования с помощью паяльной трубки (Берцелиус, 1820).
Фундаментальную основу для разделения смесей и систематического качественного анализа дали обобщения, сделанные Генрихом Розе (1829).
Для технических анализов все более уверенные шаги сделали титриметрические методы, особенно успешные у Гей-Люссака (1832).
Надежным инструментом для химиков-органиков стал метод элементного органического анализа по Либиху (1831).
Колоссальное значение для развития химического анализа в целом, и в частности для получения надежных количественных аналитических данных, сыграли проведенные в тот же период работы Берцелиуса.
В последующие годы происходили углубление, совершенствование и обобщение упомянутых аналитических методов. Важный перелом наступил во второй половине 1850-х годов. К этому времени Ф. Мор обобщил разрозненные титриметрические методы в систему «анализа мерой» (1856); Р. Бунзен и Г. Кирхгоф превратили оптический спектроскоп в аналитический прибор (1860); наконец, К. Фрезениус, систематизировавший методы качественного и количественного анализа, выявил специфические аспекты аналитической химии как науки об аналитических мето-
148
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
ттах. В 1862 г. появляется основанный Фрезениусом первый в истории специализированный журнал по химическому анализу, и эта дата открывает новый период в развитии аналитической химии.
Разработка принципиальных методических основ химического анализа '
Начало XIX в. отмечено деятельностью многих крупных химиков, глубоких теоретиков или тонких экспериментаторов. Но и среди них возвышается величественная фигура Йенса Якоба Берцелиуса (1779—1848), подлинного лидера химиков этого периода, ученого, в котором счастливо соединились изощренная наблюдательность, глубина и проблемность мышления, способность к широким обобщениям, изобретательность и искусство практического химика.
Я. Берцелиус исходил из того, что химик, как и любой ученый, должен иметь дело с достоверными данными, а потому много сил отдал разработке и совершенствованию аналитических методов. Как и во многих других случаях в истории химии, к углубленной разработке аналитических проблем побудило Берцелиуса то, что в 1807 г. он приступил к написанию учебника для студентов-медиков, которым тогда начал читать курс химии. Стержень для возможных обобщений Берцелиус усмотрел в работах И. Рихтера и решил экспериментально продолжить их на более высоком аналитическом уровне. Позднее, приняв атомистическую концепцию Дальтона, Берцелиус сосредоточил свои усилия на определении не только химических пропорций, но в первую очередь их основы—атомных весов. В 1811—1818 гг. он определил соединительные и атомные веса 45 элементов и процентный состав 2000 соединений. Исследования в этом плане пронизывают все научное творчество Берцелиуса, и приводимые им в разные периоды значения атомных весов характеризуют достигнутую точность аналитических определений.
Ученик Берцелиуса, сам видный аналитик, Генрих Розе свидетельствовал, что Берцелиус радикально изменил существовавшие до него методы анализа, а знаменитый стереохимик и историк химии П. Вальден специально подчеркивал громадное историческое значение созданных Берцелиусом приборов.
Для повышения правильности анализов Берцелиус тщательно совершенствовал аналитические приборы и процедуры. Будучи Искусным стеклодувом, он имел свободу выбора, так как не зависел от мастера. Избрав стекло в качестве основного материала, он сам изготавливал свою специальную лабораторную посуду, ^читается, что именно Берцелиус изобрел стеклянную пробирку и изготовил делительную воронку. После ряда опытов он установил для обычных воронок для фильтрования оптимальный угол
149
ГЛАВА ЧЕТВЕРТАЯ
60°, который до сего дня является стандартным. Берцелиус код. струировал и использовал лабораторные штативы и держатели к ним, изобрел хитроумный, укрепляемый на штативе, механиче-ский прибор для постепенного сливания раствора из стакана ц0 палочке на фильтр. Любопытно, что в те времена у стаканов еще не делали носиков и сливание было нелегкой процедурой.
Придавая важное значение приборам для нагревания, Берце-лиус сконструировал печь на каменном угле с поддувом, в которой достигалась температура, достаточная для плавления платины. Он усовершенствовал и использовал лампу Митчерлиха с поддувом, дававшую высокую температуру, и изготовил ряд оригинальных спиртовок. Для равномерности нагрева он широко использовал различного рода бани: водяную, масляную, песчаную, металлическую со сплавом Розе и придуманную им самим баню из раствора хлористого олова, обеспечивавшую температуру 160° С.
Для сплавлений или сожжений, не связанных с образованием свободных металлов, он использовал платиновые тигли. Для сушки осадков применял самодельный эксикатор с серной кислотой или хлористым кальцием. Для изготовления фильтров Берцелиус рекомендовал непроклеенную бумагу, изготовленную из длинноволокнистой массы, желательно зимой, чтобы приготовленную еще влажную бумагу подвергнуть замораживанию, что, по его мнению, увеличивало имеющиеся в бумаге поры. Применявшаяся им бумага из Лессебу (Южная Швеция) имела зольность менее 0,2%.
Сильнокислые растворы Берцелиус фильтровал через трубку, наполненную битым стеклом или волокнистым асбестом. Осадки промывал до тех пор, пока капля промывной воды при высушивании не оставляла более следа на пробной стеклянной пластинке (сравните с пробой Бергмана на вкус).
Применявшиеся Берцелиусом весы имели рейтер массой 5 мг. Аналогичные весы, описанные его современником X. Г. Пфаффом, при нагрузке 10 г имели чувствительность 1 мг. Разумеется, этой величиной не следует обольщаться, так как чувствительность весов и гарантируемая точность взвешивания, к сожалению, не экви валентны.
Один из основателей физической химии Вильгельм Оствальд (1853—1932), посетив в Стокгольме музей Берцелиуса и рассмотрев аналитические весы, с помощью которых с необыкновенной для того времени правильностью были определены атомные веса многих химических элементов, впоследствии вспоминал: «Мне стало необыкновенно ясно, как мало зависит от прибора и как много от человека, который перед ним сидит» [47]. Отдавая должное и Берцелиусу, и Оствальду, не следует, разумеется, забывать, что аналитическая химия — область методическая, и в ней даже небольшие приборные или методические усовершенствования играли иногда решающую роль.
150
Развйтие МЕТОДОВ хймйческого АНАЛИЗА
В отличие от предшественников и современников, использовавших для анализа навески 5 г и выше, Берцелиус ограничивался навесками 2 а, а если анализируемый материал содержал глинозем, то не более 1 а, поскольку «осадки сей земли бывают обыкновенно весьма значительного объема. Часто высшая степень точно* сти,— писал он,— достигается разложением малых количеств. Кроме того, относительный объем употребляемых реагентов, а также время, требуемое для процеживания, промывания и выпаривания, возрастают с увеличением веса разлагаемого вещества» [39, с. 40].
Изучив течение анализа, Берцелиус пришел к выводу, что при этом неизбежны ошибки. Наилучшими он считал такие методы, при которых правильность результатов менее всего зависит от искусства аналитика. «При всех разложениях,— писал он,— случаются неизбежные потери... Они у опытных химиков редко превышают 2%, а при разложении двойных или тройных соединений не превышают 0,5%. Потеря сия не должна быть поправляема через надбавления недостающего к количественному содержанию...» [39, с. 94].
Для повышения надежности результатов анализ следовало повторять несколько раз.
Чтобы понять сущность методической позиции Берцелиуса, лучше всего послушать его самого.
«Изо всех занятий химика,— писал он,— наиболее труден количественный анализ... Чтобы добиться правильных результатов, необходимо в совершенстве овладеть манипуляциями, используемыми в аналитических процедурах, что достигается многократными упражнениями. Однако одних упражнений недостаточно: необходимо еще искреннее желание к постижению Природы путем изучения состава отдельных ее компонентов.
Люди, посвящающие себя химии, должны понимать, что, пе овладев приемами количественного анализа, они пе будут пригодны ни к какому научному химическому исследованию. Необходимо научиться и привыкнуть взвешивать возможно более точно, уметь переливать жидкости из одного сосуда в другой без потерь и так, чтобы даже самая последняя капля не стекала по наружной поверхности сосуда. Необходимо обращать внимание на самые мелочные обстоятельства, пренебрегая которыми, можно обратить в ничто многодневный утомительный труд» [39, с. 38].
Строгой схемы анализа Берцелиус пе приводит. Будучи сам искусным и изобретательным экспериментатором, он считал, что «...В отношении сложных разложений... занимающиеся оными Должны сами, соображаясь со свойствами ископаемых... придумывать свои специальные способы...» [39, с. 93]. Впрочем, в приводимых им учебных примерах и в его собственных научных исследованиях мы можем усмотреть контуры системы. Так, он начинает с растворения минерала в царской водке с последующим упариванием досуха. Затем удаляет кремнезем, выщелачивая осадок соля-
757
ГЛАВА ЧЕТВЕРТАЯ
пой кислотой, после чего последовательно осаждает компоненты раствора различными реактивами, отделяя выпадающие осадки. В качестве реактивов осаждения он последовательно применяет аммиак, сульфид аммония, карбонат аммония, а для обработки осадков — едкое кали и соляную кислоту.
Более интересны разработанные Берцелиусом некоторые общие химические приемы и частные реакции. Отметим среди первых разложение силикатов фтористоводородной кислотой: тонко-измельченную навеску силиката смешивают в платиновом тигле с двойным количеством порошкообразного плавикового шпата и нагревают, причем фтористый кремний улетучивается. Важен способ разложения веществ хлором; его осуществляли, пропуская ток хлора через стеклянную трубку с раздутым шариком, в который, наполняя его на 2/3, помещали разлагаемое вещество. Шарик подогревали. Улетучивавшиеся продукты реакции поглощали в промывной склянке. Продолжительность разложения достигала 12 час. Этот способ Берцелиус применял для отделения серы, мышьяка, титана, олова и некоторых других элементов от металлов, хлориды которых не столь летучи: меди, свинца, серебра и др. Берцелиус также широко использовал приемы дробной кристаллизации и дробного осаждения.
Среди частных приемов отметим использование кислого сульфата калия в качестве флюса, вариант пробы на мышьяк в духе Марша путем восстановления углем или цианидами с образованием «мышьякового зеркала», отделение платины в виде сульфида, высаживание вольфрамовой кислоты с помощью нитрата ртути с последующим получением трехокисп вольфрама.
Искусство Берцелиуса-аналитика ярко проявилось также в открытии им новых элементов: церия, селена и тория.
В развитии методов химического анализа в первой трети XIX в. важное место занимают достижения Ж.-Л. Гей-Люссака [48]. Его вклад в развитие методов химического анализа многогранен. Пожалуй, важнейшая область — это титрцметрический анализ, о котором будет сказано ниже, но Гей-Люссак сделал многое и в других аналитических областях. Он добился высокого совершенства в анализах газов, стандартизации условий и правильности измерения объемов. Открытый им закон объемного расширения газов при нагревании (1802) послужил научной основой для всего последующего газового анализа. Именно это позволило Гей-Люссаку открыть столь важный в истории химии закон объемных отношений (1805-1808) 3.
В 1810 г. Гей-Люссак совместно с Л. Тенаром опубликовал разработку нового прибора и методики определения углерода и водорода в органических соединениях, для чего был радикально усо-
3 См. главу седьмую.
152
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
BepineHCTB0BaH спос°б Лавуазье сожжения навески анализируемого вещества в замкнутом объеме.
Принципиально важное для общих методов анализа исследование было опубликовано Гей-Люссаком в 1811 г.
Выше уже говорилось, что к началу XIX в., в частности трудами Бертолле, были заложены принципиальные основы для ис- * пользования в химическом анализе разделения с помощью сероводорода. Однако для превращения этих принципиальных основ в аналитический метод потребовалось разобраться в условиях осаждения сульфидов и свойствах получаемых осадков. Примечательно, что за исследование это взялся ученик Бертолле, весьма возможно, что по идее бывшего учителя, с которым Гей-Люссак навсегда сохранил близкие отношения.
Повторив исследование Бертолле, Гей-Люссак надежно установил, какие металлы высаживаются сероводородом даже из сильнокислых растворов, а какие лишь из щелочных или нейтральных. Универсальной для осаждения оказалась аммпачная среда. Гей-Люссак отметил восстановительное действие сероводорода па растворы некоторых металлов и образование при этом серы. Только после работ Гей-Люссака сульфидный метод начинает входить в широкую практику химиков-аналитиков, поскольку преимущества, которые он давал, оказались существеннее неудобства работы с сероводородом.
Важное аналитическое значение имели исследования Гей-Люссака по химии иода, открытого в 1811 г. Б. Куртуа. Однако иод-крахмальную реакцию, сыгравшую впоследствии существеннейшую роль в йодометрическом титровании, обнаружил в 1815 г. не Гей-Люссак и не Куртуа, а известный аналитик, геттингенский профессор химии Ф. Штромайер (1776—1835), два года спустя открывший кадмий.
Методические достижения Гей-Люссака в химическом анализе за первые полтора десятилетия XIX в. нашли отражение в курсе химии Л. Ж. Тенара.
Луи Жак Тенар (1777—1857) происходил из крестьянской семьи. Он не кончал высшего учебного заведения, а химическое образование получил, начав с 17 лет работать лаборантом У Л. Воклена, чей путь в науку начинался таким же образом. Уже в 1798 г. Тенар становится ассистентом в Политехнической школе, а позже — профессором в Сорбонне. С 1810 г. он член Парижской Академии паук. До этого времени, в течение ряда лет работая совместно с Гей-Люссаком, выполнил ряд аналитических исследований. С 1813 по 1817 г. вышло в свет первое издание его «Руководства по теоретической и практической химии» [49, а]. Последний, пятый, том этого сочинения посвящен химическому анализу. Эта книга не сыграла этапной роли в распространении или систематизации методов химического анализа. Между тем, в пей мы паходим ряд интересных приемов и частных реакций.
153
ГЛАВА ЧЕТВЕРТАЯ
И не случайно в течение первых 20 лет все «Руководство...» вьк держало шесть изданий!
В семи главах пятого тома Тенар последовательно излагает 1) общие приемы, 2) анализ газов, 3) анализ горючих тел 4) анализ окисленных тел, 5) анализ минеральных солей, 6) анализ минеральных вод, 7) способы установления природы тел. В качестве приложения дается таблица эквивалентов. В 1829 г. этот том был переведен на русский язык [49, б].
Из общих методических приемов Тенара отметим высушивание в откачанном сосуде над хлористым кальцием, в ряде случаев—на горячем песке. До тех пор вакуумные эксикаторы не описывались. Интересна практическая рекомендация ускорения кристаллизации магний-аммонийфосфата потиранием стенки стеклянного сосуда стеклянной палочкой. Этот, до того времени мало известный, прием был впервые рекомендован У. Волластоном еще в 1795 г.
Классически выглядит описание манипуляций с газами.
Из химических способов разделения отметим отделение меди от цинка растворением гидроокиси последнего в избытке щелочи. Оба эти элемента рекомендовалось количественно определять, взвешивая их окислы. Тенар полагал в окиси меди на 100 ч. металла 25,27 ч. кислорода, что весьма близко к современным значениям.
Щелочноземельные металлы рекомендовалось разделять, используя различную растворимость их солей в спирте. Так, из смеси карбонатов спиртом извлекались карбонаты стронция и кальция, а карбонат бария оставался. Из смеси нитратов извлекалась только соль кальция.
Для аналитических операций с марганцем и церием рекомендовалось прокипятить растворы с сахаром, т. е. перевести элементы в низшие степени окисления. В некоторых случаях предлагалось предварительное восстановление анализируемого раствора цинком, например, для выделения тяжелых металлов. В качестве групповых аналитических реактивов использовали аммиак, сероводород, карбонат аммония.
По мнению историков химии, наибольшее значение в этот период имело появившееся в 1821 г. «Руководство по аналитической химии» X. Г. Пфаффа [50].
Христиан Генрих Пфафф (1773—1852) получил медицинское образование в Штутгарте и Геттингене, с 1801 г. руководил кафедрой в Университете г. Киля и занимался в основном фармацевтической химией. С 1808 по 1824 г. он опубликовал семь томов справочного характера сочинения «Система медицинских веществ по химическим принципам». Работа с веществами такого рода естественным образом вывела Пфаффа к химическому анализу, а чтение курса студентам побудило к написанию руководства. Стремясь использовать все лучшее, Пфафф, тем не менее, отбирал
154
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
материал, публикуя лишь главное и практически лишь то, что проверял в собственных экспериментах. Он излагал материал, используя все основные теоретические достижения науки, в том числе атомистическую концепцию, приводя атомные и молекулярные 4 веса по шкале Берцелиуса О = 100.
Руководство Пфаффа состоит из двух томов. Оно содержит подробный перечень реактивов для анализа с указанием их внешнего вида, свойств, химических формул по Берцелиусу, состава в процентах и способов применения для анализа. В числе реактивов Пфафф упоминает почти все новое, что появилось в литературе к тому времени, включая иод и сероводород, не говоря уже о сульфиде аммония, нитрате одновалентной ртути для определения аммиака и осаждения хромата или хлорной платины для определения калия. Он рекомендует осаждение магния в виде аммонийфосфата и кальция в виде оксалата. Странным образом мы не встречаем здесь роданидов, описанных в 1809 г. английским ученым Р. Порретом и благодаря исследованиям Т. Гротгуса уже известных к 1817 г. как возможные реактивы для определения железа [51].
В приборах и процедурах Пфафф в основном следует Берцелиусу, но Шабадвари [2, с. 176] отмечает важную деталь: главную часть осадка Пфафф перед прокаливанием удаляет с фильтра, фильтр озоляет отдельно и лишь потом соединяет остаток с основной массой вещества. Таким образом он избегал нежелательного, но возможного восстановления осажденной аналитической формы.
Все металлы Пфафф разделял на три группы в зависимости от отношения их к сероводороду: 1) неосаждающиеся; 2) осаждающиеся в кислой среде; 3) осаждающиеся в слабощелочной среде.
В схеме анализа у Пфаффа просматривается определенная система, связанная с последовательным применением групповых реактивов, но это еще только зародыш системы. Историки химии отмечают, что руководство Пфаффа послужило образцом для всех последующих аналитических руководств XIX в. [1—3]. Такое суждение справедливо лишь отчасти, поскольку в руководствах по аналитической химии решающую роль играла не форма изложения, а система анализа, формирование которой последовало в ближайшее десятилетие с момента выхода в свет руководства Пфаффа.
Становление системы химического анализа
С развитием химических знаний к концу первой четверти XIX в. накопилось много сведений о частных реакциях веществ. Однако для систематизации их необходима была общая концепция, которая могла бы объяснить, что именно происходит при
4 Пфафф называет их «смешанными» (Mischungsgewicht).
155
ГЛАВА ЧЕТВЕРТАЯ
к
взаимодействии различных веществ. Такой концепцией послулщл берцелиусовскпй электрохимический дуализм.
«...Каждое сложное тело,— писал Берцелиус в 1819 г.,— незц, висимо от числа его компонентов, может быть разделено па две части, положительно и отрицательно заряженные» [55, с. 98] Первоначально он полагал, что в солях пары составляются кислотного и основного окислов, но в начале 1830-х годов у^е в равной мере предполагал возможность существования в солях электрохимической пары металл—кислотный остаток.
Электрохимический дуализм Берцелиуса очень скоро вступил в противоречие с фактами из области органической химии, по для реакций солей в водных растворах он объяснял многое, в первую очередь реакции обмена. Именно отсюда могла возникнуть теоретически обоснованная, а не просто эмпирическая, общая концепция групповых аналитических реактивов. И не случайно, что ее реализовал непосредственный ученик Берцелиуса Г. Розе.
Генрих Розе (1795—1864) происходил из семьи потомственных химиков-аналитиков. Его дед — аптекарь и химик — был учеником Маргграфа; отец, также химик и фармацевт, воспитанник Клапрота, немало преуспел в химическом анализе, предложив, в частности, методы разложения силикатов и определения мышьяка. Розе изучал химию сначала в Данциге, затем в Берлине, а с 1821 г.— в Стокгольме у Берцелиуса. Получив докторскую степень в Кильском университете, где работал Пфафф, Розе с 1823 г. и до конца жизни был профессором Берлинского университета. Он опубликовал немало научных статей, разработал и усовершенствовал многие частные аналитические методы, открыл в 1844 г. новый элемент ниобий, но более всего прославился именно системой анализа, а его руководство по качественному химическому анализу, впервые вышедшее в 1829 г., при жизни автора выдержало много изданий и неоднократно переводилось па все европейские языки, включая русский [56, 57].
Особо следует подчеркнуть, что Розе, пожалуй впервые в истории аналитической химии, четко отделил качественный анализ от количественного; в издании 1829 г.— это две части одной книги, а в последующих изданиях — два отдельных тома.
Качественный анализ Розе начинает с подробного аналитического описания сначала оснований, а затем кислот. Внимательный читатель может заметить, что порядок рассмотрения оснований не случаен: он определяется их отношением к шести основным реактивам — аммиаку, едкому кали, соде, сероводороду, сульфиду аммония, карбонату аммония. Последовательность получалась такой: аммиак, калий, натрий, литий, барий, стронций, кальций, магний, алюминий, бериллий, иттрий, торий, цирконий, церий, марганец, цинк, кобальт, никель, железо(II), железо(III), кадмий, свинец, висмут, уран (II), уран (VI), медь, серебро, ртуть (I), ртуть(II), платина(II), платина(IV), палладий, родий, иридий,
156
Развитие методов химического анализа
осмий, золото, олово (II), олово (IV), сурьма (III), молибден, вольфрам, ванадий, хром, теллур.
Определенную закономерность можно заметить и в расположении кислотообразующих элементов: тантал, титан, сурьма (V), мышьяк (V), мышьяк (III), сера, селен, азот, хлор (I, III, V, VII), . бром, иод, фосфор, бор, кремний, углерод, фтор, циан.
В качестве групповых реактивов на кислоты использовались аммиак, хлорид бария, ацетат свинца, нитрат серебра.
Изучая этот список, нетрудно заметить, что элементы, образующие основания, расположились в порядке, позволяющем разделить их на привычные аналитические группы. Но Розе не сделал этого и даже не объяснил, на каком основании ои избрал именно такой порядок, предоставляя читателям догадываться самим. Далее Розе повествует о реактивах, употребляемых при качественном анализе, о приборах, общих правилах, а затем касается порядка, т. е. последовательности, которой должно следовать при анализе веществ разной степени растворимости, минеральных вод, газов. Особое внимание уделяется чистоте реактивов, и рассматриваются примеси, которые могут содержаться в них.
Г. Розе не только последовательностью элементов упорядочил химический анализ; он сделал большее — упорядочил сам ход анализа и особо подчеркнул необходимость придерживаться его. Анализ растворов начинается у Розе с прибавления соляной кислоты, отчего осаждаются серебро, ртуть(II) и свинец. В кислый фильтрат вводят сероводород и отделяют образующийся осадок, который обрабатывают аммиачным раствором сульфида аммония, извлекая золото, сурьму, мышьяк и олово. Остаток, содержащий сульфиды кадмия, меди и, может быть, свинца и ртути, обрабатывают азотной кислотой, растворяя все, кроме сульфида ртути.
Фильтрат после первого введения сероводорода обрабатывают аммиачным раствором сульфида аммония, отчего выпадают осадки, содержащие железо, кобальт, никель, марганец, цинк и алюминий. При обработке их соляной кислотой не растворяются только сульфиды кобальта и никеля, переводимые в раствор отдельно с помощью азотной кислоты.
Фильтрат от сульфида аммония обрабатывают карбонатом аммония, осаждая кальций, стронций и барий, а в растворе определяют магний в виде аммонийфосфата и калий в виде хлороплатината.
В издании 1829 г. всему качественному анализу было уделено чуть более 130 страниц (количественному — 460). В этом кратком изложении система анализа занимала около трети объема, и имен-Но в ней была воплощена суть. Частных реакций «оснований» и «кислот» — катионов и анионов — приводилось сравнительно мало. Возе сознавал это как недостаток; к тому же перед ним был обра-3еЦ в виде фундаментального двухтомника Пфаффа... Уже во втором издании (1831) Розе почти впятеро увеличил объем раздела
157
ГЛАВА ЧЕТВЕРТАЯ
о качественном анализе, главным образом за счет частных реак~ ций и рекомендаций.
Отличие первого издания книги Розе от второго и последую, щих изданий настолько велико, что многие химики и историки химии ведут их счет не с 1829, а с 1831 г. Различие заметно во всем, начиная от названия основных частей. В первом издании это было «Руководство к качественному (количественному) ана-лизу», во втором — «Учение о качественных (количественных) исследованиях». Логика изложения нигде не отражала логики системы, и при отсутствии теоретического обоснования сама систе-ма тонула в громадном количестве подробностей, частных реакций и аналитических возможностей.
Как реакция на эту неприятную особенность руководства Розе, оказавшегося своеобразной аналитической энциклопедией, стали появляться публикации, выявляющие систему. Характерен в этом отношении перевод книги Розе на русский язык, выполненный в 1837 г. капитаном корпуса горных инженеров П. И. Евреино-вым [57]. В этом переводе вся первая часть книги Розе (300 страниц!) сведена в 10 таблиц, где элементы, перечисленные в принятой Розе последовательности, охарактеризованы реакциями с групповыми реагентами. Такой подход, правда имевший предшественников, оказался очень полезным. Впоследствии сам Розе изменил изложение и даже отдельно опубликовал свою систему, но время было упущено: в 1841 г. вышла в свет новая систематическая книга по качественному анализу К. Фрезениуса, открывшая новый этап в развитии химического анализа.
Карл Ремигий Фрезениус (1818—1897) происходил из семьи адвоката. По окончании школы некоторое время был аптекарским помощником, затем учился в Боннском университете. С 1841 г. он —ассистент Ю. Либиха в Гиссене. С 1845 г. и до конца жизни он профессор в Высшей сельскохозяйственной школе в Висбадене, где вскоре после своего прихода организовал частную аналитическую лабораторию. Единственным ассистентом в ней был сначала Эмиль Эрленмейер, впоследствии известный химик-органик, в 1847 г. руководивший всего пятью студентами. В 1855 г. там обучалось уже 50 студентов. Лаборатория имела тесные связи с промышленностью, перестала быть частной, а в дальнейшем, объединившись с некоторыми другими подразделениями, обрела статус института [58]. Столь быстрое и эффективное развитие лаборатории связано было не только с запросами науки и промышленности, но в первую очередь с личностью руководителя.
Свою первую книгу «Руководство к качественному химическому анализу» [59, 60] Фрезениус написал еще будучи студентом. В Боннском университете, где он учился, не было ни обязательных лабораторных работ, ни специальной студенческой лаборатории. Фрезениус, еще с детства увлекавшийся химическими превращу ниями, стал заниматься в частной лаборатории профессора
158
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
g Маркварта, где он по сути дела был предоставлен самому себе. Ознакомившись с литературой, он составил руководство — авто-ннструкцию для проведения систематического химического анализа. Когда рукопись Фрезениуса попала в руки Маркварту, тот посоветовал на ее основе написать книгу [61]. Успех этой книги превзошел все ожидания: за первые 10 лет она выдержала только • в Германии 7 изданий, а всего до конца века —16. Ее сразу же начали переводить на все европейские языки и продолжали переродить последующие издания.
Причина успеха коренилась не просто в доступности и систематичности изложения, но главным образом в том, что юный студент Фрезениус сформулировал систему анализа, ту систему, которой почти без изменений пользовались химики на протяжении более 100 лет. Не удивительно, что на протяжении всех 16 прижизненных изданий своей книги Фрезениус только дополнял материал или корректировал изложение, но не вносил никаких существенных изменений.
Если Розе только подразумевает берцелиусовский электрохимический дуализм, то Фрезениус сознательно и отчетливо ему следует. В качестве классификационной единицы он рассматривает, как это делал Берцелиус вначале, не металлы, а соответствующие им «основания», т. е. окислы металлов. Он делит их на шесть групп по характеру взаимодействия с групповыми реактивами:
1. Калий, натрий, аммоний. Их сульфиды, карбонаты и окислы растворимы в воде, причем последние носят характер щелочей.
2. Окислы бария, стронция, кальция и магния. Их сульфиды растворимы хорошо, окислы плохо, карбонаты и фосфаты нерастворимы.
3. Окислы алюминия и хрома (при более широком рассмотрении он включает в эту группу также бериллий, цирконий, церий, титан и др.). Окислы нерастворимы, сероводород сульфидов не осаждает, а при действии сульфида аммония выпадают гидроокиси.
4. Окислы цинка, марганца, никеля, кобальта, железа. Их сульфиды не выпадают в сильнокислой среде, частично выпадают в нейтральной и полностью в щелочной среде.
5. Окислы серебра, ртути, свинца, висмута, меди, кадмия. Их сульфиды выпадают при любой кислотности среды. В этой группе выделяются две подгруппы, первая имеет нерастворимые хлориды (серебро, ртуть).
6. Окислы золота, платины, сурьмы, олова, мышьяка. Их сульфиды осаждаются под действием сероводорода в кислой среде, но Растворяются в избытке сульфида в аммичной или щелочной сре-Де- В этой группе также рассматриваются две подгруппы в зависимости от растворимости сульфидов в сильнокислой среде.
При более полном рассмотрении Фрезениус расширял группы, Вводя дополнительные металлы, но для той задачи, для которой
159
ГЛАВА ЧЕТВЕРТАЯ
книга была написана, расширение числа объектов было не нужц0 Дело в том, что в отличие от предшествующих ему книг, представ лявших собой аналитические руководства, Фрезениус впервые написал учебник, рассчитанный и на студентов, изучающих химгь ческий анализ, и на среднее звено — фармацевтов и химиков-аналитиков, работающих самостоятельно, без опытного руководителя За подробностями он отсылает читателей к книгам Розе, относясь к трудам последнего с подчеркнутым уважением.
К. Фрезениус считал, что изучение качественного анализа должно сосредотачиваться на следующих четырех разделах: химические процедуры, химические реактивы и их употребление отношение к реактивам индивидуальных аналитических объектов’ систематический ход исследования.
Реактивы он разделял на «общие» (групповые) и «частные» (специфические), хорошо понимая и подчеркивая условность такого деления. Среди специфических он выделял реактивы «характеристические» и «чувствительные». Для анализа «мокрым путем» Фрезениус применял пять групп реактивов:
1) простые растворяющие средства (вода, спирт, эфир, хлороформ, сероуглерод); 2) кислоты и галогены (серная, соляная, азотная, уксусная, винная, кремнефтористоводородная, сероводородная; хлорная вода, царская водка); 3) основания металлов; 4) соли; 5) металлы, красящие и нейтральные растительные вещества.
Общее число реактивов у Фрезениуса так же ограниченно, как и набор определяемых металлов и кислот. Отметим среди них роданид аммония и цианистый калий. Применение цианидов в химическом анализе было одним из научных исследований самого Фрезениуса.
Так же как набор реактивов, ограничен у Фрезениуса и набор рекомендуемой аппаратуры: спиртовка, паяльная трубка, платиновый тигель, 3—4 платиновые проволочки, 12 пробирок, несколько химических стаканов и колб, фарфоровая чашка, несколько фарфоровых тиглей, стеклянная воронка, промывалка, стеклянные палочки, часовое стекло, агатовая ступка, пинцет.
По мнению Фрезениуса, для обстоятельного изучения качественного анализа студент должен был выполнить не менее 100 аналитических задач: первые 20 — определение отдельных металлов в водных растворах; 30 — определение металлов и кислот в водных растворах; 15 — то же, но для нескольких оснований и кислот в совместном присутствии; 15 — исследование смесей твердых солей («сухие задачи»); 20 —анализы конкретных объектов: минеральных вод, минералов, сплавов и т. д.
Столь же обстоятельно и систематично совершенствовал Фрс' зениус и весовой количественный анализ. Его первая книга по этому предмету вышла в январе 1846 г. и разошлась столь быстро,
160
ЖтсгМЙсп str r,t / rnno ШсгсГ; towcfe artcn / ft>fc$fc fdbigcn / fcnnb cine jcbe m fonbcr* beit/jrer mm mb etgmpbcfff пяф/ duffallt tHetdtt probwt / mb tm tlanem fewer (bllcn vcrffxbe werben/mit ctflerimg cclicber furnc^mot rnkffidbcn S&mdqwttc&n nn gtoflcn fewer/ au<b fcbaibung (Solbt/ Silber/ vrtnb nnbere OTte tMn/ вдтрс ешет Ьеп'фг bee Kupffer faigerne/ Ш effing brennerte/ vnnb Solpetec fi<b«|0/f4»cbtflkrjWgcntnincrifcbenproben/ vnb wasbencn gig ш funff25udxr vtrfafl/ ibergleicpm jinxem mcmale in ibrucf ton»/ men. 2lUcn h'cbbabcm her ^ewet Гйпре / jungert probtrent/ tmnb Jbcrcfleutcn $u nun/ mu fd)$nm ^iguun rn dbnf ber^nffrument/trcwh'cb vn fldfei’g ait Og geben.
ЗЭцгф/
Eajanw Srcfcitk
ITTit X&n: Rdy: tt7<: (EilAb vnb ^riuilegio.
Титульный лист первого издания книги Л. Эркера «Описание всех известных минеральных руд» (1574)
Промывание золы для капеллей и набивание их
А и С—формы и пестики; В, D, Е — готовые капелли; F — шаровидные комки золы, приготовленные для формования; G — промывание золы; Н — набивание капелли
Специальная печь для перегонки и большие медные перегонные аппараты с холодильниками (Лемери)
Части пробирных весов Эркера
— кованое коромысло весов; В — цельнокованый подвес; С — половина подвеса;
- коромысло весов с подвесом в сборе; Е — бусины, закрепляемые на подвесе; F — концы коромысла; G — прикрепление коромысла к подвесу; Н — втулки подвеса; к—узлы, прикрепляющие нити чашек; L — чашки весов с вложенными в пробирными чашечками; М — пинцет пробирера
Малые печи для перегонки (Лемери)
Приборы Бергмана для анализа с помощью паяльной трубки
*’ трубка; В — брызгоулавливатель; С — сопло; D — свеча с отдуваемым трубкой пламенем
Титриметрические приборы для объемного анализа (начало XJ ¥ в-/ Приборы Декруазиля: 1— мерный цилиндр; 2 — большая пипетка; 3 — малая 11,1 петка; I — реактивная склянка с большой пипеткой; 5 — склянка с двумя niiii','rb’<1. ми; 6 — титровальный комплект; 7 — то же, в разрезе; 3 — бюретка; 9 — мср11Ь сосуд Гёттлинга; 10— отборная трубка
।
Перегонный аппарат «пеликан» (Лемер и)
Приборы, применявшиеся Дальтоном (из английского каталога 1806 г.)
1 — алембик; 2 — штатив; 3 — небольшая лампа с горелкой типа Арганда; 4 — колба для длительного фильтрования
Прибор Гей-Люссака и Тенора Оля анализа органических веществ Анализируемый образец вводят через кран, который тут же перекрывают
Лабораторные приборы Берцелиуса
1 — трубка для фильтрования;
2 — газометр;
3 — аппарат для фильтрования;
4 — водяная баня;
5 — эксикатор;
6 — лампа с поддувом;
7 — калильная
печь;
8 — пробирки на штативе
Лабораторные приборы Берцелиуса
1 — стакан;
2 — прибор для промывания;
3 — капиллярный регулятор вытекания жидкости в приборе;
4 — штатив с воронкой для фи. 1 ьгро-ван ия;
5 — лампа на штативе для высушивания осадков;
6 — «автома гиче' скос» длптсДЬ' ное фи 1ьП)0' ванне;
7 — масляная лампа
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
т0 следующее издание повторено было уже в ноябре, а к 1857 г. появилось четвертое издание, включившее многие новые методы, том числе и объемный анализ.
Предшественником Фрезениуса в количественном анализе опять ясе был Генрих Розе. Его книга 1832 г. (т. II, 2-е изд. [56]), по мнению некоторых историков химии [2, с. 195], в методическом отношении не дала ничего нового по сравнению с работами Берцелиуса, в ней не было строгой системы, методики описаны недостаточно полно и без каких-либо ссылок па их авторов, так пто читатель не мог обращаться за уточнениями к первоисточнику. Общий объем информации в связи со множественностью объектов анализа — индивидуальных веществ и разнообразных смесей — был очень велик, и пользоваться руководством было нелегко. Читателей привлекало, однако, полезное приложение — таблицы для пересчета навески определяемого соединения на искомый его компонент, например, какому количеству окисла металла соответствует найденное в опыте количество его соли. Вычисление с помощью этих таблиц сводилось к простому сложению, и они пользовались большой популярностью.
Книга Фрезениуса [62] была совсем иного рода. Он начинает ее с определения круга задач и целей количественного анализа и излагает профессиональные и нравственные требования к химику-аналитику.
Далее рассматриваются основные аналитические процедуры: взвешивание, высушивание и определение влажности, растворение, концентрирование и выпаривание, осаждение и обработка осадков, включая фильтрование, промывание и сушку. В отдельной главе обсуждаются удобные аналитические формы для определения весовым путем. Последовательность рассмотрения металлов—по принятым шести аналитическим группам. Для каждого металла приводится несколько форм, описываются их признаки, свойства, способы выделения, химический состав в формулах и процентах для расчета. Затем в таком же порядке даны конкретные методики определения каждого из приведенных металлов в тех случаях, когда его соединение уже выделено. Количественному Разделению элементов в пределах одной аналитической группы посвящена самостоятельная глава. Первые издания книги завершались главой, посвященной анализу различных природных веществ, и аналитическими таблицами.
Очень интересны методологические позиции Фрезениуса. Он требует от аналитика теоретических знаний, экспериментального искусства и строгой добросовестности.
«Каждый, кто занимался количественным анализом, знает, что, в особенности, вначале, встречаются факты, при которых является сомнение относительно верности результатов. В таких случаях Должно иметь настолько добросовестности, чтобы начать работу
В всеобщая история химии 161
ГЛАВА ЧЕТВЕРТАЯ
снова. Кто не имеет настолько над собой воли, кто боится труда1 и довольствуется предположениями, когда дело идет о непрелотк ном убеждении в истине, тому должно быть отказано заниматься количественным анализом. Кто не питает полного доверия к своим работам, кто не может поручиться за правильность полученных результатов, тому... не должно публиковать своих работ, потому что они послужили бы не к развитию, а ко вреду науке» [63, с. 4]
«Умственная и научная задача каждой химической работы отражается в составлении и точном исполнении плана... Кто работает без заданного плана, тот не имеет права сказать, что он занимается химией: бессвязный ряд процеживаний, выпаривании прокаливаний, хотя бы отдельные операции производились весьма тщательно, не может быть назван химией» [63, с. 55].
40—60-е годы XIX в. были весьма плодотворными как для всей химии в целом, так и для количественного химического анализа. В области весового анализа важнейшая роль в этот период принадлежала Г. Розе и К. Фрезениусу. Но если Розе, чьи научные результаты много значительнее написанных им книг, ставил и научно решал отдельные конкретные аналитические и методические задачи, то Фрезениус научно подошел к проблемам анализа. Розе разрабатывает методики, а Фрезениус изучает источники ошибок, общие аналитические принципы и пути их реализации в методиках. Он обобщает понятие аналитической формы и требования к ней, формулирует требования к процессам осаждения и высушивания и проводит их углубленное изучение на конкретных примерах.
Можно сказать, что если от Т. Бергмана начинается научный подход к химическому анализу и постепенное выделение его в самостоятельную методическую отрасль в рамках общей науки химии, то Фрезениус превращает эту методическую область в науку о химическом анализе. И представляется вполне закономерным, что именно Фрезениус стал основателем первого в истории журнала по аналитической химии «Zeitschrift fur analytisclie Chemie» [64].
Развитие методов и системы объемного анализа
К началу ХТХ в. химики уже опробовали разнообразные тптрп-метрические методы, в том числе титрование осаждением, окислительно-восстановительное и кислотно-основное. В первые два десятилетия XIX в. эти методы продолжали применять в промышленности, но для довольно грубого контроля. Тому было Три группы причин.
Первая — социально-экономическая — заключалась в ограниченности спроса па простой массовый анализ в силу ограниченности И химических производств, и химических исследований. Текстиль'
162
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
даЯ же промышленность и стекловарение удовлетворялись пока существующими методами.
Вторая группа причин — технико-метрологическая. Результаты титрований были недостаточно надежны из-за отсутствия удобных приборов и сложности манипулирования, сложности и грубости градуировки мерной посуды, неточности определения израсходованного объема, грубости дозирования титровального раствора, неточности определения конца титрования. Не случайно Берцелиус критически относился к объемным методам, сам никогда их не применял и еще в 1829 г. считал, что они могут быть допущены лишь для приближенных оценок.
Третья группа причин — методологические. Несмотря на то что атомные веса и эквиваленты вошли в обиход в первом двадцатилетии XIX в., растворы для титрования калибровали условно и содержание вещества в них выражали в процентах или, что то же, в весовых единицах па вес или объем раствора. Содержание определяемого вещества в пробе вычисляли по факторам пересчета. Для того чтобы расход титровального раствора, помимо чисто расчетного, приобрел бы еще и химический смысл, надо было от процентных концентраций перейти к эквивалентным.
В первом десятилетии XIX в. в области объемного анализа выделяются два достижения, и оба принадлежат Ф. А. А. Декруази-лю. В 1806 г. он опубликовал «Заметки о товарных щелочах», содержащие объемный метод анализа поташа.
Поводом для разработки метода послужила необходимость определения качества поступавших на французский рынок товарных партий американского поташа, использовавшегося главным образом для беления тканей. Основную потребительскую характеристику — щелочность поташа — Декруазиль определял титрованием разбавленной 1 : 10 серной кислотой, разработав для этой цели бюретку, скорее, пока еще мензурку, с 18-ю крупными делениями, подразделенными каждое на пять мелких. Это была запаянная с одного конца градуированная трубка диаметром 14—16 мм и длиной 200—220 мм. Декруазиль назвал ее алкалиметром, оговорив, что она с тем же успехом может применяться Для определения кислоты.
Второе важное достижение Декруазиля, о чем он сообщил в 1809 г.,— изобретение мерной колбы, у которой на шейке было алмазным резцом нанесено кольцо, обозначающее отмеренный Уровень наполнения. Первая «мерная кружка» Декруазиля имела стандартную емкость 200 мл.
Из прочих публикаций этого периода можно отметить сообщение Дж. Дальтона (1814) об определении хлора («окисленной соляной кислоты», по его терминологии) путем окисления им Раствора сернокислого железа. «Конец титрования» Дальтон фиксировал по прекращению изменения цвета прикапываемого раствора сульфата. И хотя этим методом едва ли можно было получить 163 8*
ГЛАВА ЧЕТВЕРТАЯ
Бюретка Ахарда (1784) а'Ь' — трубка диаметром 1 линия; d'c'e'f' — баллон диаметром 3 линии; g' — конец пипетки диаметром У8 линии
удовлетворительные результаты, интерн сен сам объект — сульфат железа (Ц) который впоследствии сыграл в объемно^ анализе немалую роль.
Ближайшие интересные публикации по титриметрическим методам начали появляться лишь в середине 1820-х годов и последующие 15 лет прошли «под знаком» Ж.-Д. Гей-Люссака. В 1824 г. появилась первая публикация этого ученого [65], посвященная улучшению декру ази-левской «индигометрии». Здесь Гей-Люо сак ввел важные методические улучшения. Во-первых, он улучшил процедуру установления титра раствора индиго исходя из того количества двуокиси марганца (3,98 г), которое освобождает из соляной кислоты 1 л газообразного хлора при нормальных условиях (0° С, 1 атм). Во-вторых, он превратил отборочную шаровую пипетку Ахарда и Декруазиля в мерный инструмент, введя на ней кольцевую метку. В-третьих, он изменил методику, проводя титрование не в градуированном мерном сосуде, как Декруазиль, а в любой емкости, приливая к раствору гипохлорита стандартный раствор индиго из градуированного мерительного сосуда — бюретки. Именно в этой публикации мы впервые встречаемся с общепринятыми
теперь в химии терминами «пипетка» (от французского pipe — трубка, pipette — трубочка) и «бюретка» (от французского burette—склянка). Оба эти инструмента управлялись одним и тем же клапаном — подушечкой указательного пальца аналитика. Но если для отборочной пипетки это не вносило существенных затруднений, поскольку отбор всегда можно было повторить Д° точного наполнения, то при пользовании бюреткой относительная грубость дозирования титровального раствора снижала точность анализа. И в этом одна из причин того, что стандартные баллонные пипетки успешно используются до сегодняшнего дня> тогда как бюретка Гей-Люссака просуществовала всего 30 лет-
Внешне бюретка Гей-Люссака вызывает ассоциации с чайпН' ком: к ее длинному цилиндрическому градуированному телу прилегает столь же длинная, идущая снизу вверх, отводная тру^ка с отогнутым концом, как носик у чайника. Для титрования бюретку надо было наклонять, прижимая верхнее отверстие пальца1’ и, регулируя плотность нажима, приливать раствор через носпь
164
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
Титриметрические приборы Гей-Люссака (1824)
165
ГЛАВА ЧЕТВЕРТАЯ
в титровальный стакан. Из-за наличия отводной трубки бюретка Гей-Люссака была ломкой. Кроме того, из-за неудобства пользе-вапия ее сложно было градуировать. Практически бюретки ГеГь Люссака изготавливали лишь во Франции, и это тоже препятствовало широкому распространению метода.
В 1826 г. появилась любопытная публикация Хутона де ла Биллиардера об использовании в объемном анализе соединений иода. Автор обесцвечивал некоторое количество пода, нагревая его с содой, поваренной солью и порцией картофельного крахмала. При этом иод превращался в иодистый натрий. Приливание к полученному реактиву раствора гипохлорита приводило сначала к окислению иодида в иодат, а по израсходованию активного хлора появлялся свободный иод, чем отмечался конец титрования [66]. Метод, разумеется, был неточен и практического применения не получил: эра иодометрии началась лишь в 1840-х годах.
В 1828 г. Гей-Люссак сообщил о своих исследованиях по определению качества товарного поташа [67]. К этой задаче химики не раз обращались на протяжении предшествовавших лет по, видимо, качество анализов не удовлетворяло потребностей. Усовершенствования Гей-Люссака свелись к лучшему подбору и установлению концентрации титровальных растворов, использованию мерной колбы на 1 л для приготовления растворов, а также разработанных ранее отборной пипетки и бюретки.
В этой же публикации Гей-Люссак применяет термины «титр» и «титрование». Слово «титр» (titre) для французского языка не было новым: оно имело несколько значений, одно из которых — ювелирная проба; этот термин применяли также для характеристики качества: высокий титр — высокое качество. По отношению к оценке общего содержания растворенного вещества в растворе оно применялось с самого начала XIX в. В цитируемой работе Гей-Люссак упоминал слово «титр» в несколько ином смысле. Его «весовой титр» (titre ponderal) означал количество щелочи, подлежащей нейтрализации в 100 кг поташа. Его «алкалиметрический титр» означал количество кислоты, необходимое для нейтрализации 100 кг исследуемого поташа.
Титровальные растворы оп готовил таким образом, чтобы расход их сразу показывал процентное содержание искомой щелочи. Оп брал 100 г концентрированной серной кислоты уд. веса 1,8427 и разводил в мерной колбе водой до 1 л. Для нейтрализации 50 мл этого раствора, который он называл «нормальной кислотой» (acide normal), необходимо было 4,807 г чистой окиси калия. Бюретка Гей-Люссака имела емкость 50 мл и была градуирована 50-ю делениями по 1 мл. Во избежание больших ошибок при взятии навески Гей-Люссак брал от испытуемой партии поташа навеску 48,07 а, разводил ее водой до объема 500 мл и отбирал па титрование стандартной пипеткой порции по 50 мл этого раствора-
166
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
Титрование велось по лакмусу до резко красного перехода, хотя Гей-Люссак предусматривал и бикарбонатный переход.
Обстоятельства, связанные со следующей титриметрической разработкой Гей-Люссака, настолько поучительны, что рассказ 0 них сохранился не только в первичной публикации [68], но повторен также в первом обстоятельном руководстве по объемному анализу, появившемуся через четверть века после описываемых событий [69].
На протяжении столетий анализ серебра производили путем купеляции. Навеску серебра или его сплава сплавляли со свинцом. Полученный сплав переносили в капелль и выдерживали при высокой температуре («в светлом краснокалильном жару») до тех пор, пока не окислятся весь свинец и медь — обычная примесь к серебру. Расплавленная окись свинца и другие примеси впитывались в пористое тело капеллп. Оставшийся серебряный королек взвешивали.
Пробиреры, многократно проводившие такие анализы, давно заметили, что если этой процедуре подвергнуть даже заведомо чистое серебро, то при этом теряется одна-две тысячных взятого веса. Парижская Академия наук изучала этот вопрос еще в 1760-х годах. Во второй половине 1820-х годов во Франции была создана специальная правительственная комиссия, по плану которой выполнено исследование индивидуальной, внутрилаборатор-ной и межлабораторной правильности и воспроизводимости стандартного метода купелирования. Выяснилось, что величина потери серебра зависит от состава исследуемого сплава, количества взятого свинца, температуры выжигания и, что особенно важно, от плотности набивки капеллей. Максимальные относительные потери наблюдались при анализе сплавов с содержанием 900—500%о серебра.
Внутрилабораторные анализы, производившиеся семью пробирерами Парижского монетного двора, обнаружили при опробовании стандартных сплавов следующие средние значения:
Стандарт, %0 950 900 800
Средний результат анализа 947 896,5 795
Средняя погрешность -3 3,5 —5
При аналогичной проверке, произведенной в пробирных палатах 11 французских городов, обнаружилось:
Стандарт, %0 900 900 800
Средний результат анализа 947 896 795
Средняя погрешность -3 —4 —5
Правда, особо точные анализы, проведенные наиболее опыт-пЫм аналитиком в строго стандартизированных условиях и с осо-бьш способом подготовки капеллей, показали при 6—10 опреде-
Ж
ГЛАВА ЧЕТВЕРТАЯ
лепиях средние значения 949,5—899,7—799,1%, но такая процедура была неприемлема для массовых анализов и навряд воспроизводима.
Таким образом, выяснилось, что при пробе 900% аналитические потери составляли уже три-четыре тысячных. Это означало что серебряная монета, проба которой методом купелирования отвечала норме, в действительности содержала серебра на три-четыре тысячных больше.
Для бесспорного подтверждения полученных результатов был поставлен международный эксперимент, который оказался в согласии с французскими данными [70, с. 138].
Стало очевидным, что необходим новый метод анализа, и Гей-Люссак разработал его. Сущность метода заключалась в осаждении серебра из азотнокислого раствора в виде хлорида путем титрования раствором поваренной соли. Определение расхода растворов производилось измерением объемов или взвешиванием. Титровальных растворов было два: один составлялся так, чтобы 100 мл (или 100 г для весового измерения) осаждали 1 г серебра; 100 мл другого раствора — «децимального» — осаждали 0,1 г серебра. Навеску металла, содержащую чуть больше 1 г серебра, растворяли в азотной кислоте, затем приливали 100 мл (100 г) первого — «нормального» — раствора соли и дотитровывали «децимальным» раствором до прекращения образования мути. Затем раствором, содержащим в 100 мл (100 а) 0,1 а серебра, оттитровы-вали возможный избыток поваренной соли. Расход «нормального» раствора указывал граммы, а «децимального» — миллиграммы серебра во взятой навеске, т. е. аналитик получал численное значение ювелирной пробы. Разница между отдельными определениями серебра в одном образце не превышала 0,5 жа, т. е. точность определения была минимум на порядок выше, чем при купеляционном методе.
Убедившись в преимуществах метода титрования, французская пробирная служба стала применять его, и это принесло французскому казначейству только за счет фактического содержания серебра в имеющей хождение монете неожиданный подарок в сумме не менее 2,6 млн. франков.
В середине 1870-х годов в лаборатории Ф. П. Тредвелла в Цюрихском политехническом институте ассистент Г. Озанн тщательно сравнил аналитические результаты методов определения серебра в швейцарских двухфранковых монетах купеляцией но Гей-Люссаку и по Фольгарду. Содержание серебра оказалось по улучшенному купеляционному методу 0,8349 (среднее из 59 определений), по Гей-Люссаку 0,8356 (из 113 определений), однако амплитуда разброса отдельных определений по купеляционному методу в 10 раз выше [71, с. 187].
Опубликованная в 1832 г. методика Гей-Люссака почти сразу ,же была переведена Либихом на немецкий язык, а вскоре поярй^
•и
4
РАЗЙЙТЙЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
лись ее переводы на другие европейские языки. Вскоре эту методику приняли повсеместно. Изучая архивные документы, Баталии [1, с. 198] установил, что в России определение серебра по Гей Дюссаку вошло в практику в 1830-х годах. Немалую роль сыграл в этом уже упоминавшийся ранее П. И. Евреинов, работавший в лаборатории Департамент а горных и соляных дел.
В развитие методов объемного анализа Гей-Люссак внес еще один существенный вклад. В 1835 г. он опубликовал новое исследование по определению гипохлорита. Исследователи вновь и вновь обращались к этой практической задаче, так как не удавалось найти вполне удовлетворительного решения. Дело, по-видимому, в том, что индиго было не слишком подходящим восстановителем. Гей-Люссак опробовал в качестве восстановителей мышьяк (III), в кислой и щелочной средах, желтую кровяную соль в кислой среде и ртуть (I) в виде нитрата. В первых двух случаях индикатором окончания титрования служили одна-две капли раствора индиго. В случае ртути (I) о конце титрования судили по прекращению образования осадка ее хлорида.
Ж.-Л. Гей-Люссак заложил экспериментальную техническую и методическую основу для возникновения и развития других направлений объемного анализа. Он не дал каких-либо обобщений и не сформулировал общей научной основы титриметрических методов. Время для таких обобщений пришло лишь 15 лет спустя.
Начиная с 1840 г. на базе распространившихся методов Гей-Люссака повсеместно начинают рождаться новые варианты и виды титриметрических определений. Важные публикации появляются каждый год. Отметим те из них, которыми открываются новые ветви объемного анализа.
В 1840 г. профессор химии и медицины в Лионе Альфонс Дю-паскье (1793—1848) попытался использовать для титрования сероводорода спиртовый раствор иода с крахмалом в качестве индикатора [66]. На следующий год Берцелиус, анализируя это сообщение в своем «Ежегоднике», отметил, что спирт реагирует с иодом и лучше было бы использовать раствор иода в иодистом калии.
В 1843 г. парижские фармацевты А. Жели и М. Фордо описали титрование раствором иода сернистой кислоты и гипосульфита [66]. Эта публикация не имела какого-либо резонанса. В 1845 г. профессор фармацевтической химии в тогдашнем Бреславле Адольф Дюфлос (1802—1889) сообщил о новом способе определения трехвалентного железа. К железосодержащему раствору ои Добавлял точно отмеренное избыточное количество йодистого калия и выделившийся иод оттитровывал раствором хлористого олова. Дюфлос использовал для титрования растворы, содержащие в 1 л эквивалентные количества реагентов: 5,9 г олова и 1,29 г иода, е. нормальные в сегодняшнем смысле слова (см. [2, с. 239]). ^се это были, однако, лишь отдельные методики.
169
ГЛАВА ЧЕТВЁРТАЯ
Иодометрию как общий метод сформулировал в 1853 г. Роберт Бунзен [72]. Оценив восстановительную способность подпетого калия и возможность его взаимодействия с разнообразный!ц окислителями, приводящего к образованию свободного иода, Бунзен остановился на решающем вопросе: как титриметрически определять сам иод. Обратившись к работе Дюпаскье, он фиксирует внимание на обратимости химического взаимодействия: иод + сульфит •<-> иодид + сульфат. В пренебрежении этим равновесием видит Бунзен корень неточности результатов Дюпаскье. Он находит и указывает те условия, при которых обратная реакция не идет.
Решив эту узловую задачу, Бунзен использует йодометрию как общий метод определения многих окислителей, как сильных (хлорат, хромат, озон, гипохлорит п др.), так и более мягких (же-лезо(Ш), кобальт(III), мышьяк(V)). Выбор Бунзеном сернистой кислоты для восстановления иода не вполне ясен. Уже в работе Дюпаскье фигурировал тиосульфат, более удобный в обращении и с более стойким титром. В 1853 г. такую рекомендацию сделал К. Г. Шварц, о тиосульфате упоминает в 1855 г. Ф. Мор, по по-настоящему тиосульфат входит в иодометрию лишь после опубликованного в 1858 г. исследования французского химика Л. Пеан де Сен Жиля.
Более надежным, нежели сернистая кислота, восстановителем для иода был и трехвалентный мышьяк, рекомендованный в 1853 г. Мором [69,6, с. 337].
В 1846 г. парижский химик Фредерик Маргерит предложил количественно определять железо в рудах или сплавах путем растворения навески в кислоте, переведением легким восстановлением в низшую степень окисления и затем окислением железа(II) в железо (III) титрованием раствором перманганата калия [73].
Повое зачастую воспринимается с трудом и оценивается современниками не сразу. Так произошло и с предложением Маргерита. Стареющий Берцелиус в своем «Ежегоднике» 1848 г. уделил работе Маргерита всего 12 строк [2, с. 241], но уже через 10 лет после первой публикации Мор в своей книге по объемному анализу отвел методу Маргерита более 120 страниц [69, б, с. 157—283].
Развившееся в общий метод использование в объемном анализе титрованных растворов хроматов началось в 1850 г., когда профессор из Глазго Фредерик Пенни предложил использовать хромат калия для титрования железа(II). Год спустя то же описал венский профессор Якоб Шабус. В 1854 г. сотрудник Р. Бунзена И. А. Штренг описал применение хромата для объемного определения целого ряда веществ, и основателем хроматометрии как общего метода следует признать его.
Любопытна история еще одного метода, имеющего известное применение и в настоящее время.
В 1831 г. А. Э. Беккерель, отец А. Беккереля, открывшего радиоактивность, предложил как качественную реакцию па сахара
170
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
способность восстанавливать соли меди (II). В 1846 г. парижский профессор Л. Ш. Барресвилль описал использование этой реакции для количественного определения сахаров. Щелочной раствор меди(II) он готовил с винной кислотой. Эту реакцию подхватили и охотно применяли в разных вариантах. В 1849 г. ее подробно исследовал штуттгартский профессор химии Герман Фелинг, который также несколько изменил рецептуру и способ приготовления раствора. История сохранила, однако, за реактивом имя Фелинга. «В аналитической химии,— замечает по этому поводу Шабадвари,— мы часто встречаемся с тем, что имя первооткрывателя забывается, а название метода связывается с именем позднейшего исследователя. Это такая же ситуация, как присвоение Новому Свету названия Америка!» [2, с. 243].
Важным компонентом в формировании объемного анализа как самостоятельного раздела аналитической химии послужило расширение числа методов осаждения, основателем которых надо считать Гей-Люссака. В начале 1850-х годов наиболее значительным здесь оказался вклад Ю. Либиха. Заметив, что цианид серебра растворяется в избытке цианида, Либих в 1851 г. предложил определять содержание цианида титрованием раствором нитрата серебра. Конец титрования отмечался по появлению мути, т. е. в тот момент, когда весь имеющийся в растворе цианид-ион был связан в виде Ag(CN)2+.
Аналогичный прием использовал Либих и при определении хлоридов с помощью ртути (II). Он заметил, что ртуть (II) образует в нейтральной среде осадок с мочевиной, но лишь тогда, когда в растворе нет хлоридов. На этом основании Либих в 1853 г. предложил титрование хлоридов раствором нитрата ртути (II) в присутствии мочевины. Конец титрования также определялся по появлению мути.
Выше уже говорилось о том методологическом значении, которое имел для развития объемного анализа переход от выражения концентрации растворов в процентах к эквивалентным растворам. Этот переход наметился в середине 1840-х годов.
Ф. Шабадвари [2, с. 243], указывая, что первое употребление эквивалентных растворов предложено в Англии, усматривает в этом логическую закономерность.
Во Франции, где началось и развивалось использование титрп-метрических методов, в конце XVIII в. была в ходу метрическая система мер. Концентрации, выраженные в этой системе, легко пересчитывались на содержание искомого компонента. Метрических мер придерживался и Берцелиус.
В других европейских странах, в частности в Германии, старинные меры сохранялись дольше, однако с воплощением научных результатов в практику метрическая система постепенно распространилась по Европе, особенно среди химиков и физиков. Исключение составляла лишь Англия, где десятичная система мер
171
ГЛАВА ЧЕТВЕРТАЯ
не смогла одержать победы даже до середины XX в., и химики не имевшие возможности легко пересчитывать концентрации и расход титровальных растворов в количества искомого компонента стали искать способы выражения концентрации, независимые от системы мер. По мнению химиков XIX в. и современных историков химии, первым новый принцип предложил английский химик-аналитик Эндрью Юре (1778—1857). В 1844 г. он указал, что если концентрации растворов подбирать так, чтобы в единице объема раствора содержалось бы количество вещества, отвечающее атомному (молекулярному) весу, то титриметрические расчеты значительно упрощаются. Тем не менее на практике этот новый подход впервые реализовал А. Дюфлос (1845), а вслед за ним Ф. Маргерит (1846). Их примеру не последовали, и реализация методологической основы для общей системы объемного анализа отсрочи-лась на 10 лет.
Распространение титриметрических методов сдерживалось также и чисто техническими причинами. Бюретки Гей-Люссака были не слишком удобны в обращении, и неоднократно делались попытки устроить бюретку с крапом. Конструкцию со стеклянным краном предложил, например, в 1840 г. А. Дюпаскье, а в 1846 г. французский фармацевт Э. Анри предложил стеклянную бюретку с медным краном. Оба принципа не получили распространения. Медь была не безразлична к аналитическим растворам, а стеклянные краны в то время не умели делать так, чтобы бюретка «не капала». Кроме того, использовавшиеся в середине XIX в. достаточно высокие концентрации щелочных агентов быстро повреждали применявшееся для приборов стекло, и краны выходили из строя.
Между тем, исследовательский и производственный спрос требовал усовершенствования технических приемов, а накопление титриметрических методов и их вариантов вызывало общественную потребность в их систематизации и обобщении.
Решение этих задач выпало на долю Ф. Мора, который в 1855—1856 гг. опубликовал по титриметрическим методам обширное руководство, как бы распахнувшее широкие ворота в мир объемного анализа [69].
Историки химии отмечают, что книга Мора не была первым специальным руководством по титриметрическим методам. В 1850 и 1853 гг. в Брауншвейге двумя изданиями вышла книга К. Г. Шварца «Практическое руководство по анализу мерой (метод титрования)» [74].
Карл-Генрих Шварц (1823—1890) в 1840-х годах побывал в Париже, где стажировался у Т. Пелуза в Высшей политехнической школе и познакомился там с титриметрическими методами. Во время написания руководства он был доцентом в тогдашнем Бреславле, где в начале 1850-х годов кафедрой химии заведовал 40-летний профессор Р. Бунзен.
172
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗ А
Считается, что термин «ана-,03 мерой» (Ma^analyse) впер-вые встречается у Шварца и т0 именно из этого источника п распространился далее по всему свету [2, с. 246].
Слово МаВ по-немецки означает вообще меру, измерение', одно из значений этого с юва — объемная мера. Так называлась, в частности, старинная пивная кружка объемом 1,4 л. Сущность анализа титрованием заключается в измерении количеств израсходованного для аналитической реакции вещества, причем с самого начала измеряли израсходованную массу, а впоследствии, сначала альтернативно, а затем преимущественно, стали измерять израсходованный объем. Постепенно более общее понятие «анализ мерой» заменилось ранее лишь сосуществовавшими понятием и термином «объемный анализ».
L Е Н К В U С Н
et r
CHEMISCH -AMALVTISCHEN
TITRIRMETHODE.
n дсн
EIGENEN VERSl'CHLN HND SYS f EMATISC H DARGISTELLT
VON
1)k FRIEDRICH MOHR,
f>b*t*4**< iiixcKch> Cnitesium* <i< kcbK’iil
St Kbu>el. Krbtit He, Prenftcn. niter getebilfin
u<t<j F.krcmti
F<j*
CHEMIKER. ARZTE UW PHARMACEUTEN.
BEBG- END Hi IT! EN.M A NN ER. FABRIKANTEN AGRONOMEN
METALIA RGEN. MINZBEAMTE
IN Z W E 1 А В T H F I L D N С, E V
ERSTE ABTHtlLUNG
MIIT 104 IN den text EINOEBNVIKTEN KOI.;$<Ht:iTTWtim> anoehawctkn NEKECMNUNGSTABELLEN
BRAUNSCHWEIG.
ORVCK VNO VEBLAG VOX FBIEDRICH VIEWEC UN» SOHN
1 8 5 5.
Титульный лист книги Ф. Мора «Руководство по химикоаналитическим титриметрическим методам» (1855)
Опираясь в основном на
Французские разработки, с которыми Шварц практически ознакомился, работая у Пелуза, он в своей небольшой книжке добросовестно изложил основные опубликованные методы. Его обобщение ограничилось лишь тем, что он дал упомянутое общее назва
ние методу и сгруппировал методики в трех основных главах: методы насыщения (нейтрализация), методы окисления—восстановления и методы осаждения.
Титровальные растворы Шварц предлагает, на французский ианер, готовить такой концентрации, чтобы расход раствора сразу Указывал бы процентное содержание определяемого компонента. Однако он оговаривает также возможность использования растворов, составленных из расчета на эквивалентное содержание титранта в литре раствора. Он называет такие растворы «рациональными».
К. Шварц, опередив в этом отношении всех современников, приводит объяснение протекающих при титровании химических превращений, выражая их химическими уравнениями (см. [2, с- 248]). Причем он рассматривает не вещества, находящиеся в си-Ст®ме, а то, что он понимает как реагирующие фрагменты веществ.
173
ГЛАВА ЧЕТВЕРТАЯ
Например, Маргерит в своей публикации 1846 г. выписывал дЛя перманганатометрии следующее уравнение:
МпЧ)7, КО=Мп2О2 + О5 + КО,
Мп2О2 О5 + КО + 5Fe?O3—Мп-О3 + 5Fe3O3 + КО.
К. Шварц интерпретирует его проще:
Мп2О7 10FeO=2Mn + 5Fe2O3.
Несмотря на сконцентрированную информацию и указанные достоинства, книга Шварца не пользовалась особым успехом, издана была малым тиражом и с 1853 г. более не переиздавалась, поскольку в 1855—1856 гг. химическая общественность познакомилась с книгой Ф. Мора.
Фридрих Мор (1806—1879), сын аптекаря из г. Кобленц. Учился фармации в Гейдельберге, Бонне и Берлине, где на его формирование как исследователя оказал большое влияние Генрих Розе. Вернувшись в Кобленц, Мор стал аптекарем, а в свободное время занимался различными научными проблемами. За свою жизнь он написал более 10 книг, за 7 лет до Роберта Майера высказал мнение о переходах и сохранении энергии и др. Кстати сказать, ме-муар о сохранении энергии отказались печатать редакторы нескольких научных журналов, в том числе и Ю. Либих, чьим верным почитателем, тем не менее, Мор оставался в течение десятилетий.
Из всего научного творчества Мора наибольший успех выпал на долю его книги по титриметрическим методам. Будучи фармацевтом и прекрасным практическим аналитиком, Мор, заинтересовавшись титрованием, изучил и своими руками опробовал множество методов, внеся в каждый уточнения, поправки или радикальные усовершенствования.
По совету Либиха Мор с 1847 г. стал готовиться к написанию учебника по титриметрическим методам и в связи с этим уделил много внимания аппаратуре и технике эксперимента. Необычайно изобретательный, он многое оставил современникам и потомкам. Так, он изобрел холодильник, который Либих так энергично рекламировал, что в истории за этим холодильником закрепилось имя Либиха, а не Мора. Изобретенные Мором одночашечные весы для определения плотности жидких и твердых тел тоже известны более как весы Мора—Вестфаля или просто Вестфаля. Столь привычные трубчатые сверла для пробок вовсе не носят имя изобрел тателя.
Поняв, сколь существенно для титриметрических методов точное дозирование титровального раствора и точное измерение израсходованного объема, Мор изобрел знаменитый зажим, радикально изменил форму бюретки, придав ей современный вид, и способ ее использования, изобрел устройство для градуировки бюреток делениями по 0,1 мл и градуированную пипетку,
174
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АЙАЛИЗА
60 си
Зажим Мора
подзаголо-
В третьем отделе, окислительно-восстанови-
Эти простые, казалось бы, усовершенствования, в частности прямая бюретка и 3аЖИм» сДелали методы титрования аппаратур*10 и технически доступными самым цшрокнм Кругам химиков-аналитиков. j^op же изготовил и более надежный стеклянный кран. Только Бунзену несколько позже удалось прибавить сюда столь же остроумное и простое приспособление — повсеместно используемый затвор — стеклянный шарик в резиновой трубке.
Книга Мора [69] состоит из четырех основных отделов. Те из них и отдельные главы, которые посвящены непосредственно химическим методам, носят имена основателей или наиболее ярких исследователей данного метода анализа. Так, второй отдел — алкалиметрия — имеет вок « Гей-Люссак». посвященном
тельным методам, отдельные главы называются: 1) «Маргерит» (перманганато-метрия); 2) «Роберт Бунзен» (иодометрия по сульфиту); 3) «Август Штренг» (хроматометрия); 4) «Фридрих Мор»
(йодометрия по мышьяковистой кислоте). Отдел четвертый — методы осаждения — имеет подзаголовок «Юстус Либих». Есть
подзаголовки и в более мелких разделах, но ссылок в общепринятом смысле у Мора почти нет, и большей частью создается впечатление, что мы читаем собственную разработку Мора. В какой-то мере это справедливо, так как Мор более или менее усовершенствовал своими собственными экспериментами практически все приводимые им методы.
Очень важным оказалось то, что Мор писал свою книгу как Учебник. С этим связаны логичность и обстоятельность изложения, внимание принципиальным положениям и экспериментальным возможностям.
Методологическую основу титриметрического метода составляют, во-первых, последовательное применение растворов в эквивалентных концентрациях (Мор называет эквиваленты «малыми атомами»), во-вторых, последовательное применение приема обратного титрования, в-третьих, единый подход к источникам ошибок и метрологическим характеристикам отдельных методик.
В середине 1850-х годов Ф. Мор объединил различные титрп-^етрические методы па общей методологической основе, используя
175
ГЛАВА ЧЕТВЕРТАЯ
растворы эквивалентных концентраций. Широкое распространи ние выражения концентраций в эквивалентах и молях сыграЛо впоследствии роль в становлении основных представлений физической химии растворов.
Начиная с 1840-х годов в основном трудами К. Фрезениус^ выявляются внутренние научные проблемы методов химического анализа. Формулирование этих научных проблем конкретизирует научный предмет аналитической химии и превращает последнюю из просто учебной дисциплины в отдельную методическую ветвь химической науки. Превращение аналитической химии из системы методов анализа в науку о методах усиливается введением в анализ новых мощных физических способов исследования: оптической спектроскопии и электрохимии. Этот переход формально знаменуется появлением в 1862 г. первого отраслевого химического журнала — «Zeitschrift fur analytische Chemie».
Поскольку аналитическая химия — наука о методах, то в истории ее формирования интересны те рубежи, на которых происходили качественные изменения в характере или группировании аналитических методов.
Начальным рубежом, когда отдельные приемы оформились в способ, т. е. систему приемов, можно считать первую половину XVI в.; в это время в сочинениях Бирингуччо и Агриколы появились первые сохранившиеся аналитические прописи.
Второй рубеж относится к последней четверти XVII в., когда усилиями Бойля химико-аналитическая задача формулируется как особый тип химического исследования.
Третий рубеж —конец XVIII в., когда завершается важный этап накопления различных аналитических приемов и методов и объединения методов в группы, что и приводит к появлению первых специальных, химико-аналитических, руководств и учебников.
Четвертый рубеж приходится на конец первой трети XIX в., когда в аналитической химии появляются системы методов.
Пятый, последний в нашем рассмотрении, рубеж приходится на начало 1860-х годов, когда, как уже было сказано, аналитическая химия начинает обретать статус науки об аналитических методах.
История аналитической химии свидетельствует, что именно эта область оказалась первой инструментализируемой областью химии. Начиная от инструментов разделения — перегонных аппаратов и фильтровальных устройств, аналитических печей раннего перпо' да — мы встречаем далее капелли и тигли, паяльную трубку п специальные лампы, весы, эксикаторы, сушильные шкафы, эвдиометр, газометры, приборы для органического анализа — трубки для сожжения и поглотительные аппараты, бюретки и зажимы для управления ими.
176
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА
В дальнейшем инструментализация охватила и препаративную химию, но задачи аналитического характера были и остаются основной областью инструментализации химической практики.
Совершенствование аналитических подходов и методов и успехи практического химического анализа накопили для химии многочисленные достоверные сведения о составе и свойствах химических веществ и достаточно точные значения атомных или эквивалентных весов химических элементов. Так был получен надежный материал для решения одной из важнейших проблем химии — выявления естественной системы химических элементов.
ГЛАВА ПЯТАЯ
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ ДО СЕРЕДИНЫ XIX В.
Открытие химических элементов и изучение их соединений занимает важное место в истории химии. Видный русский химик и историк химии Н. А. Мепшуткин подчеркивал, что «история химии слагается из трех главнейших отделов: 1) из истории открытия элементов и их соединений, 2) из истории методов опытного исследования, 3) из истории воззрений, руководивших собиранием и объединением опытного материала» [1, с. III].
Нельзя не согласиться с этим мнением. Изучение химических элементов и их соединений имело первостепенное значение для развития химии. Познание элементов не только давало строительный материал для получения новых соединений. На этом пути испытывались, совершенствовались и создавались новые аналитические методы, оттачивались способы химических умозаключений и химических доказательств. Здесь, наконец, накапливался материал для обобщений, приведших в конечном счете к открытию периодического закона.
Знакомство человечества с химическими элементами совершалось постепенно, в течение тысячелетий, и проходило классические ступени познания вплоть до физического синтеза элементов в ускорителях или атомных реакторах. Поэтому, воссоздавая исторический ход познания химических элементов, не следует забывать, что менялось и само представление о том, что такое химический элемент. Развитие этих представлений анализируется в разделе «Классификация химических элементов до открытия периодического закона» (см. стр. 212). В данной главе речь идет о том понятии, которое четко сформулировано Д. И. Менделеевым и по существу принято в настоящее время: химический элемент — это определенный тип атома; вещества, сложенные атомами одного типа, называются простыми, а состоящие из атомов разных типов-называются сложными. Такой уровень представлений начал фор' мироваться со времен Лавуазье.
Постижение природы химических элементов на атомном уровне стало доступным лишь в XX в. В XIX же веке пределом было по^ лучение элемента в виде простого вещества. Однако представлено0
Примечание. Литературу см. стр. 406—408.
178
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
0 том, что данный элемент существует, рождалось иногда задолго до того, как этот элемент удалось выделить в виде простого вещества. Классический пример — фтор. Мнение о его существовании было настолько прочным, что с 1818 г. во всех таблицах при-водится довольно точно вычисленный его атомный вес, но сам. фтор как простое вещество получен впервые в 1886 г. А. Му-ассаном.
В других случаях элемент, выделенный в виде простого вещества, не считали таковым. Классический пример — хлор. Около 30 лет полагали его кислородным соединением, пока в 1808— 1810 гг. Ж.-Л. Гей-Люссак, Л. Тенар и Г. Дэви не доказали элементарную природу хлора.
Анализируя общий ход познания человечеством химических элементов, мы можем выделить 11 его этапов:
1. Предсказание.
2. Обнаружение в космосе.
3. Обнаружение на Земле.
4. Обнаружение в виде физических сигналов (оптические спектры, радиоактивность и др.).
5. Обнаружение или выделение индивидуальных соединений.
6. Понимание того, что в данном соединении или в их группе содержится некий незнакомый элемент.
7. Выделение в виде простого вещества.
8. Описание физических и химических свойств простого вещества.
9. Понимание того, что это вещество состоит из одного элемента.
10. Синтез элемента.
11. Химическая идентификация синтезированного элемента.
Историческая последовательность этих этапов для отдельных групп известных элементов не одинакова; для некоторых элементов ряд этапов вообще не реализован, а до середины XIX в. познание элементов складывалось лишь из этапов 5—9. Обращаясь к этому периоду, не следует забывать, что более ясное понятие о химических элементах возникло лишь после ниспровержения флогистонной теории, для которой первоосновой вещества был, например, не металл, а его «известь» — основание металла.
Далее, мы несколько подробнее рассмотрим историю открытия четырех групп элементов: щелочных и щелочноземельных метал-л°в, галогенов, элементов группы платины, редкоземельных элементов, открытых до формулирования периодического закона.
История открытия химических элементов изложена и проана-чнзпровапа в ряде обобщающих и частных публикаций отечест-Вениых и зарубежных авторов. Ниже мы, специально не ссылаясь, опираемся на материалы этих публикаций [2—9], а также на дру-гие обзорные и оригинальные публикации химиков, изучавших по* элементы.
179
ГЛАВА ПЯТАЯ
НАКОПЛЕНИЕ СВЕДЕНИЙ ОБ ЭЛЕМЕНТАХ
Щелочные и щелочноземельные металлы
Знакомство человека с солями натрия и калия уходит в глубокую древность. Уже на заре цивилизации человек начал выделять и технически применять индивидуальные соединения этих металлов. В I в. н. э. Плиний описывает поваренную соль, природную египетскую соду (натрон), селитру, квасцы, винный камень. Позже стала известна бура.
Стеклоделы, использовавшие соединения натрия и калия для изготовления стекла, очень давно умели выделять зольный щелок. Так, например, до 1240 г. поступали на Руси, куда стеклоделие пришло из Византии. Европейские стеклоделы первоначально использовали непосредственно золу растений; получать зольный щелок в этом регионе начали лишь по примеру арабов в конце X — начале XI вв. С этого времени постепенно накапливаются наблюдения о различии соды и поташа не только по их происхождению (sal aegypti, sal vegetable), но и по свойствам.
В XVII в. начинается и синтетическое получение солей щелочных металлов. Так, Ф. де ла Бое Сильвий (1614—1672) из прокаленного винного камня и соляной кислоты получил хлористый калий — «сильвиеву соль» (sal febrifugum Silvii), а Р. Глаубер (1604—1670) из поваренной соли и серной кислоты получил сульфат натрия — «чудесную глауберову соль» — мирабилит (sal mira-bili Glauberi).
Различие наблюдалось, но не обобщалось в правило. Четкая дифференциация наступила лишь в XVIII в., когда на различие солей натрия и калия обратили внимание сначала Г. Шталь (1703), затем Дюамель де Монсо и, наконец, С. А. Маргграф. Последний экспериментально показал, что в природе существуют два различных «огнепостоянных» щелочных начала — растительное (калий) и минеральное (натрий). Рядом превращений Маргграф выявил общность поваренной соли, соды, натриевой селитры, буры, глауберовой соли и физико-химические отличия соответствующего ряда солей калия. Именно Маргграф впервые описал окраску пламени, создаваемую солями калия. После работ Маргграфа понятия об «основаниях» этих солей приобретают конкретный характер.
Из «щелочных земель» человеком с древнейших времен была получена и практически использовалась негашеная известь — окись кальция. Установлено было, что ее можно получать из разных источников: мела, известняка и мрамора. Известен был я сульфат кальция в виде алебастра и волокнистого гипса —селенита. Древние римляне умели готовить жженый гипс и знали его схватывающие свойства.
Минералы, содержащие магний: карбонаты (магнезит и доломит) и силикаты (нефрит, змеевик, асбест, тальк) также были из-
180
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
еСтны издавна, но в отличие от соединений кальция индивидуальные соединения магния не выделяли и не использовали. Лишь Б последнем десятилетии XVII в. Н. Грю выделил сернокислый загний из минеральной воды Эпсомского источника (Англия). Эта соль так и вошла в историю химии и медицины под названием «эпсомская горькая соль», или «английская соль». В те же годы аналогичную соль в водах источников вблизи Галле обнаружил немецкий химик и врач Фридрих Гофман. В начале XVIII в. из эпсомской соли получили карбонат магния — «белую магнезию» (magnesia alba), а затем и окись магния — «жженую магнезию», или «горькозем» (magnesia usta, Bittererde). В 1756 г. Дж. Блэк установил и описал различия «известковой и горькой земель», т. е. окислов кальция и магния, а также различия их солей.
Из природных соединений бария первым — в 1602 г.— был найден и описан сульфат. В 1774 г. И. Ган и К. Шееле исследовали этот минерал, названный тяжелым шпатом, и выделили из него особую «землю», по свойствам напоминавшую негашеную известь. Ее назвали барит от греческого барос — тяжелый.
Согласно флогистонной теории, то, что мы называем сегодня окислами, а в XVIII в. называли «землями» или «основаниями» солей и металлов, считалось простыми телами. Для щелочных земель соответствующие металлы известны не были, но это не мешало относить их к числу простых тел. Основания соды и поташа — так называемые постоянные щелочи — многие химики тоже относили к числу простых тел, но в данном случае дело обстояло сложнее.
Выявленные исследователями первой половины XVIII в. различия щелочных металлов и их сходство с аммонием навели на мысль о близости не только свойств солей, но и природы их «оснований». Возникло предположение, что щелочи не пресуществуют в веществе, но образуются при термическом разрушении материалов, например при озолении растений.
Кислородная теория в отношении щелочей положения не прояснила. Оно еще более осложнилось после того, как К. Бертолле в 1784 г. определил, что аммиак состоит из азота и водорода, находящихся в соотношении 807 : 193 (в действительности 82,35% N; 17,65% Н). В 1789 г. Лавуазье по этому поводу писал: «По аналогии можно предположить, что азот является принципиальной составной частью всех щелочей, чему мы имеем подтверждение в случае аммиака... Но в случае потассии и содии мы имеем только Легкие предположения, которые еще не были подтверждены ни °Дним доказательным опытом» [10, с. 170].
Свои теоретические представления о химической природе веществ Лавуазье воплотил в построенной им системе химических ^единений. Ее основу составляла таблица простых веществ 1см. с для отбора веществ в эту таблицу Лавуазье в сущ-°сти отказался от абсолютного понятия химический элемент,
181
ГЛАВА ПЯТАЯ
а использовал понятие простые вещества (substances simples). ()l( ввел для них определение такого типа, какой мы называем сегодця операциональным. Лавуазье связывал понятие простое вещество с достигнутым пределом химического разложения вещества, ого. вариваясь, что этот предел относителен, и то, что сегодня счц. тается простым, завтра может быть разложено на составные части.
Такое определение было в то время плодотворным, поскольку ясно формулировало экспериментальную задачу химика, стремя-щегося постигнуть сущность веществ, давало ему опорный ур0. вень делимости вещества, но не ставило абсолютных пределов.
Усматривая химическую аналогию между «землями» и окисла-ми известных металлов, Лавуазье высказал определенное предположение, что так называемые «простые землистые вещества, способные давать соли»: негашеная известь, жженая магнезия («основание эпсомской соли»), барит, глинозем и кремнезем представляют собой кислородные соединения, которые надо еще разложить. Пока же все эти вещества он операционально отнес к числу простых.
Оснований соды и поташа Лавуазье не включил в таблицу простых тел, еще раз оговорив, что он считает эти основания сложными веществами, не содержащими неизвестных ему составляющих.
После выхода в свет книги Лавуазье «Начальный курс химии» [10], к концу XVIII в., семейство «щелочных земель» пополнилось двумя новыми представителями.
В XVIII в. в свинцовом руднике вблизи селения Стронцпап в Шотландии был обнаружен новый минерал, впоследствии названный стронцианитом (SrCO3). В 1787 г. этот минерал исследовали английские ученые А. Кроуфорд и У. Круйкшанк, которые выделили из него новую «землю», тяжелую, но отличающуюся от барита. Сообщение об этом исследовании появилось в 1790 г. В 1793 г. минерал и новую «землю» изучил М. Клапрот, который обнаружил характерное для стронция окрашивание пламени п уверенно отнес испытуемый объект к числу «щелочных земель». Несколько позже выводы Клапрота подтвердили английский исследователь Р. Кирван, французский аналитик Л. Воклеп и русский академик Т. Е. Ловиц.
В 1797—1798 гг. М. Клапрот и Л. Воклен одновременно исследовали состав минерала берилл (алюмосиликат бериллия основной формулы А12Ве3 [Si60i8]), но лишь Воклен усмотрел в осадке гидроокисей, помимо «алюмины», еще одну новую «землю», которую за сладковатый вкус ее сульфата Воклен назвал «глюциной» (от греческого гликос — сладкий). Этот результат подтвердили 11 другие аналитики. Таким образом, число солеобразующих щелочных земель достигло шести, и предположение, что в состав этп* земель входят неизвестные металлы, укрепилось. Однако это пред-
182
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ'
1
положение нуждалось в экспериментальном подтверждении, и оно было достигнуто электрохимическим путем.
После того как Л. Вольта 20 марта 1800 г. распространил сооб* щение о сконструированной им электрохимической батарее — «столбе», в некоторых странах начались опыты по химическому действию электричества. В 1800 г. В. Николсон и А. Карлейль наблюдали химическое разложение воды, а У. Круйкшанк (1745—1800) наблюдал электрохимическое выделение меди из раствора медного купороса, серебра из раствора его нитрата и свинца из раствора ацетата. В 1801 г. У. Волластон наблюдал электрохимическое отложение меди на серебряном электроде а В. Бекман выделил олово. В 1803 г. И. Ган тем же путем выделил из растворов металлические молибден, никель и железо, а в 1804 г. Л. Бруньятелли опубликовал работы по электрохимическому выделению из растворов золота, ртути, кобальта, мышьяка и платины. Электролизом солей металлов занимались также Я. Берцелиус и В. Хизингер.
С 1800 г. начал свои электрохимические исследования одни из виднейших английских ученых начала XIX в. Гэмфри Дэви (1778—1829). Сначала он проводил электролиз водных растворов солей щелочных и щелочноземельных металлов или содержащих их твердых веществ (барит, стронциан, лепидолит и др.) в водной среде, получая растворы соответствующих оснований. Ни щелочи,
ни щелочные земли разложить не удалось: разложению подвергалась только вода. Тогда Дэви предпринял электролиз едкого кали,
расплавляемого кислородно-спиртовым пламенем, направленным на поверхность. При этом электролиз происходил, но образовывавшееся вещество тут же сгорало. Успеха удалось достигнуть только тогда, когда электрический ток был использован и для раз-
ложения, и для нагревания.
В качестве источника электроэнергии Дэви использовал Три медно-цинковые «вольтаические батареи», состоящие из: 1) 24 пары пластин площадью по 12 кв. дюймов; 2) 100 пар пластин площадью по 6 кв. дюймов: 3) 150 пластин площадью по; кв. дюйма. Соединенные последовательно, эти батареи обладали значительной ЭДС. Электролитом служили растворы квасцов и азотной кислоты.
Удача пришла, когда Дэви использовал составную батарею из ^0 пар пластин. Кусочек твердого едкого кали толщиной менее 1 см несколько секунд выдержали на воздухе, чтобы его поверхность, захватив влагу, стала электропроводной, и поместили на ватиновый диск, соединенный с отрицательным полюсом бата-платиновую проволоку, соединенную с положительным полю-с°м, сконтактировали с верхней стороной кусочка. Вскоре кали плавиться с обеих сторон в зоне контакта с электродами, Ричем у верхнего (положительного) наблюдалось бурное выде-1111е газа, а у нижнего (отрицательного) появлялись маленькие
183
ГЛАВА ПЛГлЯ
। nr i'll и тип! ь, .. r«r.r - - - • . .....—-.. - ill . ... i ....... -Ь » i ,brt - < —
шарики с металлическим блеском. Некоторые из них сейчас ;Ке после образования сгорали со взрывом и с появлением яркого цпд мени [11, с. 103-104].
Таким же образом в 1807 г. Дэви получил натрий.
Выделение щелочных металлов в виде простых веществ сыгрц, ло важную роль в формировании представлений о химически^ элементах. «Сходство между свойствами земель и металлических окислов,—писал Дэви в 1807 г.,—было замечено уже в ранний период развития химии. Ядовитость и высокий удельный вес бц. рита и стронциана натолкнули Лавуазье на мысль о металлической природе этих тел. Но никто не предполагал, что металлы могут содержаться в нелетучих щелочах. Вследствие сходства этих тел с аммиаком обычно считали, что они содержат азот и водород. Удивительно, что раньше всего удалось доказать идентичность с металлическими окислами в случае именно тех тел этой группы, которые, казалось бы, менее всего на них похожи» [11, с. 152].
Получение щелочных металлов электролизом побудило химиков к поиску химических путей выделения этих металлов из их соединений. Бэкеровская лекция, на которой Дэви сообщил о своих результатах, состоялась 19 ноября 1807 г. [11, с. 99]. Уже в 1808 г. Гей-Люссак и Тенар получили калий и натрий в препаративных количествах, накаливая в железной трубке смесь чистых щелочей с железными опилками, а немецкие ученые X. Бухгольц и И. Тромсдорф — путем восстановления соды и поташа накаливанием их смесей с углем и льняным маслом.
Я. Берцелиус, также занимавшийся электролизом, в своих опытах использовал ртутный катод, на котором в 1808 г. получил амальгаму аммония. Дэви воспользовался ртутным катодом в следующей модификации: из слегка увлажненных водой щелочных земель он изготавливал чашечки наподобие капеллей. Чашечку, наполненную ртутью, помещали в закрытом сосуде па платиновый диск, а сверху в ртуть опускали платиновую же проволоку. Такое устройство позволило Дэви получить в препаративных количествах амальгамы кальция, стронция, бария и магния. Отгоняя затем ртуть, он получал металл.
Получение щелочных и щелочноземельных металлов оказало сильнейшее влияние на сознание химиков. Стало ясно, что все «земли», не соединяющиеся с кислородом, на самом деле являются уже соединениями кислорода, как это и предполагал Лавуазье. Вновь поднялось значение понятия химический элемент. Химики получили теоретическое основание и возможность трактовать получаемые ими новые незнакомые вещества как соединения новых элементов, и таким образом были определены окислы и соли многих еще не выделенных к тому времени металлов, напрпмоР иттрия, циркония и др.
Первые шаги в электрохимическом получении индивиду щелочных и щелочноземельных металлов были медленными
184
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
трудными, а зачастую и неуспешными. Бериллий, например, уда-дось восстановить лишь химическим путем. Это осуществили в 1828 г. Ф. Вёлер и, независимо, А. Бусси, действуя на хлорид бериллия металлическим калием. Зато литий, о существовании соединений которого узнали лишь в 1817 г., уже через год тем же Дэви совместно с X. Брандесом был электролитически выделен
________ —— V ш- л » Rg-W X~v R R' » 4 — R Ш- X-. — ~ xR. . .. xr. в L
I
(Li, Na, H) [AlSiiOio] «——* ** •* **
в I____
свободном состоянии, правда в очень малых количествах.
Содержащие литий минералы — петалит основной формулы 1 и сподумен (Li, Na) Al[Si2O6] — находили Швеции в 1800 г. и позже. В 1817 г. ученик Берцелиуса J0, А. Арфведсон (1792—1841) проанализировал образец петалита и первоначально не свел баланс, потеряв около 4% вещества. Несколько раз повторяя анализ, Арфведсон получал тот же результат. Тщательные поиски позволили из раствора, содержащего сульфаты щелочных металлов, выделить заметно более растворимую соль, из которой был получен новый, дотоле неизвестный тип «огнепостоянной щелочи». По предложению Берцелиуса, это вещество назвали «литион» от греческого литое — камень. В 1818 г. Л. Гмелин обнаружил, что литий придает пламени характерную окраску.
К 1825 г. литий был обнаружен еще в нескольких минералах и минеральных водах. Препаративный способ получения лития путем электролиза расплава его хлорида разработан в 1855 г. Р. Бунзеном и А. Матиссеном и несколько позднее улучшен Троо-стом, предложившим усовершенствованный электролизер.
Вопрос о том, почему следующие два щелочных металла — рубидий и цезий — были открыты более чем через 40 лет после лития, заслуживает особого обсуждения.
Рубидий нельзя отнести к редким элементам. Даже в списке элементов, содержащихся в организме, он занимает 14-е место. На 70 кз веса человек содержит 245 г калия, 105 г натрия и 1,2 г Рубидия (1,7- 1О“зо/о). По весовому содержанию в земной коре Рубидий (ат. вес 85,47) занимает 17-е место в общем списке элементов (0,03%), опережая среди щелочных металлов литий (нт. вес 6,94), занимающий 36-е. и цезий (ат. вес 132,9), занимающий 43-е место. В атомных процентах соотношение несколько иное, но атомов рубидия в природе больше, чем атомов мышьяка, °лова, свинца или ртути. И тем не менее названные элементы и соединения известны за тысячи лет до обнаружения рубидия. Же относится и к цезию, число атомов которого в природе дольше, чем, например, атомов сурьмы или висмута.
Чтобы понять причину сложности обнаружения рубидия и це-ЗИя, полезно сопоставить атомные проценты прочих щелочных ^пталлов. Оказывается, что на 1 атом цезия в земной коре содержится 14 атомов рубидия, 200 атомов лития и 6400 атомов калия. иРИ этом рубидий в минералах изоморфно замещает калий; при-Мерпо тдкор же геохимический характер цезия. Литий же геохи-
ГЛАВА ПЯТАЯ
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Первый спектроскоп Бунзена и Кирхгофа (1860)
В полую стеклянную призму 1 залит сероуглерод. Призму поворачивают ручкой 2. Угол поворота отсчитывают по удаленной шкале, наблюдаемой через зеркало 3. 4 — горелка Бунзена
Усовершенствованный многопризменный спектрограф Кирхгофа (1862)
мически близок не щелочным металлам, а магнию и закисному железу.
То, что рубидий и цезий сопровождают калий, практически не образуя сколь-нибудь заметного количества собственных минералов, приводит к тому, что исследователь, ищущий рубидий п цезий, должен обнаруживать в основном веществе крайне малую примесь, весьма близкую по свойствам к основному веществу. Влияние, которое оказывает такая примесь, определяется в первую очередь не весовой, а ее мольной долей в исследуемом веществе. Мольная же доля при равном весовом содержании тем выше, чем ниже атомная (молекулярная) масса. Весовое содержание и лития, и рубидия в наиболее богатых ими минералах составляет 3—4%. Но если для лития, например в лепидолите1 * *, это отвечает содержанию более 6 ат.%, то для рубидия в том же лепидолите такое же весовое содержание соответствует около 0,5 ат.%.
Все эти особенности привели к тому, что открытие рубидия и цезия произошло только на рубеже нового этапа в развитии аналитической химии,— когда на помощь органам чувств человека
1 Лепидолит, или литиевая слюда, относится к группе фторалюмосилит<а
тов: K2(Li, Al)5-e(Rb, Cs) [Si6—tA12—i02o] (OH, F)4. И хотя из щелочнЫ4
металлов в нем обычно преобладает калий (4—13% К2О), содержал^
1д2О лежит в пределах 3—7% от массы минерала.
пришли не только механические, но и более сложные физические приборы, в частности оптический спектроскоп.
Испытывая на конкретных природных объектах созданный ими аналитический спектроскопический метод, Р. Бунзен и Г. Кирхгоф, работавшие в Гейдельберге, исследовали минеральные воды расположенных сравнительно близко от Гейдельберга источников Бад-Дюркгейма и Бад-Крёйциаха. При исследовании первых же проб в 1860 г. Бунзен обнаружил не известные ранее две голубые спектральные линии, усиливавшиеся при концентрировании пробы.
Р. Бунзен был убежден, что эти линии принадлежат новому химическому элементу из группы щелочных металлов, который назвали цезием за небесно-голубой цвет (лат. cesium) его характерной спектральной линии.
Несколько позже, уже в 1861 г., изучая спектры солей, выделяемых при концентрировании вод Бад-Дюркгейма, Бунзен зафиксировал и фиолетовые линии, до тех пор не известные, в том иисле две линии высокой интенсивности. Такие же линии он обнаружил и в спектрах саксонского лепидолита. За наличие в спект-Рах этого элемента нескольких темно-красных линий (лат. rubis) его назвали рубидием.
Поскольку выяснилось, что новые элементы химически подобны калию, их смесь осаждали в виде хлороплатинатов и далее Раскристаллизовывали, используя небольшие различия в растворимости. Насколько кропотливым было это разделение, можно судить но величинам растворимости хлороплатинатов.
186
187
ГЛАВА ПЯТАЯ
Хлороплатинат калия рубидия цезия
Растворимость, е/100 а Н?О
0° с 100° с
0,478 5,03
0,014 0,33
0,0047 0,0915
Для выделения новых элементов Бунзену пришлось перерц-ботать более 40 м3 минеральных вод, в результате чего было выделено 17 г хлористого цезия и несколько большее количество хлористого рубидия. Из 150 кг лепидолита также было выделено некоторое количество цезия и ощутимое количество рубидия, химию которого удалось довольно хорошо изучить. Все стадии выделения и кристаллизации контролировали спектроскопически, что позволило найти паилучшие условия и избежать потерь искомых веществ. Вся эпопея открытия рубидия и цезия оказалась первым триумфом нового аналитического метода — оптической спектроскопии.
Металлический рубидий впервые получил в 1863 г. Р. Бунзен восстановлением гидротартрата рубидия. С выделением металлического цезия было немало сложностей, поскольку он немедленно воспламеняется при контакте с воздухом. О получении достаточно чистого металлического цезия впервые сообщил в 1882 г. К. Зеттерберг, проводивший в особых условиях электролиз расплава смеси цианидов цезия и бария.
Г ало гены
Из числа галогенов хлор и фтор достаточно широко представлены в природе, занимая место во втором десятке элементов земной коры, как по общей массе, так и по числу атомов. Их геохимическое поведение обусловлено степенью растворимости галогенидов. Так, большинство фторидов нерастворимо и фтор входит в состав многих силикатов и фосфатов. Большая часть остальные галогенидов растворима, и основная часть хлора, брома и иода земной коры содержится в соляных отложениях, соленых грунтовых водах или водах мирового океана.
Хлор представляет собой неотъемлемую и значительную по массе часть живых организмов (на 70 кг веса человека приходится около 105 г хлора), но чрезмерно не концентрируется, тогда кая иод заметно накапливается, особенно в морских водорослях.
Из числа стабильных галогенов только для брома неизвестно определенное биологическое значение, хотя и он избирательно усваивается некоторыми моллюсками и водорослями. По содержа' пию в земной коре бром занимает место в шестом, а иод — в сеДь' мом десятке химических элементов. Все эти особенности предопр6' делили характер открытия элементов группы галогенов.
Как уже упоминалось, обыкновенную поваренную соль челонс
188
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
знал и использовал с глубокой древности. В античные времена соль добывали путем выпаривания морской воды, позже — путем вЬ1Варки из минеральных вод.
С Х1П в. в Европе приобретает значение каменная соль, которую добывали шахтными разработками.
Получение хлористого водорода и соляной кислоты историки химии относят к XVI в. Упоминание о ней впервые встречается в 1595 г. у А. Либавия, а Р. Глаубер в XVII в. уже дал подробное описание получения соляной кислоты из поваренной соли и купороса или серной кислоты. Однако еще ранее, по крайней мере с начала XIII в., европейским алхимикам стала известна царская водка, которую они готовили из купороса, селитры и соли. Имея дело с царской водкой, они, в сущности, уже имели дело с хлором, по не могли его отделить и идентифицировать.
С появлением в химической практике соляной кислоты стало возможным получать различные индивидуальные хлориды. Так, Глаубер и позднейшие исследователи имели дело с хлоридами сурьмы и при их термической обработке, безусловно, наблюдали выделение хлора, но не улавливали его: методические основы обращения с газами появились лишь в конце XVII в. после работ Р. Бойля. Не созрели также и методологические установки: настоящий интерес к газообразным веществам развился только во второй половине XVIII в. И тут же был открыт хлор.
К. В. Шееле держит в истории химии абсолютное первенство по числу открытых им элементов пли индивидуальных соединений ранее неизвестных химических элементов — шесть! С его именем
связано также открытие хлора и фтора.
В 1774 г. Шееле изучал природу пиролюзита (МпО2), в кото
ром предполагалось присутствие какого-то металла. Действуя на пиролюзит соляной кислотой, Шееле наблюдал выделение какого-то газа. Предполагая, что этот газ входит в состав пиролюзита, Шееле решил изучить его подробнее. Он поместил смесь пиролюзита с соляной кислотой в реторту, к оттянутому концу которой был подвязан пустой бычий пузырь. При нагревании реторты на песчаной бане началось выделение газа, заполнившего пузырь. Как сообщал Шееле, газ имел желто-зеленую окраску и резкий запах, как у горячей царской водки. Шееле наблюдал и
описал действие полученного газа на корковые пробки, листья и Цветы растений, растительные красители, бумагу, металлы и др.
К. Шееле полагал, что полученный газ — это дефлогистирован-цвя соляная кислота. Т. Бергман, латинизируя терминологию, взывал соляную кислоту acidum muriaticum (от лат. muria — Рассол, морская соль). Повое вещество назвали acidum muriaticum aephlogystorum. Французские сторонники вскоре появившейся кислородной теории считали хлор пе просто «окисленной соляной кислотой», но имели в виду, что он —сложное тело, в состав которого входит кислород (acide muriatique oxy gene). Эта точка зре-
189
ГЛАВА ПЯТАЯ h—....... ..... ....... — ----------'
ния основывалась на теории кислот Лавуазье, считавшего, чщ кислотный характер вещества определяется входящим в его состав кислородом. Поэтому кислород уже предполагался даже в исход, ной соляной кислоте. Сам Лавуазье, полагая, что бескислородная основа соляной кислоты должна существовать, включил гипотетический «радикал соляной кислоты» (le radical muriatique) в свою таблицу «простых веществ» 1789 г. Он включил в эту таблицу д «радикал плавиковой кислоты» (le radical fluorique), в которой также предполагал наличие кислорода.
Хотя фтор входит в состав достаточно часто встречающихся минеральных веществ — фосфоритов и апатита, первое и достаточно широкое применение из его природных соединений получил флюорит — плавиковый шпат (Fluflspat), использовавшийся в качестве флюса в металлургии. Описание его имеется в XVI в. у Агриколы, Либавия и в трудах легендарного Василия Валентина. В 1670 г. нюрнбергский мастер Генрих Шванхардт обнаружил, что смесью плавикового шпата и серной кислоты можно травить стекло, и использовал этот прием для нанесения узоров на стеклянную посуду. Однако характер действующего начала оставался неизвестным вплоть до исследований Шееле 1771 г.
К. Шееле выделил, хотя и не очень чистую, плавиковую кислоту и доказал, что именно она растворяет стекло. Он доказал также, что плавиковый шпат представляет собой соединение «известковой земли» с этой кислотой.
Предположение о том, что в состав соляной и плавиковой кислот входит кислород, продержалось до конца первого десятилетия XIX в.
В 1808—1809 гг. Ж.-Д. Гей-Люссак и Л. Тенар исследовали хлор, соляную кислоту (сухой хлористый водород) и плавиковую кислоту (фтористый водород). Последнюю они впервые получили в достаточно чистом виде, отгоняя из смеси плавикового шпата с серной кислотой в свинцовом сосуде. Задача заключалась в том, чтобы обнаружить в исследуемых веществах кислород. Гей-Люссак и Тенар пропускали газ через раскаленную фарфоровую трубку, наполненную кусочками древесного угля. Ни уголь, ни хлор ие подвергались видимым изменениям. Г. Дэви, повторив эти опыты, получил тот же результат. Он попытался затем разложить хлор электролитически, но, разумеется, безуспешно.
Столь же безуспешны оказались попытки электролитического разложения газообразного сухого хлористого (впоследствии и фто' ристого) водорода. Зато действуя хлором на металлы и окисл# металлов, Дэви получил их хлориды, идентичные солям, из которых можно было выделить соляную кислоту. Водные раствор# последней он также разлагал, получая хлор. Однако при взаиМ0' действии хлора и водорода, которое параллельно изучали Дэви Гей-Люссак с Тенаром, получались хлористый водород и соляяаЯ кислота, но не получалась вода. Все эти опыты привели ДэвЯ
190
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
убеждению об элементарной природе хлора и бескислородной природе хлористого водорода.
В то же время известный французский ученый Андре Мари Ампер (1775—1836) сопоставил поведение и характер соляной и плавиковой кислот и показал их аналогию. Дэви, признав аргументы Ампера, попытался разложить электролитически и плавиковую кислоту. Несмотря на неудачу, он не сомневался, что в этой кислоте существует начало, подобное началу соляной кислоты.
По предложению Дэви, «начало соляной кислоты» получило название от греческого хлорос — зеленоватый (лат. хлорум означает зеленый).
В 1811 г. немецкий ученый И. Швейгер предложил называть хлор галогеном, т. е. солеродом — порождающим соли, а хлориды металлов — галогенидами, т. е. солеподобными. Оба эти термина впоследствии привились как групповые.
В этом же 1811 г. был открыт третий элемент из группы галогенов — иод.
Открытие иода связано с решением практической задачи производства черного пороха — в то время основного взрывчатого вещества. Громадная наполеоновская армия нуждалась в большом количестве пороха, население было обложено натуральной селитряной податью, и во всей Франции развивалось промышленное и кустарное производство селитры. Технология его, описанная еще в XVI в. Эркером, заключалась в приготовлении увлажняемых мо-пой навозно-земляных компостов, в которые замешивали также золу или известь. Через некоторое время нитрифицирующие микроорганизмы превращали азотистые соединения в нитраты, которые затем вымывали из компостов водой. Для превращения нитрата кальция в необходимый для пороха нитрат калия — калийную селитру — полученные щелока обрабатывали поташом, осаждая карбонат кальция. Этот процесс требовал значительного количества поташа, источники которого — древесина — были во Франции уже ограниченны.
Одним из производителей селитры был ученик А. Фуркруа и Л. Тенара умелый химик Бернар Куртуа (1777—1838), который, кстати, за 12 лет до Сертюнера выделил из опия кристаллическую соль морфина. Для получения поташа Куртуа пытался использовать золу морских водорослей, которую жители Бретани издавна иримепяли в качестве моющего средства. Проводя концентрирование зольных и селитряных щелоков, Куртуа заметил, что его бедные котлы быстро корродируют. Обнаружению причины способствовал случай: кошка разбила склянку с серной кислотой, ко-т°рая попала в золу, вызвав бурное выделение фиолетовых паров.
Б. Куртуа довольно подробно изучил новое вещество: его кристаллическую форму, физические свойства, взаимодействие ^ Фосфором, металлами, водородом и аммиаком. О своем открытии ^Уртуа сообщил Парижской Академии наук, где Гей-Люссак про-
ГЛАВА ПЯТАЯ
вел опубликованное в 1814 г. и ставшее классическим образцом обстоятельное исследование нового вещества, попутно доказав его элементарную природу [12]. Аналогичное исследование провел и Дэви.
За свое открытие Куртуа получил от Академии премию 6000 франков, а новый элемент, подобно хлору, получил название в соответствии со своей окраской: иодес по-гречески означает фиалковый.
Таким образом, к 1814 г. большинство химиков признали существование трех элементов—галогенов и элементарную природу хлора п иода, что окончательно опровергло кислородную теорию кислот Лавуазье.
Открытие четвертого элемента этой группы — брома — было связано уже не только с чисто практическими, а и с систематическими научными исследованиями. После открытия иода оно было неизбежно. На бром должны были натолкнуться при попытках найти в природе или промышленно получить иод, поскольку распространенность брома в земной коре на порядок выше распространенности иода. Все зависело лишь от внимательности исследователя.
В 1825 г. знаменитому в будущем Ю. Либиху (1803—1873) была прислана на экспертизу проба бурой жидкости, полученной действием хлора на маточный раствор промышленного производства иода. Молодой химик, незадолго до того принявший кафедру в Гиссенском университете, посчитал присланное вещество смесью хлора и иода.
Летом 1825 г. студент Гейдельбергского университета Карл Лёвих (1803—1890) исследовал минеральную воду Бад-Крёйцнаха. Пропуская в нее хлор в поисках иода, он обнаружил окрашивание воды и появление красно-бурых капель. Экстрагируя эфиром, Лёвих выделил некоторое количество брома и показал его своему профессору Леопольду Гмелину. Было решено накопить большее количество вещества для исследования, но к этому времени появилась публикация А. Ж. Балара.
Фармацевт по образованию, Антуан Жером Балар (1802— 1876), в то время препаратор па кафедре химии Университета в Монпелье, изучал содержание иода в растительных и животных тканях. С этой целью он озолял материал, а затем обрабатывал золу или зольный щелок хлорной водой, контролируя появление пода по крахмалу. Изучая таким образом золу южнофранцузских водорослей, Балар заметил, что поверх синего слоя крахмала, соединившегося с иодом, имеется еще какой-то ярко-желтый слои, обладающий резким запахом. Балар сначала извлекал вещество этого слоя эфиром, а затем разработал способ прямого получения красно-бурой жидкости, воздействуя на соответствующие соли сер' ной кислотой или двуокисью марганца.
Еще не проведя обстоятельные исследования, Балар припЮл
192
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
к выводу, что полученное вещество — новый неметаллический элемент, единственный жидкий элемент из неметаллов, подобно тому, как ртуть — единственный жидкий металл. Он усмотрел сходство 110вого элемента с хлором и иодом и дал ему название мурид от латинского muria (см. выше).
О своем открытии Балар 30 ноября 1825 г. написал статью во французские «Annales*> и закрытое заявочное письмо в Парижскую Академию наук. После публикации статьи Академия создала специальную комиссию в составе Гей-Люссака, Тенара и Воклепа. 3 августа 1826 г. они наблюдали, как известный химик-аналитик Д’Арсе на их глазах проделывал пробы, указываемые Баларом. Месяц спустя открытие нового элемента было признано официальным документом. Комиссия только отклонила авторское название п предложила свое — от греческого слова бромос, что значит зловонный.
Впоследствии Балар стал академиком и заведовал кафедрой химии в Коллеж де Франс. К. Я. Лёвих впоследствии тоже стал профессором в тогдашнем Бреславле. Его научное имя частично связано с бромом, поскольку в 1832 г. он впервые получил бромоформ.
Ю. Либих после публикации Балара внимательно исследовал присланную ему на анализ жидкость и убедился в поверхностности своего первоначального заключения. Впоследствии насмешливо, по своему обыкновению, замечая, что не Балар открыл бром, а бром открыл Балара, Либих с горечью писал по этому поводу: «Не может быть большего несчастья для химика, как неспособность освободиться от предвзятых мнений и стремление объяснить наблюдаемые явления лишь на основе этих мнений, а не на опыте» [13].
Элементы группы платины
По содержанию в земной коре платиновые металлы находятся в последнем десятке стабильных элементов периодической системы. Довольно инертные химически, опи в основном находятся в природе в самородном состоянии и, обладая высокой плотностью, ’Скапливаются в россыпях при разрушении коренных пород. Будучи в самородном металлическом состоянии, платиновые металлы °бычпо совмещены друг с другом. Как правило, в индивидуальном Минерале преобладает какая-либо пара металлов. Так, ферропла-?ина содержит 70—80% Pt, до 4,5% 1г, до 0,5% Pd и 15—20% Fe. Палладистая платила содержит, помимо Pt (55—91%), до 40% Pd 11 некоторое количество золота. Зато иридий и осмий совмещаются Друг с другом в самых различных соотношениях, образуя таким °бразом несколько минералов. Палладий нередко встречается 11 ^единениях с золотом, свинцом и оловом и мышьяком, а руте-’htii ___ с серой. В качестве спутников встречаются также никель, МеДь и хром.
^Се0бщая история химии 193
ГЛАВА ПЯТАЯ
Внешне платиновый шлих имеет серовато-белый цвет и содер. жит белые и серые зерна собственно платиновой руды и обычно такого же цвета твердые плоские зерна пли гексагональные кристаллы осмий-иридия, по плотности близкие к зернам платины так что простой промывкой их отделить нельзя.
В Европе платина стала известна после открытия Америки. Возможно, что с платиной встречались и ранее, но никто не описьь вал ее как самостоятельный характерный металл. Первое описание платины как тугоплавкого металла, который можно расплавить только с помощью «испанского искусства», принадлежит итальянскому врачу Скалигеру (1557). Более подробно платину описали в 1748 г. испанский ученый и мореплаватель де Уоллоа, в 1750 г.— англичанин У. Уотсощ а в 1752 г.— шведский химик Хенрик Теофил Шеффер.
Начальное изучение платины и открытие каждого из элементов ее группы было связано с решением задач чисто прикладного характера. Исследователи пытались получить из платиновой руды — «сырой платины» — ковкий металл, пригодный для механической переработки. Исследования в этом плане на рубеже XVIII—XIX вв. проводили две группы ученых: французские, преимущественно А. Фуркруа и А. Воклен, и английские — У. Волластон и С. Теннант.
Уильям Хайд Волластон (1766—1828) учился в Кембридже, где получил медицинское образование, был затем практикующим врачом, даже доктором медицины, но проявлял особый интерес к химии и физике. В 1793 г. он был избран членом Королевского общества, а в 1800 г. полностью оставил практическую медицину и занялся исключительно наукой. Не будучи материально обеспеченным, Волластон решил получить средства для продолжения научной деятельности, разработав технически приемлемый спосоо изготовления изделий из платины и, видимо, уже к 1800 г. он, в принципе, овладел таким способом.
Находясь в дружбе и финансовых отношениях с другим английским ученым Смитсоном Теннантом (1761—1815), Волластон разделил с ним область научных исследований сырой платины. Теннант изучал черный остаток от растворения сырой платины в царской водке, а Волластон — сам раствор, причем особенно внимательно ту часть, которая остается после осаждения платины хлоридом аммония. Насколько серьезны и масштабны были я* намерения, можно судить по тому, что Волластон п Теннант, ирп-ступая к работе в 1800 г., приобрели и по частям растворили в цяр' ской водке 7000 унций сырой платины, т. е. около 200 кг.
У. Волластон первым добился успеха и, судя по сохранившимся черновикам, открыл палладий уже в июле 1802 г.
Не желая обнаруживать направление своих прикладных исследований, но не желая также утратить приоритет открытия ттовог0
194
ОТКРЫТИЕ И КЛАССИФИКАЦИЙ ХИМИЧЕСКИХ ЭЛЕМЕПТОЙ
металла, Волластон прибегнул к своеобразному способу информации, о котором рассказано ниже.
Работы французских ученых по исследованию платины и ее спутников тоже непосредственно связаны с получением платиновых изделий. В 1800-х годах Воклену удалось разработать надеж-’ ный путь очистки соединений платины, на основе которого ювелиры Жанетти сумели воспроизвести французский безмышьяко-вый способ получения металлической ковкой платины.
Последний из элементов платиновой группы — рутений — был найден русским ученым К. К. Клаусом также при попытке решить технико-экономическую задачу: добиться более полного выделе ния платины из ее руд.
Успех открытия металлов платиновой группы был обеспечен не только практической направленностью основных исследований с побочным выходом в виде новых химических элементов. Дело в том, что, несмотря на близкое содержание этих элементов в земной коре, только платина была обнаружена в концентрированном самородном виде, привлекательном для переработки. Однако в богатых платиновых рудах ее спутники содержатся в малых количествах и неравномерно, так что обнаружение этих «минорных компонентов» оказалось возможным только потому, что технической переработке подвергалось большое количество платиновых руд и при этом накапливалось достаточное количество остатков.
Собственно история такова (см. схемы 1 и 2).
В своей книге «История платины» Д. Мак-Дональд [44] пишет, что еще немецкий посол во Франции К. Зикинген (1737—1791), стремясь получить ковкую платину, пытался исследовать черный остаток, образующийся при растворении платины в царской водке. Он сплавлял его с бурой, но исследование не завершил. В 1801 г. Ж. Пруст сообщил, что этот остаток составляет 2—3% от взятой сырой платины и не является ни графитом, ни свинцом (см. схему 1).
Исследователи, осаждавшие платину хлоридом аммония из растворов в царской водке, многократно отмечали, что осадки от Раза к разу отличаются по цвету, который меняется в широком Диапазоне — от желтоватого до темно-красного. Эту особенность исследовал французский горный инженер А. В. Колле-Декотиль (1773-1815), первым опубликовавший свои результаты в 1803— 1804 гг. Он обратил внимание на то, что черный остаток частично Растворяется в царской водке, особенно при повышенном содержании азотной кислоты. Он заметил также, что цвет осадка хлороплатината аммония зависит от степени растворения черного остатка. j*eM более его растворилось,, тем осадок хлороплатината краснее. ^олле-Декотиль заметил также, что если окрашенный осадок хлороплатината аммония прокалить и полученную платиновую губку Растворить в царской водке, то при этом снова остается черный остаток. Если полученный раствор обработать хлоридом аммония,
195
9*
f ЛАВА ПЯТАЙ
ОТКРЫТИЕ ЭЛЕМЕНТОВ
1804 Волластон
196
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Схема 1
гГуППЫ ПЛАТИНЫ
(следы)
7Р7
ГЛАВА ПЯТАЯ
198
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
54
т0 новый осадок хлороплатината оказывается более светлым, педели исходный. Было замечено, наконец, что при наличии в растворе платины восстанавливающих веществ с хлоридом аммо-яИЯ выпадает только желтый осадок. Колле-Декотиль пришел к выводу, что наблюдаемые явления обусловлены наличием в исходном материале какого-то неизвестного металла (см. схему 1).
Современный читатель понимает, что роль восстанавливающих веществ заключается в переводе иридия(1У) в иридий(Ш), который с хлоридом аммония осадков не образует, но этот вывод сумел сделать лишь К. К. Клаус 40 лет спустя (см. схемы 1 и 2).
В то же время (1803—1804) Фуркруа и Воклен опубликовали проведенное ими исследование платиновых руд. Изучая свойства черного остатка, они обнаружили, что он полностью сплавляется с едким кали и (или) селитрой. Плав растворяется в воде, образуя зеленый флуоресцирующий раствор и зеленый же остаток. Исследователи подозревали присутствие в растворе хрома, а остаток растворяли в кислотах и различным образом испытывали, но определенного результата не получили. Тем не менее они высказали убеждение, что имеют дело с новым, ранее неизвестным металлом, присутствие которого обусловливает окраску высаживаемой аммонийной соли. Тщательнее изучая характер ее осаждения, Фуркруа п Воклен установили, что сначала выпадает светло-желтый, потом оранжевый, а последним — красный осадок. Если осадок хлороплатината аммония несколько раз подвергать последовательным циклам прокаливание—растворение—осаждение, то можно добиться полного отделения неизвестного металла от платиновой соли. Другой путь — растворение красной составляющей в кипящей воде, лучше с прибавлением едкого кали. Желтый осадок платиновой соли при этом не растворялся. Щелочной экстракт подкисляли, причем выпадал зеленый хлопьевидный осадок. Его промывали, сушили и прокаливали, получая новый металл.
Фуркруа и Воклен нашли этот металл и в платине, очищенной по мышьяковому способу Жанетти. Чтобы лучше изучить свойства нового металла, они намеревались накопить большее его количество (см. схему 1).
В то же время в Англии исследованием черного остатка записался уже упомянутый Смитсон Теннант. Осенью 1803 г. он также убедился, что этот остаток отнюдь не свинец, по некий новый металл. Несколько позже, изучив появившиеся к тому времени пУбликации Колле-Декотиля и Фуркруа с Вокленом, Теннант при-к выводу, что все они имели дело не с одним новым металлом, а с Двумя. Проводя разделение по Воклену попеременным действием щелочи и кислоты, Теннапт выделил второй неизвестный Рапее металл из щелочного раствора, в котором Воклен предпола-Гал лишь наличие хрома. Сначала оп пе мог подтвердить этого, 110 впоследствии установил, что после подкисления щелочного Раствора из пего можно отогнать летучий окисел. Это бесцветное
ГЛАВА ПЯТАЯ
вещество, обладающее резким и характерным запахом, копдепсц-ровалось в виде густой жидкости, застывавшей в полупрозрачную массу. Теннант предложил назвать новый металл осмием (от греческого осмэ — запах).
Второй металл, выделенный Фуркруа и Вокленом, Теннант предложил назвать иридием (от греческого ирис — радуга) в связи с разнообразной окраской, которая возникает при растворении его соединений, в частности в морской воде (см. схему 1).
С. Теннант опубликовал своп исследования в 1804 г., доложив результаты в Английском королевском обществе 21 августа; французские ученые без дискуссии признали его приоритет, хотя сам Теннант всегда отдавал должное французским коллегам, считая, что иридий открыли они.
Между тем, еще в апреле 1803 г. в витрине небольшой лавки в Сохо (Лондон) появилось объявление о том, что у ее владелицы, г-жи Форстер, можно приобрести образцы нового благородного металла под названием палладий. В восьми пунктах этого объявления, отнюдь не для случайных покупателей, а для компетентных ученых, перечислялись физические и химические свойства нового металла. Его серебристая фольга продавалась по цене 1 шиллинг за 1 гран (английский граи 0,0648 г) 2.
Напечатанные типографским способом объявления такого же (содержания были анонимно разосланы многим английским ученым. Началась дискуссия о природе нового металла, который английский химик Р. Ченевикс полагал сплавом платины со ртутью, а всю рекламную затею — розыгрышем. Однако Воклен, которому были посланы образцы, проведя анализы, подтвердил индивидуальность испытанного металла.
24 июня 1804 г. Волластон представил Английскому королевскому обществу доклад, в котором сообщил, что палладий в небольших количествах содержится в «платиновой руде» и что из этого же источника им выделен еще один повый металл, которому Волластон дал название родий (от греческого родон — роза) за розовый цвет его солей. Более подробные сведения о выделении этих элементов опубликованы Волластоном в 1805 г. (см. схему !)•
Исследуя раствор, из которого хлоридом аммония высажен осадок, содержащий платину, Волластон ввел туда железо. В результате появился осадок — шламм, который растворялся лишь в цар-ской водке. Из полученного раствора хлоридом аммония удалось высадить дополнительное количество платины. Маточный раствор снова совместили с железом, в результате чего был получен «второй металлический осадок» (см. схему 1).
2 Для сравнения заметим, что в 1809 г. Волластон ценил производимую 1|А( ковкую платину по 16 шиллингов за тройскую унцию (31,1 ?/’ а в 1821 г.— уже по 5 шиллингов. На рынке она стоила заметно дороже.
200
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
PALLADIUM;
OR,
NEW SILVER, Has «nese Properties.amongst other» that shew it to be A NEW NOBLE METAL.
red “ P“rc Spirit Of Nitre> and makes a dark
2» Green Vitriol throws it down in the state of a regulus from this solution, as it always does Gold from Aqua Rtgia.
X If you evaporate the solution you get a red calx that dissolves tn Spirit of Salt or other acids»
4. It ts thrown down by Quicksilver and by allthemetab ’ but Gold, Platina, and Silver,
* 5. Its Specific Gravity by hammering was only 11.3, but
by flatting as much as 11.8.
. 6. In a common fire the face of it tarnishes a little and
turns blue, but comes bright again, like other noble metals on
v being stronger heated. *
1 7* Тнж g^atest heat of a blacksmith’s fire would hardly v
J melt it; 7
i 8; BuT y<>u touch it while hot with a small bit of Sul-
phur it runs as easily as Zinc»
1 П is SOLD ONLY BY
Mr, FORSTER, at No, 26, GERRARD STREET, SOHO,
I 1
I nj e c Five Shillings, Haifa Guinea, & One Guinea each.
J. Moore, FrirHet, Drury Laoc
Объявление о продаже нового благородного металла под названием палладий
Волластон обработал этот осадок сначала разбавленной азотной кислотой, удалив медь, а затем и концентрированной азотной Золотой, которая растворила еще часть осадка3, окрасившись при Эт°м в темно-красный цвет. Капля полученного раствора оставляла На чистой медной пластинке черный осадок, который можно было йовь растворить в азотной кислоте; при этом получался темнокрасный раствор (см. схему 1). Затем Волластон прибавил к ос-
Установил^ позже сам Волластон, не растворявшаяся в концентриро-анной азотной кислоте часть осадка — это иридий.
201
ГЛАВА ПЯТАЯ
новной массе азотнокислого раствора некоторое количество ртутц и нагревал, помешивая, до тех пор, пока ртуть не приобрела вид амальгамы. Извлекши ее, Волластон нагреванием до температуры красного каления отогнал ртуть и получил белый металл, который не плавился под паяльной трубкой. Этот металл растворялся в азотной кислоте, образуя темно-красный раствор, из которого хлорид аммония не выделял никакого осадка.
Полученный новый металл Волластон и назвал палладием по имени незадолго до того открытого астероида Паллада (см. схему 1).
Неоднократно повторяя процесс, Волластон заметил, что сколько ни прибавлять ртути, окраска раствора полностью не исчезает. Он стал совершенствовать процесс осаждения палладия и заметил, что прекрасным реактивом для этой цели оказалась цианистая ртуть, осаждавшая цианид палладия, который при прокаливании превращался в металл. Однако окраска маточного раствора полностью не исчезала. Изучая его, Волластон сумел выделить еще один новый металл, который и назвал родием.
В 1819 г. месторождения платиновых элементов обнаружились па Урале. Был найден осмиридий, содержавший 60% 1г, 30% Os, 2% Pt и 5% Fe, а некоторое время спустя —и богатые руды платины. В 1826 г. в лабораторию Департамента горных и соляных дел поступило на исследование полпуда сырой гороблагодатской платины. Ее изучением занялись русские металлурги В. В. Любарский (1795—1852) и обер-берг-пробирер П. Г. Соболевский (1782—1841), который самостоятельно разработал производственный процесс выделения очищенного до нужной степени хлороплатината аммония и получения высококачественного ковкого металла из платиновой губки; его коммерческая цена была определена 4—5 руб. за золотник (около 1 руб. за 1 а). На лондонском рынке в то время 1 тройская унция платины стоила 25—28 шиллингов (приблизительно 12—14 руб.).
Между тем, тогдашний русский министр финансов и глава Горного департамента Е. Ф. Канкрин разослал образцы русской платиновой руды иностранным научным обществам и отдельным химикам с просьбой изучить материал и внести предложения по его использованию. Он выслал, в частности, 2 фунта во Францию, 1 фунт в Англию, 1,5 фунта А. Гумбольдту, по 0,5 фунта Волластону и Берцелиусу и др. Одновременно 4 фунта получил профессор химии Дерптского университета Г. Ф. Озанн (1796—1866). z
В 1826—1828 гг. Озанн опубликовал несколько сообщений ею исследовании полученной им платиновой руды. Первоначально оп обнаружил в ней три (!) новых металла, образцы которых посла# Берцелиусу. Берцелиус, который сам проанализировал образцы русской платины и не нашел ничего нового, внимательно проверь пробы Озанна. Две из них — поллиний и рутений — оказались сплавами окислов кремния, титана, циркония и иридия, а третий"'
202
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
пЛюраний — Берцелиус признал индивидуальным и новым для себя телом. Однако Озанн не смог получить дополнительную порцию эТОго нового вещества и вскоре отказался от своего анонсированного открытия.
Новые результаты были получены только через полтора десятка лет.
Между тем, в 1828 г. Канкрин решил чеканить из платины монету, и на Монетном дворе по методу Соболевского стали перерабатывать по 3 пуда платины в неделю. Этого оказалось мало, и к 1834 г. научились очищать по 1 пуду в день, правда, за счет некоторого снижения качества очистки. Тем не менее на Петербургском монетном дворе накопилось значительное количество платиновых остатков, т. е. того, что остается после растворения платиновой руды в царской водке.
В 1837 г. заведование химической лабораторией Казанского университета принял Карл Карлович Клаус (1796—1864), в прошлом работавший и учившийся в Дерпте. Он начал свою научную деятельность исследованием Сергиевских минеральных вод на Урале. Совершая там поездки, он побывал также на платиновых приисках и решил подробнее изучить выделение платины из уральских руд, имея в виду повысить степень ее извлечения. С этой целью он обратился к Соболевскому, который в 1840 г. прислал ему 2 фунта остатков. В этой порции содержалось около 10% неизвлеченной платины, и Клаус, желая отработать способ извлечения платины из остатков, запросил добавочное их количество.
В 1842 г. Клаус получил новую значительную порцию — 15 фунтов очень бедных платиной остатков. Первоначальная цель отпала, и Клаус предпринял просто обстоятельное изучение состава полученного материала. Переработка материала потребовала больших усилий. Подобно Воклену, Клаус сплавлял остатки со щелочью и селитрой, затем выщелачивал сплав водой, полученный раствор концентрировал, кристаллизовал и исследовал. Основную часть кристаллов составлял осмат калия. Из маточного раствора после подкисления азотной кислотой отгонялся остаток осмиевого ангидрида и выпадали осадки кремниевой и титановой кислот. Клаус отделял их, промывал соляной кислотой и исследовал маточник и промывные воды. Оказалось, что при прибавлении хлористого калия из пурпурно-красного солянокислого раствора выпадает так же окрашенная двойная соль, которую Клаус сначала полагал с°единением иридия.
Остаток селитряного плава, пе растворившийся в воде, Клаус Растворял в царской водке, раствор выпаривал, сухой остаток промывал и растворял в растворе поташа. После нейтрализации соляной кислотой выпадала все та же окрашенная двойная соль.
В одном из вариантов обработки Клаус хлорировал остатки су-хлором, а затем выщелачивал. Его насторожило, что серово-Д°родная проба неожиданно дала синее окрашивание, а осаждеп-
203
ГЛАВА ПЯТАЯ
ная из раствора хлоридов гидроокись железа, простояв несколько дней, почернела.
Все признаки свидетельствовали о наличии какого-то ранее неизвестного металла; Клаус выделил и изучил его (см. схему 2)
В 1844 г. Клаус опубликовал результаты своего двухгодичного исследования, назвав новый металл рутением (от латинизированного названия России — Ruthenid). После некоторой дискуссии Верцелиус, а за ним и другие химики признали результаты Клауса.
Узнав об открытии рутения, Озанн, к тому времени переехавший из Дерпта в Германию, предъявил претензии на приоритет, полагая, что рутений Клауса — это тот плюраний, который ему не удалось повторно выделить. Однако Клаус объяснил, что Озанн не мог иметь в руках рутений, так как исследовал только твердые остатки после кислотного осаждения, тогда как в его условиях рутений оставался в растворе.
То, что Озанн, не слишком изощренный в химическом эксперименте, мог допустить такой просчет, нет ничего удивительного. Но как мог ошибиться великий химик Берцелиус, хорошо знавший повадки металлов платиновой группы? Сам Клаус объяснил, что «двойную соль» хлоридов калия и рутения трудно отличить от аналогичного соединения иридия. Однако, по мнению Клауса, скорее всего, та порция уральской платиновой руды, которую Кан-крин прислал Берцелиусу, могла не содержать заметных количеств рутения.
Действительно, относительное содержание платиновых металлов в их рудах может существенно колебаться даже в пределах одного месторождения. Открытие Клаусом рутения при всей трудоемкости этой работы было в известной степени предопределено, так как Клаус имел дело не со случайной выборкой руды, а с остатками, полученными в результате переработки по меньшей мере сотен килограммов руды, что статистически усредняло состав. Впрочем, успехи Волластона и Теннанта в открытии элементов группы платины также связаны с накоплением остатков в связи с технической переработкой значительного количества «сырой платины».
Редкоземельные элементы
Исторически в группу редкоземельных элементов (РЗЭ) объединяли все новые элементы, которые обнаруживали в «редких землях» — цериевой и иттриевой. По этому геохимическому критерию к числу редкоземельных относят, помимо лантана и лантаноидов, также иттрий и скандий.
Самое замечательное в истории открытия РЗЭ это то, что три четверти их числа впервые обнаружено в двух минералах: иттер* бите (гадолините) и тунгстене (церите), а еще три —в самарските-Причина этому — необычайная близость химических свойств РЗЭ,
204
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Схема 3
ПОСЛЕДОВАТЕЛЬНОСТЬ ОТКРЫТИЯ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ
эта же причина затрудняла их химическое разделение, а следовательно, и открытие. В результате открытие элементов этой группы происходило ступенчато: выделяли смесь соединений, признавая ее за индивидуальное вещество, и давали название предполагаемому элементу. Через некоторое время выяснялось, что предшествующее исследование имело дело не с химическим индивидом, а со смесью, и процесс познания повторялся. Как отмечают историки пауки, выявление отдельных РЗЭ и их групп сопровождалось множеством ошибок, число которых с годами не уменьшалось, а увеличивалось. Так, только с 1878 по 1910 г. зарегистрировано не менее 90 ложных открытий РЗЭ [8, с. 37].
Последовательность открытия редкоземельных элементов представлена на схеме 3.
История РЗЭ начинается с открытия двух минералов.
В 1750 г. А. Кронштедт изучил и описал красноватый минерал, найденный на медно-висмутовом месторождении Бастнес. За высокий удельный вес Кронштедт назвал этот минерал тунгстеном (по шведски — «тяжелый камень»).
Когда К. Шееле в 1781 г. описал природный вольфрамат кальция, из которого выделил трехокись вольфрама, Т. Бергмап пред-,Г()Жид для пего латинское название лапис пондерозус — «тяжелый камень», т. е. по-шведски опять-таки тунгстен, что затем не раз
Ж
ГЛАВА ПЯТАЯ
приводило к путанице. Впоследствии название тунгстен в ряде языков закрепилось за вольфрамом, а вольфрамат кальция получил название шеелит. Между тем, К. Шееле и Д’Эльгуяр исследовали тунгстен Кронштедта и не обнаружили в нем вольфрама.
В 1787 г. минералог К. Аррениус в заброшенном карьере возле селения Иттербю, недалеко от Стокгольма, нашел необычный тяжелый черный минерал, который назвал иттербитом. Высоким удельным весом этот минерал напоминал и вольфрамовые руды и тунгстен Кронштедта.
В 1794 г. финский химик Юхан Гадолин (1760—1852) произвел химический анализ иттербита и не нашел вольфрама, но обнаружил окислы кремния, железа, как он предполагал, магния, а также 38 вес. % неизвестного ранее вещества, нерастворимого в воде, имевшего характер основания и растворявшегося в кислотах с образованием солей4 5.
В 1797 г. шведский химик А. Экеберг повторил анализ этого минерала, получившего новое название — гадолинит, и подтвердил результаты Гадолина, но нашел, что нового вещества, названного им иттриевой землей, содержится 55%. Дальнейшие исследования, в которые включились французские и немецкие химики, не прояснили положения: по Воклену, иттриевая земля составляла 35%, а по Клапроту — 59,75%.
В 1803 г. М. Клапрот и, независимо от него, Я. Берцелиус и В. Хизингер обратились к изучению тунгстена Кронштедта. Схема разложения у них оказалась примерно одинаковой. Минерал растворяли в кислоте, причем отделялся кремнезем. Затем подщелачиванием осаждали гидроокись, растворяя и отделяя глинозем. Не содержащий железа белый остаток гидроокисей составлял до 50% от исходной навески. При прокаливании он становился коричневым, хотя и не содержал железа. Дело было, конечно, в содержании окрашенного окисла празеодима, малейшие примеси которого придают смеси окислов РЗЭ желтовато-бурую окраску.
М. Клапрот предложил назвать новое вещество охроитом от греческого охрос — желтовато-коричневый. Берцелиус был смелее. Он считал, что имеет дело с окислом нового, ранее неизвестного металла, для которого предложил название церий по имени открытой в 1801 г. малой планеты Церера. Минерал, из которого было выделено новое вещество, тунгстен Кронштедта, стали называть церитом*.
После того как Г. Дэви в 1807—1808 гг. успешно выделил металлы из щелочей и щелочных земель, Берцелиус и другие хп-
4 По современным данным, гадолинит имеет основной состав МгОз* •3(Ве, Fe)O-2SiO2, где М — редкоземельный элемент. Обычно содержится 31—47% иттриевых и 3—8% цериевых РЗЭ.
5 По современным данным, церит имеет основной состав 2СаО-3(М, А1)гОз’ •6SiO2-3H2O, где М — редкоземельный элемент. Содержание цериевых РЗЭ в церите достигает 58—69%.
206
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
мики уверились, что и иттриевая земля — кислородное соединение металла, который стали называть иттрием.
В 1826 г. Ф. Вёлер и К. Мозапдер пытались получить металлический церий, восстанавливая его хлорид металлическим натрием. В процессе подготовки материала была замечена химическая неоднородность цериевой земли.
Карл Густав Мозандер (1797—1858), с 1832 г.—преемник Берцелиуса на кафедре в Медико-хирургическом институте, вернулся к обстоятельному исследованию церита лишь 10 лет спустя. В конце 1838 г. Мозандер определенно пришел к выводу, что в церите содержится «нечто новое». Мозандер «дал мне понять,—писал Берцелиус Вёлеру 1 февраля 1839 г.,—что то, что мы с Хизингером называли церием, было смесью двух окислов, ни один из которых не обладает свойствами смеси» [14]. Через две недели между Берцелиусом и Мозандером речь шла уже о новом элементе — лантане. *
Новооткрытый лантан просуществовал недолго. Уже в 1841 г. Мозандер разделил лантановую землю на два компонента. Один из них сохранил название лантан (от греч. ланта — скрытый), а другой назвали дидим от греческого дидомос — близнец, один из двойни. Мозандер не предвидел тогда, что через 60 лет выяснится, что в его дидиме содержалось пять (!) редкоземельных элементов.
Выделив лантан и дидим, Мозандер обратился к иттриевой земле. В 1842—1843 гг. он разделил ее на три компонента, которые назвал иттриа, тербиа и эрбиа,— все три названия образованы от имени селения Иттерби. Две из этих земель — иттриевая и тербиевая — действительно содержали по одному РЗЭ, но в эрбиевой земле, как выяснилось впоследствии, содержалось семь (!) редкоземельных элементов.
Указанные особенности открытия редкоземельных элементов дают основание для установления следующей датировки их открытия до 1870 г., т. е. до формулирования периодического закона: церий — 1839 г., лантан — 1841 г., иттрий — 1843 г., тербий — 1843 г. Все последующие элементы этой группы были обнаружены начиная с 1879 г., т. е. после открытия периодического закона.
ОТКРЫТИЕ ЭЛЕМЕНТОВ И РАЗВИТИЕ ХИМИИ
Динамика открытия. Точную динамику открытия элементов трудно представить в наглядной форме в силу рассмотренной выше многостадийности процесса открытия. Связанные с этим процессом события, относящиеся к рассматриваемому в настоящем томе историческому периоду, сведены в таблицу.
Как видно, наибольшее число событий, связанных с открытием элементов, приходится на период около полустолетия, точнее,
207
ГЛАВА ПЯТАЯ
Динамика открытия химических элементов с 1735 по 1863 г.
Годы 0 1 2 3 4 5 6 7 8 9
173 Co
4 H Pt
5 Ni
б H N
7 О F N Cl, Mn Ba • ()
8 Мо, W Те Выводы Лавуазье C, Cl, F, В, Ca, Mg, Ba, Al, Si v, Zr
9 Ti Sr Y Ti Cr Be
180 Nb, V Ta, I’d Ce Rh, Ir Os K, Na Mg, Ca, Ba, Zr, В Sr
1 CZ, F J, Si Cd, Li Se
2 67 Br Th
л 3 V Ce
КЗЧГ- 4 La V,Tb, Er, Di Nb, Ru
5
6 Сз | Rb, Tl 1 1 1 1 1 1
Примечание. В левой колонке таблицы указаны десятилетия, начиная с 1730-х годов (XVIII в.) и кончая 1860-ми годами (XIX в.). Прочие колонки таблицы, от 0 до 9-й, отвечают отдельным годам каждого десятилетия. Таким образом, каждая клетка таблицы соответствует конкретному году. Например, клетка на пересечении строки 173 и колонки 5 отвечает 1735-му году, а на пересечении строки 180 и колонки 0 — 1800-му году. Таким образом, никель открыт в 1751 г., а индий —в 1863 г.
Жирным шрифтом элементы обозначены в те годы, которые целесообразно считать датами открытия. Светлым шрифтом элемент обозначается в год предсказания или осознания существования элемента. Курсивное обозначение помещается в год подтверждения.
с 1771 по 1818 г. Из 49 элементов, открытых с 1735 по 1863 г., только четыре достоверно открыты до начала и девять — по окончании этого периода; в 1830—1840-х годах также подтверждено открытие четырех элементов, правильно предположен одни элемент (эрбий) и неправильно — другой (дидим).
208
ОТКРЫТИЕ и кттассйфйКация химических элементов
Сопоставляя упомянутые пятьдесят лет с хронологией истории развития аналитической химии, мы видим,
р рергмана и заканчивается обобщающими методическими рабо-
j839" 1844 г., бесспорно, связан с обобщающими и методическими работами Генриха Розе, который сам в это время заново открыл ниобий.
Видимо, этими открытиями исчерпываются возможности клас
что этот временной интервал начинается обобщающими методическими работами 'р Бергмана и заканчивается обобщающими методическими работами Я- Берцелиуса и X. Пфаффа. Новый всплеск открытий
[
аботамп Генриха Розе, который сам в это время заново открыл
сического чисто химического анализа, поскольку все последующие открытия химических элементов, начиная с цезия и рубидия, достигнуты с помощью различных спектрально-аналитических методов.
О национальном вкладе ученых различных стран. Принято считать, что за рассматриваемые 135 лет ученые Швеции открыли 19 элементов, Англии—14, Германии —8, Франции —5, ученые Дании, России и Венгрии —по 1. С 14-ю элементами, известными до XVII в., это составляет 63 достоверных химических элемента, которыми оперировал Д. И. Менделеев при открытии периодического закона [8]. Такой подход формален, так как в числе «общепринятых дат» фигурируют и получение простых веществ, как. например, для алюминия, и получение окислов, как для иттрия, урана, циркония, и др. При таком подходе полностью исключается роль Лавуазье, признавшего наличие неизвестных еще элементов, входящих в состав некоторых кислот и «земель». Для правильной оценки научной активности ученых и их вклада в аналитическую и препаративную химию мы должны считаться с параллельными, почти одновременными открытиями хрома во Франции и Германии, церия в Швеции и Германии, иридия во Франции и Англии И др. Кроме того, необходимо учитывать вклад ученых разных стран в осознание элементарной природы выделенных веществ, как это было с кислородом у Лавуазье, с хлором и фтором у Гей-Люссака и Дэви и др. С учетом всех этих обстоятельств, ученые Разных стран приняли следующее участие в открытии химических элементов до 1869 г.:
Швеция Англия Франция Германия
19 16 14 12
Таким образом, выявляется картина не только национального с°перничества, но и международного сотрудничества ученых, ко-тТт2₽Ое особенно ярко проявляется в научных контактах Я. Бер-
Вместе с тем мы можем усмотреть определенную специфику, * — -----_________________________^„л:, открытия
кГЛИЙских химиков в основном связаны с исследованием газов °Дород, кислород, азот), платиновых металлов и электрохимиче-^ими
Цедиуса
___________________ ......г Л ЛЛ ЧЛ ЛЛ J^Z V Л. ЪУ Л л. ЛЛ J ЛЧ/
РИсущую некоторым группам исследователей. Так
/ "^ИСпих химиков в ОС
Г^°Р°Д1 кислород, азот),
- исследованиями Г. Дэви. Исследования французских уче-
209
ГЛАВА ПЯТА}!
—.М,,, .................. - - - --
ных поровну распределяются между исследованиями газов (А.
вуазье), металлов и минералов (Л. Воклен) и солей и вод, нрц ведших к открытию пода и брома. Исследования немецких химд. ков сосредотачиваются в основном па минералах, ио обращаются также и к минеральным водам (Р. Бунзен). Открытия шведсщц ученых сделаны исключительно при исследовании минералов ц однажды, продуктов их переработки (селен). Скандинавия — Мц’ нералогическая сокровищница Европы — предоставила матерцал для открытия всех природных элементов, и дело было только за химиками. Не случайно поэтому элементы, открытые шведскими химиками, принадлежат к семи группам периодической системы
Таким образом, если открытия английских ученых сделаны главным образом на повой методической основе (пневматическая химия, электрохимия), то открытия шведских химиков — в основном на базе классических методов химического анализа. Новые методические разработки внесли вклад и в открытия элементов немецкими химиками (спектроскопия), а для французских характерны также новые теоретические установки (А. Лавуазье).
Индивидуальный вклад отдельных ученых6. В чисто экспериментальном плане «абсолютное первенство» по числу открытых элементов, как уже указывалось, принадлежит К. Шееле, принявшему решающее приоритетное участие в открытии шести элементов: фтора, хлора, марганца, молибдена, бария и вольфрама.
Я. Берцелиус получил в чистом виде кобальт, цирконий, молибден, первично открыл церий, селен, торий и идентифицировал кремний, т. е. всего четыре, не говоря о многих других, где оп был первым рецепзентом-эксперпментатором, или вдохновителем, или руководителем работы (литий, ванадий и др.).
Г. Дэви непосредственно открыл два элемента: калий и натрий, но он подтвердил реальность существования кальция, стронция, бария и магния, выделил металлический литий, принял решающее участие в доказательстве элементарной природы хлора и фтора, т. е. его имя связано с открытием не двух, а девяти элементов.
Г. Клапрот приоритетно открыл три элемента: уран, титан, цирконий, но также принимал участие в открытии хрома и церия, т. е. связан с открытием пяти элементов.
К. Мозандер выделил в чистом виде лантан, тербий и иттрий, смесь, которую назвал эрбием, и другую смесь, которую назвал дидимом. Таким образом, он фактически связан с открытием четЫ-рых элементов.
Забегая вперед, в следующий исторический период, интересно отметить вклад У. Рамзая, открывшего пять элементов: аргон, ге' лий, криптон, неон, ксенон, и П. Лекока де Буабодрана, открыв' шего четыре элемента: галлий, самарий, гадолиний и диспрозий’
6 Этот аспект более подробно рассмотрен в специальной работе [8].
210
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Названные ученые являются авторами открытия 29 элементов, е третьей части стабильных элементов, открытых на Земле [8, с-163].
рассматривая личный вклад ученых в открытие химических элементов, нельзя не отметить немецкого ученого Ф. Вёлера, который получил в металлическом состоянии несколько элементов, вКлючая бериллий и алюминий, впервые в ощутимых количествах. Еще до П. Сефстрёма он открыл ванадий, но не опубликовал результаты; в начале 1840-х годов он выделил ниобиевую кислоту л соответствующий окисел, убедился в том, что это соединение нового металла, ио уступил первенство Г. Розе.
Открытие каждого нового элемента — это химико-аналитическое исследование, и оно, видимо, лучше удавалось типичным аналитикам по складу мышления и основному направлению деятельности. Аналитиками были Шееле, Ган, Клапрот, Воклен, Берцелиус, Балар, Розе, Штромейер и др. В сущности аналитиком был и Дэви. Вёлер же, наиболее прославившийся работами по органической химии, был склонен скорее к препаративным, синтетическим исследованиям, а потому ему лучше удавалось получение чистых веществ и выделение элементов в свободном состоянии.
Ложные открытия7. Ошибочно было бы думать, что открытие элементов имело лишь отмеченные в истории успехи и не сохранившиеся историей неудачи, не венчавшиеся приоритетными открытиями. В действительности на всем протяжении поиска элементов этот процесс сопровождался объявлениями о предполагавшихся открытиях, которые при проверке оказывались ложными. Объявления о ложных открытиях делали разные химики, в том числе столь видные аналитики, как Генрих Розе и Я. Берцелиус. За рассматриваемый период сделано около 40 таких ошибочных объявлений.
Это упоминание — не упрек исследователям и не усмешка пс поводу их неудач. Химия — наука экспериментальная, где истина Добывалась и добывается тяжелым, кропотливым, а зачастую и опасным трудом. К. Шееле и Г. Дэви получили тяжелые отравления, ставшие в конечном счете причиной их преждевременной смерти. Ж.-Л. Гей-Люссак получал нелегкие отравления и травмы при опасных экспериментах, что также тяжело сказалось на его Здоровье. То же можно сказать и о многих других, менее известных, исследователях. На их опыте — и положительном, и отрицательном — учились их ученики и коллеги. Жизнь состоит не из одних удач, и неуспехи химии поучительны для химиков не менее, нем ее успехи.
7 Этот аспект рассмотрен Н. А. Фигуровским и подробнее В. П. Мельнико НЬ1М [7, 9].
211
ГЛАВА ПЯТАЯ
КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ ДО ОТКРЫТИЯ ПЕРИОДИЧЕСКОГО ЗАКОНА*
Первая попытка классификации элементов, точнее - просты^ тел, принадлежит А. Лавуазье. В своем «Начальном курсе химии» (1789) он привел таблицу простых веществ [10, с. 192], в которой все известные в то время простые тела были разбиты на четыре группы: «1. Простые вещества, представленные во всех трех царствах природы, которые можно рассматривать как элементы 2. Простые вещества неметаллические, окисляющиеся и образующие кислоты. 3. Простые вещества металлические, окисляемые и дающие кислоты. 4. Простые вещества солеобразующие и землистые» (см. главу третью, стр. 116—117).
В первой группе объединены пять «веществ»: свет, теплота, кислород, азот и водород. Видимо, только эти три газа и два «невесовых флюида» Лавуазье считал подлинно элементарными телами природы; об остальных «простых веществах» Лавуазье судит не столь определенно.
Вторую группу составляют сера, фосфор, углерод и три гипотетических «радикала кислот»: соляной, плавиковой и борной. К третьей относятся 17 известных к тому времени индивидуальных металлов, включая открытые в 1780-х годах молибден и вольфрам. Четвертая группа включает пять «земель», которые, как уже указывалось выше, Лавуазье подозревал сложными веществами, но содержащими, кроме кислорода, какой-то неизвестный компонент.
По-видимому, таково же было отношение Лавуазье ко всем 23 объектам, внесенным в таблицу: если они сами, кроме первых пяти, могли оказаться пе элементарными, то имели, по его мнению, свой оригинальный элементарный корень. Как уже указывалось, основание соды и поташа он ие включил в эту таблицу, считая их производными известных простых тел.
Признание множественности элементов — важный шаг, совершенный Лавуазье. Он сам хорошо понимал это и так сказал во Введении к «Начальному курсу химии»:
«Отсутствие в настоящей книге главы, посвященной элементарным частям, составляющим тела, неминуемо вызовет удивление, но я позволю себе заметить, что стремление считать все тела природы состоящими из трех или четырех элементов происходит от предрассудка, перешедшего к нам от греческих философов. Предположение о четырех элементах, которые в различных пропорциях составляют все известные нам тела, было высказано задолго до того, как появились первые экспериментальные данные физики и химии... Теперь же, когда мы собрали факты, мы должны
* Данный раздел написан Н. А. Фигуровскпм.
212
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
оТвергнуть эти элементы, поскольку они пе отвечают нашим нынешним представлениям».
Отмечая, что авторы последующих сочинений включали в число элементов серу и соль, увеличив число элементарных принципов д0 шести, а Бехер признавал элементарными еще три «земли», Лавуазье продолжает:
«Все, что можно сказать о числе и природе элементов; ограничивается чисто метафизическими суждениями; это неопределенная проблема, допускающая бесчисленное множество решений, из которых скорее всего ни одно конкретное не согласуется с природой. Я лишь скажу, что если мы будем понимать под элементами простые и неделимые частички, образующие тела, то, вероятно, мы таких не знаем; если же, напротив, с понятием элемента или начала мы свяжем представление о последнем пределе, достигаемом анализом, то все вещества, которые мы еще не смогли никаким способом разложить, окажутся для нас элементами. Однако мы не можем быть уверены, что вещества, которые мы считаем простыми, не состоят па самом деле из двух или большего числа начал. Но так как эти начала никогда не разделяются, лучше сказать, что мы не смогли их разделить, мы можем рассматривать такие тела как простые, и мы же должны считать их сложными до тех пор, пока это не будет доказано опытом и наблюдениями» [10, с. XV—XVII].
Добавим к этому, что Дж. Дальтон, обогативший содержание понятия «элемент» (точнее — понятия «атом»), в сущности в основном лишь повторил в 1808 г. определение понятия «элемент», данное Лавуазье: «Под элементарными веществами, или простыми телами, мы понимаем такие, которые еще не были разложены. Мы не знаем ни про одно из веществ, называемых элементарными, что оно абсолютно неразложимо, но его следует называть простым до тех пор, пока его не удастся подвергнуть разложению...» [15, с. 221-222].
Представления Лавуазье и Дальтона об элементах и простых телах безоговорочно признавались большинством химиков и физиков XIX в. вплоть до 1860-х годов — времени окончательного признания атомно-молекулярного учения. Д. И. Меделеев, невидимому, первый сформулировал па основе атомно-молекулярного Учения четкие определения обоих понятий (1871). «Понятия и слова простое тело и элементписал он,— нередко смешиваются между собою, подобно тому, как до Лорана и Жерара смешивались названия частица, эквивалент и атом, а между тем для ясности химических идей и эти слова необходимо ясно различать. Простое тело есть вещество, металл или металлоид, с рядом физических признаков и химических реакции... Под именем элементов Должно подразумевать те материальные составные части простых 11 сложных тел, которые придают им известную совокупность физических и химических свойств. Элементу отвечает понятие об
213
ГЛАВА ПЯТАЯ
атоме. Углерод есть элемент, а уголь, графит и алмаз суть тетт простые» [16, с. 102]. а
Большинство попыток систематизации химических элементов в середине XIX в. было предпринято на фоне неопределенности понятий «элемент» и «простое тело». Дело в том, что даже вид нейшие химики того времени, такие, как Я. Берцелиус [17, с. 104] Ю. Либих [18, с. 111—112] и др., следуя логике Лавуазье, пред" почитали пользоваться термином «простое тело», вместо термина «элемент».
Эта ясная и недвусмысленная позиция отчетливо отражается в литературе того времени. Так, публикуя в своем журнале таблицы атомных и молекулярных весов, И. Поггендорф озаглавливает соответствующую часть не термином «элементы», а словами Un~ zerlegte Kbrper — перазложенные тела (см., например, [19]). Описанные в 1839—1843 гг. К. Мозандером разделения на несколько компонентов цериевой и иттриевой «земель», считавшихся индивидуальными, только подтвердило справедливость осторожности химиков. Вместе с тем попытки систематизации простых веществ как элементов, особенно систематизации па основе атомного веса, вызывали естественное недоверие химиков, для которых в то время физической реальностью были не элемент, а простое вещество, и не атомный вес, а эквивалентный (или соединительный) вес. Неправомерность отождествления последних также была ясна химикам, а потому недоверие вызывали самые правдоподобные обращения с эквивалентными весами, как с атомными.
Как мы увидим в дальнейшем, такое смешение понятий при классификации элементов в таблицах в значительной степени обесценивает научное значение этих таблиц.
«Таблица простых тел» Лавуазье классифицировала простые тела по единственному известному в то время признаку общих химических свойств (металлы, неметаллы, земли). С введением в 1803 г. Дж. Дальтоном понятия «атомный вес», значительно обогатившим содержание понятия «атом», внимание исследователей, естественно, обратилось к осознанию значения его в теории и практике дальнейшего изучения состава и строения вещества.
Признание множественности химических элементов в духе Лавуазье и различного веса атомов в духе Дальтона породило новые проблемы.
Попытки ответить на эти вопросы привели к появлению нескольких гипотез о бесконечной делимости атомов, о первичной материи и др. Наибольшее значение получила гипотеза У. Праута (1816) о водороде как первичной материи. Привлекательность этой гипотезы, ее соответствие с представлениями Лавуазье сделали ее не только исключительно популярной, но и важной для всего дальнейшего развития учения об элементах [18, с. 116—137]. 1
Гипотеза Праута появилась всего 13 лет спустя после созданий Дж. Дальтоном атомной теории. Первая, анонимная, статьй
214
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Праута, опубликованная в 1815 г. в журнале «Annales of Philo-^nhv», издававшемся T. Томсоном, была озаглавлена «Об отноше-цИИ между удельными весами тел в их газообразном состоянии и весами их атомов». В ней путем сопоставлений значений удель-bUX весов ряда простых тел и соединений в газообразном состояний Праут сравнил вычисленные по методу Дальтона величины аТомных весов с атомным весом водорода, равным единице, и кислорода, равным 10 (как это принимал У. Волластон). Резюмируя расчеты и данные своих таблиц. У. Праут писал: «Все элементарные числа, если за единицу принимать водород, делятся на 4 за исключением углерода (Праут принимал атомный вес углерода равным 6.—Я. Ф.), азота и бария, которые делятся на 2, что, как будто, указывает на то, что они ориентируются большим числом, чем единица, или водород. Есть ли это другое число 16, или кислород? И состоят ли все вещества из этих двух элементов?» [20, с. 288].
В другой статье, опубликованной в 1816 г. и имевшей целью исправление ошибки, допущенной в первой статье, У. Праут писал: «Если взгляды, которые мы решились высказать, правильны, то мы почти можем считать, что крсогт; бкт] древних (т. е. первичная материя.— Н. Ф.) воплощена в водороде — точка зрения, впрочем, не совсем новая. Если мы будем считать, что это действительно так, и далее примем, что удельные веса тел в их газообразном состоянии выражают собой числа объемов сконденсированных в один, то, другими словами, это будет означать, что кратные по весу количества абсолютного веса одного объема первичной материи г; (о ' 7 , который тела содержат, весьма вероятно, всегда
соответствуют их кратному по объему» [20, с. 293].
Отметим здесь интересное с исторической точки зрения обстоя-тельстсво, что Праут пользуется для аргументации существования первичной материи не только кратностью величин атомных весов (простых и сложных атомов), но и объемными отношениями газообразных тел в соответствии с законом объемов Гей-Люссака.
Гипотеза Праута нашла многочисленных сторонников, стремившихся подтвердить ее экспериментально путем многочисленных повторных определений атомных весов. Многим химикам XIX в. казалось, что признание за атомами водорода роли первичной материи само по себе решает задачу установления «состава» и даже строения атомов всех элементов. Отсюда задача классификации элементов казалась сама собою решенной. Действительно, признание существования первичной материи предопределяло наличие тесной связи между степенью «сложности» атомов Различных элементов и их физическими и химическими свойствами. Однако гипотезу Праута в XIX в. не удалось подтвердить экспериментально, т. е. не удалось установить, что атомные веса Всех элементов действительно кратны единице — атомному весу ВоДорода. Тем не менее в попытках установления связи между эле-
215
ГЛАВ \ ГШДлЯ
ч.
ментами, т. е. в попытках систематизации элементов, гипотез Цраута не просто получила отражение, но лежала в основе соот ветствующих сопоставлений.
Более примечательной с исторической точки зрения, хотя ц 11е столь эффектной в сравнении с гипотезой Праута, представляется систематизирующая попытка химика И. Дёберейнера, сопоставившего численные значения атомных весов некоторых элементов и установившего существование так называемых «триад», т. е. групп состоящих из трех химически сходных элементов с определенный соотношением атомных весов.
Йенский профессор Иоганн Вольфганг Дёберейнер (1780—1849) был одним из первых последователей атомистической теории Дж. Дальтона в Германии. С 1813 г. он занимался стехиометрическими исследованиями и в 1816 г. опубликовал таблицу «Относительные числа элементов, образующих химические соединения» [21]. Во втором издании таблицы (1823) он привел символы, названия и атомные веса 52 элементов. Целью его изысканий уже тогда было установление «определенных и неизменных отношений элементов друг с другом, выражаемых числами», т. е. атомными весами.
Сравнивая атомные веса («стехиометрические числа», пли эквиваленты) элементов, Дёберейнер нашел, что для многих, широко распространенных в природе, элементов эти числа довольно близ кп, а для Fe, Со, Ni, Сг, Мп они практически одинаковы8.
В 1817 г. Дёберейнер установил [22], что эквивалентный вес окиси стронция (50) должен быть равен среднему значению эквивалентных весов окиси кальция (27,5) и окиси бария (72,5):
Sr О =
СаО + ВаО 2
27,5 + 72,5 2
Кроме того, Дёберейнер полагал, что существует подобное отношение между удельными весами сульфатов этих элементов:
SrS04 =
CaSO4 + BaSO4 2
И. Дёберейнер заметил также (1817), что Fe, Со и Ni, имеющие близкие эквивалентные веса, имеют много общих химических свойств.
В 1829 г. в статье «Опыт группировки элементарных веществ по их аналогиям» [23] Дёберейнер привел несколько других триад и расчетов атомных весов промежуточных в триадах элементов:
8 В таблицах Дёберейнера [21] приведены следующие значения атомных весов: Fe=27,13; Со=29,52; Ni=29,58; Сг=29,00; Мп=28,46. В то время атомные веса этих элементов, действительно, можно было считать тождественными.
216
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Литий Кальций Хлор Сера Натрий Стронций Бром Селен Калий Барий Иод Теллур
Марганец Хром Железо
Например, атомный вес брома Дёберейнер вычислил как сред-йес арифметическое атомных весов хлора и иода:
35,47(С1)+126,47(1)
---------к---------= oU,42(br).
Интересно отметить, что Дёберейнер не ввел во вторую триаду магний (видимо, не желая нарушать принципа триад введением тетрады), очевидно считая его «изолированным» элементом, не входящим в триаду. Что же касается фтора, то, хотя в то время он и считался простыл! телом, его признаки как простого вещества не были известны, так как он еще не был выделен в свободном состоянии.
И. Дёберейнер пытался составить и другие триады из элементов, которые обнаруживали химическое сходство или простые отношения между атомными весами, но для ряда предполагаемых триад ему недоставало третьего элемента, например в случаях В и As, Sb и Bi, Sn и Cd. «Если бы он не искал во что бы то пи стало триад,— отмечал позднее Л. Мейер,— то, конечно, заметил бы связь между собой всех четырех элементов: Р, As, Sb и Bi» (цит. по [24, с. 48]). В этом, как мы сейчас понимаем, сказалась ограниченность его систематизирующего подхода.
Не касаясь здесь других подробностей построений Дёбереппе-ра, описанных в литературе [25, с. 64 и др.], отметим, что дальнейшие попытки систематизации элементов и установления взаимосвязей между их физическими и химическими свойствами заключались главным образом в сопоставлениях численных значений атомных весов с целью установить «правильности» в этих значениях.
Остановимся прежде всего па построениях немецкого химика-атомиста, ученика Я. Берцелиуса,—Леопольда Гмелипа (1788 1853), первого автора широко известного справочника «Gmelins Handbuch der anorganischen Chemie». В третьем издании (1827) этого труда Гмелин предложил две триады, первая из которых была составлена Дёберейнером еще в 1817 г.:
Са 4- Ba Li + К
-—2-----= Sr и-----2--= ^а.
Кроме того, Гмелин сделал попытку ввести в триаду (Са, Ва, Sr) магний, чего, как мы видели, не сделал Дёберейнер, но при этом Гмелину пришлось отступить от формального «закона Триад»:
12(Mg)4-68,6(Ba)
4
= 20,15(Са).
217
ГЛАВА ПЯТАЯ
Расположение химически сходных элементов по Гладстону (1853) на основе классификации Гмелина
О N 14 H 1
8*
F С1 Вг J Li Na к
19 35 80 127 6 23 39
S Se Те Mg Ca Sr Ba
16 39 64 12 20 44 68
Р As Sb G3* Y Jj.4* Ce Di La
31 75 129 5 —- — — 47 50 47
С в Si Zr Th Al
6 11 21 22 60 14
Ti Та Nb Ре2* W Sn Cd Zn
25 184 — — 92 58 60 53
Мо V Сг и Mn Co Ni Fe
46 69 27 60 28 29 30 28
Bi Pb Ag Hg Cu
208 104 108 100 32
Os Ru 1г Rh Pt Au
100 52 99 52 53 197
* Атомный вес согласно’’Годичному очерку” Либиха за 18511,
•2* Пелопий.
3* Глюций (бериллий) . 4* Тербий.
В четвертом издании своего «Справочного руководства» (1843) [26] Гмелин выступил с концепцией о существовании, помимо триад, групп химически сходных элементов. Он писал: «Существуют группы элементов, обладающих сходными физическими и химическими свойствами. Должны ли эти группы состоять только из триад элементов, как это полагал Дёберейнер, сгруппировавший элементы в триады, остается сомнительным» (цит. по [25, с. 69—70]). Но тут уже сомнения были отброшены, и Гмелин приводит свою таблицу групп элементов, расставленных в порядке возрастания атомных весов и состоящих не только из триад, по и тетрад, пентад и гексад. В таблицу вошли не все известные в то время элементы. В дальнейшем (1853) Дж. Гладстон (1827— 1902) несколько расширил таблицу Гмелина, включив в нее редкие и вновь открытые элементы [25, с. 71].
Представляет интерес, что в верхней части таблицы Гмелин поместил «базисные» элементы — кислород, азот и водород. Видимо, это сделано не столько с целью демонстрации «типических» монад, сколько для характеристики электрохимических свойств элементов в свете электрохимической теории Берцелиуса.
Таким образом, систематизирующая мысль разорвала искусственные ограничения группировки элементов только лишь
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
триадам, но не смогла продвинуться дальше по пути систематики самих групп.
Опуская менее важные с исторической точки зрения детали построений Гмелина и Гладстона, обратимся к дальнейшим попыткам систематизации и классификации элементов.
В 1850 г. ученик Ю. Либиха М. Петтенкофер (1818—1901) опубликовал таблицу диад, триад и тетрад сходных элементов (см. [27]). Одним из поводов для этой публикации послужило установление в 1840-х годах гомологических рядов органических соединений. Петтенкофер сделал попытку найти подобные же соотношения у элементов, рассматривая их как сложные образования из нескольких «первичных» частиц. Таким образом, речь шла об установлении «гомологической разницы» в отдельных группах химически сходных элементов.
В 1851 г. известный французский химик Ж. Б. Дюма (1800— 1884) в своем докладе Британской ассоциации содействия наукам также отметил существование правильных соотношений между атомными весами у элементов, подобных тому, как это имеет место в гомологических рядах [25, с. 75]. Он сделал далеко идущие выводы из этого и даже высказал гипотезу, что атомы элементов возможно разложить на некоторые «субэлементарные частицы».
В 1857 г. Дюма опубликовал найденные им численные соотношения между эквивалентами простых тел, близких по химическим свойствам:
Т=19 = 19
Cl=19-f-16,5 =35,5
Вг=19 + (2Х16,5)4-28 = 80
> J = 19 4-(2X16,5)+(2 x28) 4-19=127
О—8 = 8
S=8+8 = 16
Se=8 + (4x8) =40
1те=8 + (7х8) = 64
’ N=14 = 14
Р=14+17 = 31
< As=14+17+44 = 75
Sb=14+17 + (2x44) =119
Bi=14+17 + (4X44) =207
Mg=12 = 12
Ca=12+8 = 20
- Sr=12+(4X8) = 44
Ba=12+(7X8) = 68
Pb=12+(10x8) =104
В 1854 г. Дж. Кук (1827—1894), профессор химии и минералогии в Гарвардском университете (США), ссылаясь на
Б. Дюма, лекции которого он слушал в Париже в 1848 г., приписывая Дюма открытие триад *, расположил все известные еА1У элементы (простые тела) в шести группах по величине их
* Дж. Кук, видимо, имел в виду учебник химии («Traite de chemie, applique aux arts». P., 1828) Ж. Б. Дюма, в котором автор составил три группы элементов: F. Cl, Br, J; О, S, Se; N, As, Р.— Прим. отв. ред.
219
ГЛАВА ПЯТ VI
атомных весов [27, с. 885]. Значения атомных весов элементов в каждой из групп определялись следующими отношениями:
1) 8+п9; 2) 8 + п8 или 4 + ^8; 3) 8-Ьлгб;
4) 6 + п5; 5) 4 + /г4 -или 2-Ь/г4; 6) 1Д-лгЗ,
где п — целое число [25, с. 79].
При расчете атомных весов по этим формулам оказывается что некоторые элементы попадают сразу в несколько групп К первой группе Кук отнес О, F, Cl, Br, J, (CN); ко второй-О, S, Se, Мо, Те, V, \\, Та; к третьей — О, N, Р, As, Sb, Bi; к четвертой — С, В, Si; к пятой — Mg, Al, Ca, Ti, Cr, Mu, Fe, Nj’ Co, Cu, Zu, Sr, Ba, Cd, Pt, Hg, Ag, Au и к шестой — H, Li К, Na.
В 1857 г. 20-летний ассистент К. Р. Фрезениуса Э. Ленсеи опубликовал таблицу [28], включавшую 20 триад, в том числе также и триады редкоземельных элементов. Эти триады показывали связь не только между атомными весами, но и между свойствами элементов. Ленсеи брал округленные значения атомных весов, принятых в то время, п сопоставлял рассчитанные атомные веса с экспериментально установленными числами. Кроме того, Лепсен предложил метод расчета атомных весов некоторых элементов из «эннеад» (ev —по-гречески девять), т. е. на основании данных, полученных из трех триад. Например, он объединил в эннеаду три триады:
Калий Барий Кадмий
Натрий Стронций Цинк
Литий Кальций Магний
и нашел по методу Дёберейнера атомные веса для натрия — 23,03 (23,00), стронция — 44,29 (43,67) и цинка —33,8 (32,5) 9. Взяв затем округленные значения атомных весов этих трех элементов, Ленсеи составил из них новую, вторичную, триаду — эннеаду и па ее основе получил для цинка атомный вес, равный 33. Такой прием двойного расчета атомных весов элементов привел Ленсена к признанию «закона триад». На основе этого «закона», пользуясь как триадами, так и эннеадами, Лепсен делает попытку расчета неизвестных еще атомных весов редкоземельных элементов, составляя триады, объединяющие сходные по химическим свойствам элементы [25, с. 84].
Наблюдения Дюма и Ленсена относительно правильностей ъ величинах атомных (эквивалентных) весов исключали случайность и свидетельствовали о наличии какого-то еще не понятого закона. Д. И. Менделеев по этому поводу писал: «...между всеми— учеными, которые раньше меня занимались сравнением величин атомных весов элементов, я считаю, что обязан преимущественно
9 В скобках приведены принятые в то время экспериментально определенные значения атомных весов.
220
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Триады Ленсена {1857) и рассчитанные на их основе атомные веса в сопоставлении с найденными экспериментально
Рассчитанные атомные веса Экспериментально установленные атомные веса
1 К + Li 23,00 6,95
J ’ NTq 9Q OQ 39,11
2 — in a —
2. Ba4-Ca -5— = Sr = 44,29 Zu 68,59 43,67 20
3. Mg + Cd _ 2 •— — 33, 8 12 32,5 55,7
4. Mn 4- Co r — Fe — 28,5 Zu I 27,5 28 29,5
5. La + Di 2 CuC 4:0,3 47,3 47 49,6
6. Yt Er Tb 7. Tb Norium Al 32,2 • 1
8 Be + U 59,5 ? 13,7
°* 7т* QQ 33,6 п с\
2 — ZjT ——• oo, о 7 60
9 Cr + Cu -s = Ni = 29,3 Zu 26,8 29,6 31,7
IP. Ag + Hg • = Pb = 104 Zu 108 103,6 100
11. 0 + c J -N-7 Л1+1!__в„_,2>2 Zu 8 15 7 11 6 9,5*
13. ’Cl 4-j 9 _ Br — 40,6 Zu 17,7 40 63,5
14. S + Te — Se — 40,1 Zu 16 39,7 64,2
15. P + Sb 9 — As — 38 Zu 16. Ta-j-Ti 9 — Sn —58,7 Zu 16 92,3 37,5 59 60 25
17. W-J-Mo -L = V = 69 Zu 92 68,5 46
18. Pa + Rh _ Ru — 52,2 La 53,2 52,1 51,2
19. Os + Ir X = pt —98,9 la 99,4 99 98,5
20. Bi + Au 2 —Hg-101,2 104 юо** 98,4**
* тт v 130 г 1 ~. 11 -f* IV . г
при двойном атомном весе фтора триада ---------=э ы =-----— =>15.
В оригинальной таблице эти два атомных веса переставлены.
221
ГЛАВА ПЯТАЯ
26,2, Мп = 26,7, Fei’
двум: Ленсену и Дюма. Я изучил их исследования, и они побудили искать действительный закон» [29, с. 288]. я
Попытки объединения и классификации элементов по триада тетрадам и т. д. в 50-х и 60-х годах XIX в. были довольно мц ’ гочисленны. Так, П. Кремере, работавший в частной лаборатордп в Бонне и затем в Кёльне, в серии работ, посвященных изучение растворов, неоднократно обращался к триадам и на основе рас четов атомных весов формулировал выводы о свойствах одинак0-вых солей металлов, входящих в эти триады [27, с. 885].
В 1859 г. немецкий химик Л. Штреккер (1822—1871) расположил все известные в то время элементы в ряд серий. Например одна из серий объединяла элементы: Сг = 26,2, Мп = 26,7, Fe =’ = 28, Ni = 29, Со = 30, Си = 31,7, Zn = 32,5. «Вышевыставленные отношения между атомными весами (или эквивалентами) химически сходственных элементов, конечно, едва ли могут быть приписаны случайности,— писал ученый.— Но ныне мы должны предоставить будущему отыскивание законности, проглядывающей между указанными числами» (см. [29а, с. 350]).
Помимо рассмотренных выше и некоторых других попыток установления связей между свойствами, в первую очередь между атомными весами отдельных элементов, в 60-х годах XIX в. появились сопоставления несколько иного рода. Стали сравнивать не
только отдельные элементы, но и группы химически сходных элементов. Такой подход привел к составлению таблиц, объединявших все (или почти все) известные элементы и носивших уже классификационный характер. Цели авторов таких таблиц были довольно различны. Видимо, одни исходили из стремления проиллюстрировать генезис элементов из «первичной материи», другие интересовались исключительно численными соотношениями значений атомных весов в группах химически сходных элементов. Рассмотрим в этой связи таблицы, составленные отдель
ными учеными.
В 1857 г. оксфордский профессор химии В. Одлинг (1829— 1921) опубликовал работу, в которой представлена таблица и3 49 элементов, разбитых на 13 групп сходных элементов [30]. В 1861 г. Одлинг в своем «Руководстве по химии» предложил таблицу из 57 элементов [31]. Приведенные в таблице атомные веса (эквиваленты) вдвое меньше истинных.
Таблица Одлинга — один из примеров сопоставления элементов, имеющих аналогичные химические свойства, однако без каких-либо попыток сделать хотя бы скромные обобщения и*110 выводы. Обращает на себя внимание в ряде случаев несообразность подбора элементов. Очевидно, что Одлинг лишь чисто Ф°Р' мально придерживался «закона триад» Дёберейнера, т. е. при0' ципа, согласно которому в триаде атомный вес среднего элемента равен полусумме атомных весов крайних. У Одлинга даже 06 всегда выполнено условие химического сходства элементов 0
222
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Таблица Одлинга (1861)
п Hydrogen 1 В Boron 11 Ni Nickel 29
р г Fluorine 19 Li Lithium 7 Co Cobalt 30
г сл Chlorine 35,5 Na Sodium 23 Cu Copper 31,7
Rr Bromine 80 К Potassium 39 Al Aluminum 13,7
т Iodine 127 Ca Calcium 20 Zr Zirconium 33,5
о Oxygen 16 Sr Strontium 44 Ce Cerium 46
s Sulphur 32 Ba Barium 68,5 La Lanthanum 47
Ss Selenium 80 G Glucinum 4,7 D Didymium 48
т Tellurium 128 Y Yttrium 32 U Uranium 60
N Nitrogen 14 Th Thorium 59,5 Mo Molybdenum 48
Р Phosphorus 31 Mg Magnesium 12 Vd Vanadium 68,5
As Arsenic 75 Zn Zinc 32,5 W Tungsten 92
sb Antimony 120 Cd Cadmium 56 Au Gold 197
Bi Bismuth 208 Hg Mercury 100 Ro Rhodium 52
С Carbon 12 Pb Lead 103,5 Ru Ruthenium 52
Si Silicon 28,5 Ag Silver 108 Pd Palladium 53
Ti Titanium 48,5 Cr Chromium 26 Pt Platinum 98,5
Sn Tin 118 Mn Manga ne.( e 27 Ir Iridium 98,5
Ta Tantalum 138 Fe Iron 28 Os Osmium 99,5
триадах (например, триады: ртуть, свинец, серебро; бериллий (глиций), иттрий, торий и др.). Впрочем, подобные же примеры можно найти и у Ленсена, которому, по-видимому, отчасти следовал Одлинг.
В октябре 1864 г. Одлинг опубликовал статью «О пропорциональных числах элементов» [32], написанную, по-видимому, не без влияния построений Ньюлендса (см. ниже). В этой статье приведены две таблицы. Одна —список из 61 элемента, расположенных по возрастанию атомного веса в тех значениях, которые были приняты в то время. Из значительных отложений отметим атомные веса ванадия, иттрия, урана, тантала, ниобия. Тремя месяцами ранее такая же таблица была опубликована Дж. Ныо-лендсом.
Значительно большего интереса заслуживает приводимая в той Же статье Одлинга другая таблица, представляющая собой тго-пъгтку построения системы элементов, В этой таблице не все ясно. В пяти ее колонках размещены не 61 элемент, приведен-яь!с в предшествующем списке, а только 57: не нашлось места Дидиму, лантану, ниобию и иттрию. В таблице усматривается стремление, хотя и не явное, сохранить старые триады, тетрады 13 Др. Общее число горизонтальных «групп» — 17. Среди них мы встречаем в одном ряду натрий, калий, рубидий и цезий; пунктиром «подсоединены» к ним литий и водород, по литий помещен
223
ГЛАВА ПЯТАЯ
Таблица Одлинга (октябрь 1864 г. )
f Ro 104 Pt 197
< Ru 104 Ir 197
[Pt 106,5 Os 199
II 1 99 99 Ag 108 Au 196,5
59 99 Zn 65 Cd 112 Hg 200 ---
г L 7 *9 99 99 Tl 203
: G 9 99 99 »> Pb 207 --
11 Al 27,5 99 и 120 99
: ; с 12 Si 28 *9 Sn 118 —- - 99 — —
: N 14 P 31 As 75 Sb 122 Bi 210 ;
О 16 S 32 Se 79,5 Те 129 — - 99 1 1 1
: F 19 Cl 35,5 Br 80 J 127 *9 1 1 1
— - Na 23 к 39 Rb 85 Cs 133 99 1 1 1
Mg 24 Ga 40 Sr 87,5 Ba 137 --- • 99 . w . 1
Ti 50 Zr 89,5 Ta 138 Th 231,5 '
99 Ce 92
Cr 52,5 Mo 96 f V 137 --- " *
Mn 55 I w 184
Fe 56
< Co 59
Ni 59
Cu 63,5
в одном ряду с таллием. В одном ряду находятся углерод, кремний и олово; пунктиром «подсоединен» к ним торий, который находится в одном ряду с титаном, цирконием и... танталом.
Не останавливаясь здесь па других особенностях таблицы Одлинга, отметим лить, что если расположение элементов в первых трех колонках в общем соответствует порядку возрастания атомных весов, то в следующих двух колонках имеются необъяснимые несоответствия. Например, положение олова или ванадия. Вместе с тем нельзя пе отметить стремление Одлинга опираться в первую очередь на химическое сходство. Так, он поместил теллур иа одной горизонтали с кислородом, селеном и серой, золото, считавшееся тогда более легким,— после металлов платиновой группы.
В. Одлпиг, видимо, полагал, такое размещение более соответствующим порядку расположения сходных элементов в горизонтальных рядах, чем расположение в порядке возрастания атомного веса. Загадочными остаются кавычки в таблице. Обозначают ли они еще пе открытые элементы, так сказать, вакантные места» Одлппг не комментировал их, так же как и пунктир.
224
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Таблица Одлинга (1865)
Mo 97 Pd 106,5 W 184 Au 196,5 Pt 197
^7 7 G 9 в я С 12 N 14 О 16 F 19 Na 23 Mg 24 Al 27,5 Si 28 P 31 s 32 Cl 35,5 Zn 65 As 75 Se 79,5 Br 80 Ag 108 Cd 112 Sn 118 Sb 122 Те 129 J 127 Hg 200 Tl 203 Pb 207 Bi 210
К 3 Ca 40 Ti 48 Cr 52,5 Mn 55 Rb 85 Sr 87,5 Zr 89,5 Cs 133 Ba 137 V 138 Th 231
Публикация Одлинга 1864 г. не попала в сферу внимания Д. И. Менделеева, но внешне она напоминает первый вариант таблицы самого Менделеева. Ф. Н. Савченков, который в 1867 г. перевел «Курс практической химии» Одлинга (1865) [33] на русский язык и, видимо, следил за публикациями этого автора, на апрельском (1869) заседании Русского химического общества, после знаменитого сообщения Д. И. Менделеева, обратил его внимание на таблицу Одлинга. Впрочем, сходство это чисто внешнее. Пытаясь выразить химическое сходство элементов, наблюдавшееся многими химиками, Одлинг идет на перестановку элементов, но не на изменение их атомного веса, т. е. он еще не видит системы. Он и сам писал в статье 1864 г., что его таблица представляет собой «чисто арифметическое распределение элементов по сериям в горизонтальном порядке в соответствии с обычно принятым способом группировки элементов».
В 1868 г. в «Химическом словаре» X. Уатта появилась статья Одлинга «Металлы, их атомные веса и классификация» [34, с. 975]. Там приведена таблица, которая включает 45 элементов — преимущественно металлы. Сравнение этой таблицы с прежней обнаруживает перенос некоторых элементов на новые К1^ста. Фактически сформировались будущие 2-й и 3-й периоды, 110 группы щелочных и щелочноземельных металлов разрушились. Обращают на себя внимание две вертикальные жирные ^Ииии, выделяющие основные триады и тетрады. Черточками ^означены еще не открытые элементы, существование которых иДлинг полагал «не невероятным». В 1868 г. в третьем издании
всеобщая история химии 225
ГЛАВА ПЯТАЯ
Таблица Одлинга (1868)
Триплетные группы '*****^
Н 1 Mo 96 w 184
•— A« 196,5
Pd 106,5 Pt 197
Li 7 Na 23 Ag 108
G 9 Mg 24 Zn 65 Cd 112 Hg 200
] 3 и Al 275 —— — T1 203
( и 12 Si 28 — Sn 118 Pb 207
N 14 P 31 As 75 Sb 122 Bi 210
( 3 16 s 32 Se 79,5 Те 129 -
F 19 Cl 35,5 Br 80 J 127
К 39 Rb 85 Cs 133 —. —
Ca 40 Sr 87,5 Ba 137 —*
Ti 48 Zr 89,5 — Th 231
Cr 52,5 1 1 1 V 138 ——
Mn 55
«Курса практической химии» [33, с. 226] Одлинг приводит эту таблицу, подчеркивая триадную природу организации.
Мы видим, таким образом, что Одлинг первым из рассмотренных нами исследователей, хотя и не очень уверенно, но переходит от попыток частной систематизации отдельных элементов в группы к классификационному подходу, по-новому распред е-л я я элементы в первоначально созданных им группах. Эта классификационная тенденция еще ярче проявилась у Ныолендса.
Как уже отмечалось, некоторые из таблиц Одлинга весьма близки по идее к соответствующим таблицам Ныолендса.
Джон Александер Рейна Ньюленде (1837—1898), ученик А. В. Гофмана, работал химиком в Королевском сельскохозяйственном колледже, а затем с 1864 г.— химиком-аналитиком, главным образом в сахарной промышленности.
Первое выступление Ныолендса по вопросу о соотношении атомных весов относится к 1863 г. и озаглавлено «О соотношениях между эквивалентами» [35]. Ученый привел несколько групп химически сходных элементов и констатировал разнидь1 в эквивалентных весах между элементами в этих группах. Эт°» как мы видели, делалось и ранее. Ньюленде показал, что существует большее число пар элементов с одинаковой разницей в значениях эквивалентных весов. В ряде случаев эта разность оказалась равной 8 или числу, кратному 8.
226
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Таблица Нъюлендса (1864)
Triad
Lowest Term Mean Highest
Term
I. Li 7 + 17 = Mg 24 Zn 65 Cd 112
II. В 11 Au 196
III. с 12 4 lb = Si 28 Sn 118
JV. N 14 + 17 =P 31 As 75 Sb 122 + 88-Bi 210
V. о 16 4-16 =S 32 Se 79,5 Те 129 + 70= Os 199
VI. F 19 + 16,5=0 35,5 Er 80 J 127
VII. Li 7 + 16=Na 23 + 16 =K 39 Rb 85 Cs 133 + 70 = TI 203
VIII. Li 7 + 17=Mg 24 +16 =Ca 40 Sr 87,5 Ba 137 + 70=Pb 207
IX. Mo 96 V 137 W 184
Pd 106,5 Pt 197
В июле 1864 г. Ньюленде опубликовал еще одну таблицу, в которой 37 элементов расположены в 10 рядах. В основе таблицы лежит принцип триад (как и у Одлинга), расположенных друг под другом в порядке возрастания эквивалентных весов [36]. Эквиваленты низших и высших членов триад рассчитаны из эквивалентов «типических» элементов, причем разницы в эквивалентных весах для низших членов триад и типических элементов составляют приблизительно +16, иногда +17. Разницы в эквивалентных весах для высших членов триад составляют +70, а в одном случае (сурьма—висмут) +88.
Вскоре после составления этой таблицы, 20 августа 1864 г. Ньюленде опубликовал еще одну таблицу. В ней 24 элемента, начиная с азота, были расположены в 5 рядах, обозначенных начальными буквами латинского алфавита (а —е). «Если элементы расположить в порядке их эквивалентов, присвоив водороду [номер] 1, литию — 2, глюцинию — 3; бору — 4 и т. д.,— писал Ньюленде,— то будет заметно, что элементы, имеющие последо-отельные номера, часто или принадлежат к одной или той же труппе, или занимают сходные позиции в различных группах... Иными словами, восьмой элемент, начиная с данного, является Как бы повторением первого, подобно восьмой ноте октавы в му-Зыке» [27, с. 887].
Более полную таблицу, включающую уже 62 элемента, ученый опубликовал через год—18 августа 1865 г. Она подтвержда-Ла ранее сделанное наблюдение: «элементы, принадлежащие к °Дной и той же группе, обычно появляются в одном и том же
227
10*
ГЛАВА ПЯТАЯ
Таблица Ньюлендса (1864)
№ № № №
Группа а N 6 Р 13 As 26 Sb 40 Bi 54
« b О 7 S 14 Se 27 Те 42 Os 50
« с FI 8 Cl 15 Вт 28 J 41 —
« d Na 9 к 16 Rb 29 Cs 43 T1 52
« е Mg 10 Са 17 Sr 30 Ba 44 Pb 53
Закен октав Нъюлендса (1865)
№ № № № № № № №
H 1 F 8 Cl 15 Co Ni 22 Br 29 Pd 36 J 42 Pt Ir 50
Li 2 Na 9 к 16 Cu 23 Rb 30 Ag 37 Cs 44 Tl 53
G 3 Mg 10 Ca 17 Zn 25 Sr 31 Cd 38 Ba V 45 Pb 54
Bo 4 Al 11 Cr 19 Y 24 Ce La 33 и 40 Ta 46 Th 56
C 5 Si 12 Ti 18 In 26 Zr 32 Sn 39 W 47 Hg 52
N 6 P 13 Mn 20 As 27 Di Mo 34 Sb 41 Nb 48 Bi 55
0 7 S 14 Fe 21 Se 28 Ro Ru 35 Те 43 Au 49 Os 51
горизонтальном ряду» и «номера сходных элементов, как правило, отличаются или на 7, или на некоторое число, кратное семи». Ньюленде назвал замеченную им закономерность «законом октав» [37].
25 августа 1865 г. Ньюленде опубликовал еще одну краткую статью, в которой «попытался показать, что все численные соотношения между эквивалентами... включая хорошо известные триады являются просто арифметическими результатами, вытекающими из существования «закона октав» [38, с. 15—17]. Такой вывод следовал из того, что частное от деления эквивалентов на номера элементов представляло собой приблизительно постоянную величину для больших групп последовательных элементов, т. е. эквиваленты были приблизительно пропорциональны номерам элементов. Причем коэффициент пропорциональности изменялся для четырех групп элементов от 2,5 до 4.
У каждого элемента в таблицах Ньюлендса был свой номер» если элемент имел отличный от других элементов эквивалент-При равных эквивалентах элементы имели одинаковый номер* Конечно, это еще не были порядковые номера элементов, к которым мы привыкли сегодня и с которыми мы связываем вполне определенный смысл заряда атомного ядра. Для Ньюлендса сГ° номера элементов служили всего лишь способом выражения еГ°
228
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
«закона октав» — регулярного появления химически сходных цементов в их ряду, построенном по повышающимся атомным весам. Это была идея о периодичности, выраженная в самой общей и пока весьма необычной форме. По-видимому, именно такая необычная форма представления регулярности вместе с ее явными нарушениями в таблице для целого ряда элементов (реальная периодическая система элементов оказалась гораздо сложнее) стала причиной более чем холодного отношения со стороны других ученых к этому, пожалуй, первому наблюдению периодичности.
При обсуждении доклада Ньюлендса с его нумерацией элементов на заседании Лондонского химического общества в марте 1866 г. один из участников заседания Г. Фостер (профессор физики в Глазго) спросил Ньюлендса иронически, «...не пробовал ли он расположить элементы в таблице в алфавитном порядке и не заметил ли оп при таком расположении каких-либо новых закономерностей». На это Ньюленде ответил, что прежде чем прийти к представленной таблице, он пробовал несколько других схем и в результате всего проделанного им он считает невозможным установление закона октав на основе эквивалентных весов.
По его мнению, только система атомных весов, установленная на основе положений С. Канниццаро, может привести к успеху [38, с. 17—19]. В результате обсуждения доклада Лондонское химическое общество отказало Ньюлепдсу в полной публикации его сообщения. Отвечая своим оппонентам, Ньюленде убежденно писал вскоре после заседания: «Тот факт, что такое простое соотношение существует, теперь дает веское основание предполагать, что это всегда будет сохраняться, даже если будут открыты сотни новых элементов. Ибо, хотя разность в номерах сходных элементов в таком случае может измениться с 7 или кратного семи на 8, 9, 10, 20 или любого возможного числа, существование простого соотношения номеров сходных элементов не может быть менее очевидным» [38, с. 20]. Это высказывание Одновременно показывает, что ученый не придавал абсолютного значения номерам соответствующих элементов.
Размещая элементы по сформировавшимся у него группам (строкам таблицы), Ньюленде не слепо следовал своим порядковым номерам, т. е. возрастанию эквивалентного веса. Так, у него Первый «сбой» в паре хром—титан (19, 18), затем —в паре Чинк—иттрий (25, 24), а далее еще несколько раз. Мы видим, Чт° Ньюленде определенно классифицирует элементы и... утрачивает при этом систематизирующее начало — закономерное изменение атомного веса. «Закон октав» для него — форма; могут существовать и другие интервалы, но только чтобы соблюдалась Периодичность химических характеристик.
229
ГЛАВА ПЯТАЯ
27 июня 1873 г., уже после опубликования Д. И. Менделеевы^ его классических статей по периодическому закону (1871) Дж. Ньюленде обратился в Лондонское химическое общество ’ просьбой — формально подтвердить его права на приоритет в от крытии периодического закона.
В. Одлинг, ставший к этому времени председателем Общества объяснил, что сообщение Ньюлепдса 1866 г. пе было напечатано’ поскольку Общество «взяло за правило не публиковать работьт чисто теоретического содержания, так как это могло привести к появлению статей спорного характера» [39, с. 273—274]. Одлинг подчеркивал также, что в 1866 г., будучи секретарем Общества он, по его словам, имея в виду эти правила, не читал в Обществе сообщений, описывающих его собственные исследования об отношениях между атомными весами и химическими свойствами элементов, несмотря на то, что «эти статьи заслуживали серьезного внимания».
В дальнейшем Ньюленде настойчиво напоминал о своих работах 1864—1866 гг. в связи с открытием периодического закона Д. И. Менделеевым. Впоследствии (1884) он выпустил небольшую книгу, в которой были воспроизведены все его основные статьи, а также отзывы на них других ученых [38]. В 1887 г. Королевское общество все-таки присудило ему за указанные работы медаль Дэви.
Среди многочисленных авторов, занимавшихся в 60-х годах XIX в. сопоставлениями атомных весов элементов в различных таблицах и графиках, необходимо назвать французского ученого А. Шанкуртуа и немецкого химика Л. Мейера.
В 1862—1863 гг. профессор Парижской горной школы Александр Эмиль Бегие де Шанкуртуа (1819—1886) представил взаимосвязь свойств элементов в виде графика, получившего в литературе наименование «земной винт» (Vis tellurique), или «винтовая линия Шанкуртуа». В своем первом сообщении [40] Шанкуртуа сопоставил некоторые отдельные группы химически сходных элементов и сделал попытку объединить все известные элементы в одной системе. Центральная часть сообщения — описание графического изображения системы — «теллургического винта» — в первой публикации отсутствовала: издатели «Comptes Rendus», очевидно, исключили описание иллюстрации. По этой причине Шанкуртуа решил опубликовать полную статью, которая появилась в 1863 г. [41]. Он нанес на образующую поверхность цилиндра под углом 45° к его основанию атомные веса в определенном масштабе. Образующая поверхность цилиндра была разделена им на 16 частей (16 —атомный вес кислорода)’ т. е. на части, составляющие каждая 22,5° окружности. Атомные веса элементов (простых тел) были отложены в соответствующем масштабе (за единицу принят атомный вес водорода). ЕсЛ0
230
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
График ре Шанкуртуа
"Tableau bES Carriers Geometrique (dCVELOPPEMENT D'UN CYLINORE DE 0?025 OlflMerRE) 1 а ^<-и, г а *со о , WJ о « or »сО оа <я <к UJ Z ш Е (Л и аг со X о Z NOMBRtt COMPL1M I Р»й LfV»S fAC Г •A a w or Э 4 г lA UJ а e X о Z
01234 5 67 89МЙ13Ш
*4 / /
* л 'л г/ 2 2
□ я 3 Д
г ... ч> 2’
г/ Г л 5
т— 2J6
л Z Л 7 7 8
'тт <п Л / 2*1
01 7 ‘ с 5 3a 9
V с т~ 2f /0
z // //
(Йа л 51 12
£ Я /3 13
б Л P7 14
J1 35 /J
О 1 2“ 16
6 /7
2’’
9 ► 9 ‘ 19 >9
i-J 2’5 3^ 2o
2i
7n 22
V. ?3 23
к —/Vi 2"3 24
.Mg « ’ » • Т1 • S‘ 25
р\ 2>J 26
— а? — F 2*7 IL 28
— Si 1Т ?9 ?3J 3o
X 31 3/ 32
2 2J
4- 3n 33
?’7 3y
—« 37 35 36
d ->] А ГТ 2J
si \ 37 JZ J8 39 4q ‘h
— — — — Я! ?19 3)3 2*3 2^
*— —- 43 231 42 ^3
<5 г 7 ч 44
3j ‘/5
223 /6
X 5 X X '47 47
-=л 3 т
ГЛАВА ПЯТАЯ
теперь развернуть образующую поверхность цилиндра, то плоскости вместо винтовой линии получится ряд отрезков цря^ мых (под углом 45°, параллельных друг другу). Первый сверху отрезок прямой (от 0 до 16) фиксирует атомные веса для эдД ментов от водорода до кислорода (1—16), второй —от 16 до 32 третий — от 32 до 48 и т. д.
Л. А. Чугаев дал следующую оценку построению Шанкуртуа. «При таком расположении сходные элементы часто, но не всегда попадают на одну и ту же образующую цилиндра (из числа 16 „главных44 образующих, проведенных через деления окружности) а атомные веса их А могут быть приблизительно выражены общей формулой A=n+iQm)...
Таким образом, ясно выступает периодическое чередование свойств... Ясно, что в этой системе заключается уже зародыш периодического закона. Но система Шанкуртуа дает обширный простор произволу. С одной стороны, в среду элементов-аналогов попадаются нередко элементы, совершенно посторонние. Так, за кислородом и серой между Se и Те попадает титан; Мп попадает в число аналогов Li, Na и К; железо помещается на одну образующую с Са и т. д. С другой стороны, та же система дает два места для углерода: одно для С с атомным весом 12, другое — отвечающее атомному весу 44» [42, с. VI].
Таким образом, зафиксировав интересные соотношения между атомными весами элементов, Шанкуртуа, оставаясь на почве примитивной систематизации, не смог прийти к формулировке периодического закона. На основе его винтовой линии, например, нельзя рассчитать атомные веса элементов, существование которых можно было предполагать в то время, т. е. классификационная задача решена быть не могла.
Одновременно с Шанкуртуа Лотар Мейер (1830—1895) в 1864 г. опубликовал книгу «Современные теории химии и их значение для химической статики» [43]. В этой книге приведена таблица из 44 элементов (в то время было известно уже 62 элемента), расположенных в 6 столбцах в соответствии с их валентностью по водороду, причем учтены п некоторые другие свойства. В этой таблице автор фиксировал внимание на разностях атомных весов между сходными элементами в столбцах. Таблица была разбита на две части. В главной части элементы размещены в порядке возрастания атомных весов, что тогда уже не было новостью. Вторая часть может рассматриваться как извлечение и3 первой. Однако если в главной таблице в столбцах фигурирую1 тетрады и пентады, сгруппированные в целом безупречно, то во вторую таблицу внесены элементы, не укладывающиеся в пра' вильный порядок групп первой таблицы. Разности в атомных весах в горизонтальных рядах между элементами групп (тетрвД и пентад) приблизительно постоянны и составляют для элементов верхних рядов около 16 и следующих рядов — около 45 (до 49/1
232
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Таблица Л. Мейера (1864)
233
ГЛАВА ПЯТАЯ
Таблица Л. Мейера (1868)
X!—... ----------------------
234
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Описывая таблицы Л. Мейера, некоторые авторы обычно указывают, что в 1868 г. появилась вторая таблица10 этого ученого, которая будто бы предвосхищает периодическую систему элементов Д- И- Менделеева [24, с. 56, 61].
рассмотрение этой таблицы, в которой элементы размещены в 16 группах (16-я группа, однако, пустая) приводит к выводу, чт0 она представляет собой несколько усовершенствованную таблицу 1864 г. В ней фигурируют (группы VIII—XV) тетрады и пентады. Распределение по группам соответствует валентностям элементов по водороду. В таблице отсутствуют многие элементы, например водород, бор, уран, торий, ниобий. Такая таблица, естественно, не может быть признана в качестве выражения периодического закона.
Приведенный обзор важнейших попыток систематизации и классификации химических элементов до 1869 г. показывает, что периодическая законность «...имела под собой к 60-м годам подготов лепную почву и если высказана с определенностью лишь к концу 60-х годов, то этому причину... должно искать в том, что сравнению подвергали только элементы, сходственные между собою. Однако мысль сличить все элементы по величине их атомного веса до того была чужда общему сознанию, что ни «vis tellu-rique» De Chancourtois, ни «Law of Octaves» Newlands не могли обратить на себя чьего-либо внимания, хотя у них обоих, как у Дюма п Штреккера, видно еще больше, чем у Петтенкофера и Ленсена, приближение к периодическому закону и даже его зародыш...
Штреккер, де Шанкуртуа и Ньюландс стояли впереди всех на дороге к периодическому закону, и им недоставало только решимости поставить дело на подобающую ему высоту, с которой видны закон и рефлексы закона на факты» [16, с. 212—213].
Эти слова Д. И. Менделеева представляют собой краткое и вместе с тем всеохватывающее резюме целей и содержания разнообразных попыток выявить закономерные отношения между численными значениями атомных весов химически сходных эле-мсцтов, предпринимавшихся до начала 1860-х годов. Вместе с тем Д. И. Менделеев дает здесь историческую оценку значения всех этих сопоставлений для подготовки открытия периодического закона. Действительно, несмотря на остроумие и изобретательность Исследователей, оперировавших с триадами, тетрадами... эннеада-Мв, а также авторов различных приемов и формул для расчета, этомных весов в группах сходных элементов, в этот период об
10 Эта таблица была опубликовала в 1895 г., уже после смерти Л. Мейера, (то учеником К. Зейбертом.
235
ГЛАВА ПЯТАЯ
щий закон, устанавливающий взаимосвязь свойств всех элемед тов, определенно еще не был открыт. Основной причиной неуд^' чи исследователей был ограниченный, упрощенный подход авто' ров сопоставлений к решению сложной проблемы систематизации элементов. Они оперировали только с группами химически сход, ных элементов и игнорировали сопоставления элементов, различающихся по свойствам друг от друга, т. е. занимались преимущественно систематикой на низшем уровне или классификацией в отрыве от систематизирующего начала. Открытие же закона увенчалось успехом именно в сочетании систематизирующего и классификационного подходов. Это обстоятельство не обесценивает огромной работы предшественников открытия периодического закона, подготовивших почву для этого открытия.
ЧАСТЬ ВТОРАЯ
СОЗДАНИЕ АТОМНО-МОЛЕКУЛЯРНОГО УЧЕНИЯ
ГЛАВА ШЕСТАЯ
ХИМИЧЕСКАЯ АТОМИСТИКА
АТОМИСТИЧЕСКАЯ ТЕОРИЯ ДАЛЬТОНА [1]
Предпосылки создания атомистики Дальтона
Важнейшей предпосылкой для создания атомистики Дальтона явилось развитие количественных методов в химии, начавшееся в середине XVIII в. * * В связи с этим в корне менялся самый подход к изучению вещества. Когда на первом плане стояло качественное исследование, все внимание химиков сосредоточивалось на раскрытии специфических свойств, характеризующих отдельные вещества и их составные части. Теперь же, для того чтобы иметь возможность сравнивать с количественной стороны совершенно различные вещи, внимание химиков должно было в известной степени отвлечься от качественной определенности вещей как от несущественного в данном случае момента исследования и сосредоточиться па общих для всех сравниваемых вещей свойствах, каковыми являются вес и объем.
Убеждение в том, что вес — самое главное свойство материи, составило краеугольный камень механистического мировоззрения XVIII в., которое по наследству досталось естествоиспытателям XIX в. При помощи количественного критерия было установлено, что сера химически более проста, чем серная кислота, так как переход ее в кислоту связан с увеличением веса и, следовательно, представляет собой реакцию соединения. Наоборот, всякая реакция разложения ведет к образованию веществ, каждое из которых обладает меньшим весом, чем исходный продукт. Обобщая это положение, ученые пришли к выводу, что химическим элементом как пределом химических превращений является вещество, вес которого никакими химическими способами не может быть уменьшен путем разделения этого вещества на составные части.
Уже само установление факта, что определенные вещества суть химические элементы, явилось существенной предпосылкой для обоснования атомистики. В самом деле, чтобы более конкретно говорить об атомах как о мельчайших частицах, на которые распадается каждый элемент, химики должны были прежде всего
Примечание. Литературу см. стр. 408—410.
* Атомистическая теория в химии возникла не без влияния многочислен' пых предшествующих атомистических представлений, культивировавшихся первоначально в патурфилософии, а затем в физике.— Прим. отв. ред-
238
ХИМИЧЕСКАЯ АТОМИСТИКА
Умственно познать самые элементы. Иначе разве можно было «ак-либо конкретно применить понятие атома к таким туманным, ^етафизическим представлениям, как tria prima алхимиков или флогистон? Поэтому только в результате полного разгрома всех этих ложных и вульгарных представлений, на основе установления понятия о реальном элементе была расширена дорога для знеДРения атомистики в химию.
Вместе с тем доказательство при помощи весовых измерений
химической неразложимости элементов явилось необходимой предпосылкой для обоснования свойства неделимости простых атомов. Но самое главное состоит в том, что в результате чисто коли-
чественных исследований состава тел постепенно накапливался как раз тот необходимый фактический материал, отсутствие которого обусловило в свое время неудачу атомистики Р. Бойля.
Этот материал позволил химикам открыть три важных химических закона, которые явились ближайшими логическими предпосылками возникновения атомистики Дальтона.
Первой предпосылкой послужил закон постоянного, или определенного, состава тел. Уже в этом открытии наметилась более глубокая связь между качественной и количественной сторонами химических веществ и превращений. Было установлено,
что каждое качественно определенное вещество имеет всегда точно определенный количественный состав, свойственный при всех условиях именно этому веществу. Иначе говоря, элементы обра
зуют каждое вещество всегда в точно определенном количеств
венном соотношении.
Вместе с тем этот закон устанавливает один момент, очень важный с точки зрения обоснования атомистики. Если сравнивать количественный состав различных тел, то при этом обнаруживается определенная прерывность отношений. Особенно наглядно эта прерывность проявляется для тех веществ, которые состоят из одних и тех же элементов, но различаются по их количественному соотношению. Исторически эта группа веществ сыграла исключительно важную роль в обосновании атомистики.
Возьмем, например, вещества, состоящие из углерода и кислорода. Эти вещества имеют не любой произвольный химический состав, а лишь один из двух возможных: 1) углекислый газ содержит 27,3% углерода и 72,7% кислорода; 2) окись углерода содержит 42,9% углерода и 57,1% кислорода, никаких промежуточных соединений, как считалось в то время, никакого непрерывного перехода от одного состава к другому не существует; отношения здесь резко прерывны. Как будто природа вследствие Каких-то неизвестных причин отметила на непрерывной линии возможных соединений углерода и кислорода две определенные точки, и только в соответствии с этими точками она соединяет эти два элемента друг с другом. Как говорил Ж. Пруст, природа Дала химическому соединению постоянный состав и тем самым
239
ГЛАВА ШЕСТАЯ
поставила его в совершенно особое положение по сравнению раствором, сплавом и смесью. . с I
Правда, этот факт еще не является достаточным эмпириче ским обоснованием атомистики; тем не менее идея прерывистого строения вещества получает благодаря этому известное подкрец ление, поскольку явление прерывности обнаруживается и в Со^ ставе вещества.
Как известно, вокруг закона постоянства состава развернулся знаменитый спор Ж. Пруста с К. Бертолле, длившийся семь лет (1801—1808 гг.) и окончившийся победой Пруста, защищавшего идею постоянства состава тел. В то время этот спор имел огромное методологическое значение для всей химии. Подтверждение постоянства состава и, следовательно, его прерывности открывало путь атомистике. Напротив, подтверждение противоположного мнения Бертолле о переменном составе тел лишило бы в то время атомистику всякой эмпирической опоры. Вот почему Пруст и Бертолле, сами того не подозревая, спорили о важнейшей предпосылке для химической атомистики, которая в тот момент уже рождалась в работах Дальтона.
Второй логической предпосылкой для атомистики Дальтона явился закон паев, или эквивалентов. Этот закон представляет собой результат дальнейшего углубления и развития закона постоянства состава, который говорит о составе отдельно взятого вещества, о количественной пропорции образующих это вещество элементов. Закон паев раскрывает те количественные соотношения, в которых реагируют друг с другом различные вещества; тем самым этот закон переходит от рассмотрения отдельных веществ к рассмотрению пропорций между рядом веществ, сходных по химическим свойствам и составу. Основным фактическим материалом для обоснования этого закона послужили реакции взаимной нейтрализации кислот и оснований с последующим образованием солей.
В законе эквивалентов уже отчетливо выступает более глубокая связь между качественной и количественной сторонами изучаемых химических превращений. Понятие «эквивалент» является переходом от абстрактно количественных выражений — от безличных весовых единиц (грамма, грана или унции) — к новым химическим единицам.
Эти новые единицы основаны на том, что каждое качественно определенное вещество обладает своим специфическим количеством и это количество вступает в данную химическую реакцию как целый пай, т. е. как определенная единица. Правда, закон эквивалентов устанавливает не абсолютную величину новых единиц, а специфический пай для каждого вещества по отношению к ряду других веществ, способных вступать в данную химическую реакцию (реакцию нейтрализации, осаждения, окисления и т. п.)*
Выражение химических превращений через эквиваленты яв-
240
ХИМИЧЕСКАЯ АТОМИСТИКА
ддется значительным преимуществом перед их абстрактными сражениями в весовых или объемных процентах. Переход от процентов к паям соответствовал переходу химии от изучения состава отдельных тел к изучению соотношений между рядом тел, вступающих в однотипные реакции. Понятие «пай» родилось, таким образом, при переходе к изучению отношений между реагирующими веществами. Это понятие расширяет ту основу, которую лодводит под новую атомистику закон постоянства состава. Связь качественной и количественной определенности вещества выстудила в понятии «пай» как конкретное проявление «качественного количества».
С другой стороны, понятие «пай» выразило в гораздо более резкой и определенной форме прерывность химических отношений, которая здесь выступает как отношение целых чисел, поскольку каждая химическая реакция может быть представлена как реакция между целыми числами паев реагирующих веществ.
Последнее особенно важно с точки зрения эмпирического обоснования атомистики. Ведь атомистика предполагает, что атом химически неделим и что в химической реакции он участвует как целое. Поэтому если прерывность в составе отдельных веществ и в их взаимодействиях друг с другом обнаружит характер прерывности ряда целых чисел, то главные положения атомистической теории строения материи будут эмпирически подтверждены.
Этим подтверждением явилась третья логическая предпосылка новой атомистики — закон простых кратных отношений.
Мы уже отмечали особую роль, которую сыграли для обоснования атомистики вещества, состоящие из одних и тех же элементов. По сути дела закон кратных отношений представляет собой дальнейшее расширение закона паев и распространение его на сложные вещества, состоящие из одинаковых элементов, но в разных их соотношениях. Выше был приведен пример окиси углерода и углекислого газа, из которых первая содержит 42,9% углерода и 57,1% кислорода, а второй —27,3% углерода и 72,7% кислорода. Пока состав этих веществ выражен в абстрактных процентах, никакой зависимости между этими двумя рядами цифр обнаружить невозможно. Но как только мы перейдем к паям и будем рассматривать количество кислорода в обоих соединениях, взятое по отношению к одному и тому же количеству углерода (т- е. к его паю), то получим следующее: в окиси углерода па 42,9 части углерода приходится 57,1 части (не %!) кислорода; в углекислом газе на то же количество углерода приходится 114,2=2X57,1 части кислорода, так что во втором случае кислорода ровно в два раза больше. Иначе говоря, в окиси углерода 0Дин пай углерода соединяется с одним паем кислорода, а в углекислом газе — с двумя паями кислорода. Совершенно ясно, что т°лько при переходе к новым химическим единицам в виде паев
241
ГЛАВА 1ПЕСТАЙ
можно было раскрыть этот закон *. Старое же, процентное, вы ражение, как это хорошо видно на этом примере, только осложняло дело. Недаром Энгельс писал, что «старые, удобные, дрц, способленные к прежней обычной практике методы переносятся в другие отрасли знания, где они оказываются тормозом: в химии — процентное вычисление состава тел, которое являлось самым подходящим методом для того, чтобы замаскировать — и которое действительно достаточно долго маскировало — закон постоянства состава и кратных отношений у соединений» \
Особенно рельефно проявляется закон на примере соединений азота и кислорода; таких соединений имеется пять.
Помещая в первом столбце число паев азота, а во втором — число паев кислорода, получаем следующий ряд отношений:
Закись азота 2 : 1
Окись азота 1 : 1
Азотистый ангидрид 2 : 3
Двуокись азота 1 : 2
Азотный ангидрид 2 : 5
Здесь уже совершенно отчетливо выступает прерывность отношений, выраженных простыми целыми числами. Если же мы выразим эти отношения в процентах, то никакой закономерной связи между составом всех кислородных соединений азота мы не сможем выявить.
В случае закона кратных отношений наиболее отчетливо проявляется переход от старых единиц измерения к принципиально новым, которые уже сами по себе предполагают качественную определенность вещества. Каждый пай, будучи единицей измерения соответствующего элемента, имеет свою специфическую величину, выраженную определенным числом обычных весовых единиц. Но всякий раз, когда вводится новая единица измерения, предполагается установление новой меры вещей или процессов. В данном случае новой мерой явился атом, который представляет собой единство качественной определенности вещества, развитой в понятии элемент, и его количественной определенности, развитой в понятии пай, или эквивалент. Этим завершается данный этап развития химии. Познание химиков на этом этапе двигалось от чисто внешнего соотношения между моментами качества и количества в химическом исследовании к раскрытию их
* В течение августа и сентября 1804 г. Дальтон исследовал состав болотного газа (метана) и маслородного газа (этилена), открытого еще в 1794 г. Для этилена он установил формулу СН, а для метана — СН2. Эти данные подтверждали, что количественные соотношения элементов, образуют0* несколько соединений, выражаются отношением целых чисел.— При** 1-отв. ред.
1 Маркс К., Энгельс Ф. Соч. 2-е изд., т. 20, с. 608.
242
ХИМИЧЕСКАЯ АТОМИСТИКА
в11утре1Шег0 единства, к признанию атома мерой химических 11ревратЦений-
После эмпирического установления закона кратных отноше-jniii, в котором резко обнаружилась химическая недробимость определенных единиц вещества, до создания атомистики остается т0дько один шаг. Нужно лишь теоретически осмыслить эмпири-деские правила и закономерности.
Но если мы говорим, что рассмотренные выше три стехиометрических закона явились логическими предпосылками для обоснования атомистики Дальтона, то не следует при этом забывать, что не все из них представляли для нее также и исторические предпосылки. Если логически каждый последующий из трех приведенных стехиометрических законов является развитием пре-дыдуЩег°5 то процесс исторического развития химии шел гораздо более извилистым путем. Так, например, исследования в области нейтрализации исторически предшествовали спору Пруста и Бертолле, а этот спор настолько затянулся, что не был решен еще д тогда, когда атомистика Дальтона уже выступила на сцену; и не последним аргументом в пользу представлений Пруста явилось известие о работах Дальтона. Закон же кратных отношений (если не считать работ В. Хиггинса, которые остались почти неизвестными) не только не был открыт до установления атомистики, но само его открытие последовало в результате применения к химии уже готовых атомистических идей.
Таким образом, нельзя провести резких граней и сказать, где кончилось развитие предпосылок для новой атомистики и где началось ее собственное развитие. Сама атомистика выступила таким мощным теоретическим фактором исторического развития химии, что сразу же повлияла на ход отдельных экспериментальных направлений и споров вокруг более или менее частных обобщений, способствуя быстрейшей доработке необходимого для нее фактического обоснования. Поэтому только в известной абстракции мы можем говорить о логических предпосылках химической атомистики.
Создание химической атомистики
К началу XIX в. было уже накоплено множество фактов о с°ставе отдельных веществ и его изменении в результате химических реакций. Теперь требовалось каким-то образом обобщить все это, свести в единую систему.
Стехиометрические правила явились первыми чисто эмпириче-сНими обобщениями; рациональное же их содержание оставалось **0Ка еще не раскрытым. Задача химии и состояла как раз в том, Чтобы подвести общую теоретическую основу под весь эмпирически материал, раскрыв рациональный смысл эмпирических за-чтобы показать, что эмпирические данные о составе тел
243
ГЛАВА ШЕСТАЯ
вытекают из теоретических представлений о строении этих те Следовательно, эта задача заключалась в выяснении связи межи составом веществ и их внутренним строением. у
Состав и строение — вот две стороны, которые предстояло свя зать химикам в единое, цельное учение.
Можно сказать, что задача состояла в том, чтобы решить воц рос о соотношении в области химии между макроскопическим^ величинами, к которым относятся данные о составе тел, и микр0. величинами, к которым относятся представления о строении тел
Эту задачу блестяще разрешил в начале XIX в. Джон Далт^ тон * [2]. ж
Каково же было решение, данное Дальтоном? Прежде всего Дальтон развивает свои представления о строении вещества на основе старой идеи об атомах. В главе «О строении тел» своей «Новой системы химической философии» ** Дальтон показывает что уже простейшие наблюдения над агрегатными состояниями таких веществ, как вода, «привели к почти общепринятому (под-черкнуто нами.— Б. К.) заключению, что все тела, обладающие
* Джон Дальтон родился 6 сентября 1766 г. в небольшой английской деревушке Игльсфильд (Кемберленд) в семье бедного прядильщика шерсти. В раннем возрасте зарабатывал себе на пропитание частными уроками. В 1793—1799 — учитель математики в «Новом колледже» Манчестера. С 1799 — частный преподаватель математики и натуральной философии. Принимал активное участие в работе Манчестерского литературного и философского общества. С 1800 — секретарь, а с 1817 — президент этого общества. В 1822 г. избран членом Лондонского Королевского общества. Умер Дальтон 27 июля 1844 г. в Манчестере.
О жизни и деятельности Дж. Дальтона см.: Henry W. С. Memoirs of the life and scientific researches of J. Dalton. London, 1854; Кедров Б. M. Атомистика Дальтона. M.; Л.: Госхимиздат, 1949; Cardwell D. S. John Dalton and the Progress of Science. Manchester, 1968.— Прим. отв. ped.
** Дальтон весной 1807 г., после лекций в Эдинбурге и Глазго, где он впервые изложил основные положения химической атомистики, решил опубликовать свою теорию. В июне 1808 г. вышла первая часть книги «Новая система химической философии», в которой автор писал: «Одна из важнейших задач данной работы — показать важность и преимущество установления относительных весов первичных частиц простых и сложных тел» [3, с. 91].
С разрешения Дальтона Т. Томсон включил основные положения новой теории в третье издание своего учебника «Системы химии» (1807), изложив их в специальной главе «Гипотезы Дальтона относительно плотности атомов газов». В этой главе Томсон формулирует закон кратных отношений, приводит символы атомов элементов по Дальтону и таблицу атомных весов некоторых элементов и их соединений. По мнению автора «теория Дальтона есть нечто более, нежели простая гипотеза; она заслУ' живает особенного внимания и может оказать большую помощь при исследовании составных частей твердых тел» [4]. Ц
В январе — феврале 1810 г. Дальтон прочитал в Манчестере лекди0’ в которых подробно раскрыл основное содержание своей теории. Изло жение основных положений химической атомистики и ее роли в хим0 было дано им во второй части «Новой системы...», опубликований в 1810 г. [5]. Здесь он дал таблицу атомных весов для 36 элементов*" Прим. отв. ред.
ХИМИЧЕСКАЯ АТОМИСТИКА
о1ДУт11МОИ величинои? как жидкие, так и твердые, состоят из грОмного числа чрезвычайно малых частиц, или атомов, материи, связанных вместе силой притяжения» [5, с. 141].
Как видно, саму идею атомов Дальтон считает почти обще
принятой. Речь же идет о том, какими конкретными свойствами нужно наделить эти атомы, для того чтобы при помощи них А1Ожно было объяснить существующие закономерности в соста-
ве тел.
Таких основных свойств Дальтон установил четыре. Первым свойством является полное тождество атомов одного и того же вещества, что следует из факта постоянства состава тел.
Дальтон категорически отвергает учение К. Бертолле, «согласно которому химическое действие пропорционально массе,
так что во всех химических соединениях существует неощутимая градация в соотношениях составных частей» [5, с. 142].
Но если мы признали постоянство и определенность этих соотношений, то тогда не безразлично, будем ли мы считать атомы данного вещества одинаковыми по форме, весу и т. д. или же различными. Допустим, например, что атомы воды различны. «Но тогда,— замечает Дальтон,— едва ли можно понять, каким образом агрегаты могут быть однородными, если состоят они из непохожих частиц. Если бы некоторые из частиц воды были тяжелее, нежели прочие, и если бы часть жидкости по той или иной причине состояла преимущественно из этих более тяжелых частиц, то это должно было бы сказаться на удельном весе воды, а это обстоятельство нам неизвестно. Подобные наблюдения можно произвести и в отношении других веществ. Поэтому можно заключить, что первичные частицы2 всех однородных тел совершенно одинаковы по весу, виду и т. д. Иначе говоря, любая частица воды равна любой другой частице воды...» [5, с. 143].
Таким образом, соответствие между макро- и микровеличинами в химии было достигнуто прежде всего путем признания тождественности атомов каждого вещества.
Вторым свойством, каким нужно было наделить атомы для того, чтобы эмпирические правила, которым подчиняется состав тел, получили рациональное объяснение, была способность различных атомов соединяться между собой в различных соотношениях.
В главе «О химическом синтезе» Дальтон рассматривает, каким образом могут соединяться два простых тела (в данном слу
2 Согласно Дальтону, все вещества распадаются непосредственно на атомы, или частицы; если же это элементарные вещества, то они распадаются на простые атомы (частицы), если они химически сложные, то — на составные атомы (частицы), состоящие каждый из двух или более различных простых атомов (частиц). В дальнейшем в этом разделе будем придерживаться терминологии Дальтона.
245
ГЛАВА ШЕСТАЯ
чае два элемента) А и В, способных химически взаимодейств0 вать друг с другом:
1 атом А + 1 атом В — I атом С, двойной;
1 атом А-| 2 атома В = 1 атом D, тройной;
2 атома А + 1 атом В = 1 атом Е, тройной;
1 атом А + 3 атома В = 1 атом F, четверной;
3 атома А + 1 атом В = 1 атом G, четверной, и т. д. [5, с. 213].
На примере кислородных соединений азота как раз можно продемонстрировать образование «сложных атомов» двойного тройного и высших порядков, что и делает Дальтон* [5, с. 331]’
Третьим свойством атомов, с которыми связана их способность соединяться друг с другом в целочисленном отношении является абсолютная неделимость простых атомов ни в смысле их механического дробления на более мелкие части без изменения природы и свойств самого вещества, ни в смысле их химического разложения па качественно отличные от них, химически более простые составные части.
В связи с этим нужно отметить, что некоторых химиков и историков химии вводит в заблуждение следующая оговорка Дальтона: «...Под элементарными составными частями, или простыми телами, мы имеем в виду те, которые не разложены... мы не знаем, всякое ли тело из тел, названных элементарными, абсолютно (подчеркнуто нами.—Б. К.) неразложимо» [5, с. 221-222]. Эта оговорка была вызвана тем, что окислы щелочных металлов, которые вначале принимались за элементы, были впоследствии разложены Г. Дэви электролитическим путем с выделением свободных металлов.
Оговорка Дальтона при определении понятия простого атома дала повод Р. Смиту [6] утверждать, что вообще Дальтон «отнюдь не предполагал, что в наших элементах мы добрались до неделимого атома» и что «во всяком случае он воздерживается тут от суждения». Это неверно. Дальтон был убежден в том, что действительно простые атомы абсолютно неразложимы. Однако он еще не был вполне уверен в том, что среди неразлагаемых атомов нет таких, которые являются сложными и могут быть со временем разложены на более простые. «Мы не можем знать, — говорит Дальтон,— какие элементы являются абсолютно неразложимыми и какие упорно не поддаются разложению вследствие того, что мы не применяем надлежащих способов для их разложения (восстановления — reduktion)» (цит. по [7, с. 112]).
Таким образом, Дальтон соединил в одном понятии «элемен-
* В атомистике Дальтона «сложный атом» является производной системой свойства которой определяются атомами, из которых она составлена, т. е-«сложный атом» (молекула) обладает абсолютной аддитивностью. СамЫ*1 слабым местом атомистики Дальтона является то, что понятие «молекУ' ла» не применяется к простым веществам.— Прим. отв. ред.
246
ХИМИЧЕСКАЯ АТОМИСТИКА
яОе вещество» те атомы, которые являются действительно Шестыми, и те сложные атомы, которые химики не сумели еще огДа разложить на составные части.
т Приведенная выше оговорка Дальтона относится не ко всем
„ементарным атомам, а только к тем из них, которые временно Несены к этой группе и которые принципиально могут быть изложены. Поэтому не прав Смит, который полагает, что Даль-1ОН в принципе готов допустить разложимость всех атомов.
Неделимость простых атомов Дальтон связывает непосредственно с законом сохранения материи. «Химический анализ и синтез,— пишет он,— не идут дальше отделения частиц друг от друга и их обратного воссоединения. Создание материи заново пли ее разрушение не находится в пределах досягаемости химического действия» [5, с. 212]. И далее, Дальтон сравнивает попытку разрушить или создать атом водорода с попыткой разрушить одну из планет солнечной системы или создать новую планету. Но если вследствие своей недробимости атомы вступают
в соединение только целыми единицами, то тем самым строение «сложных атомов», образованных всегда из целого числа различных элементарных атомов, объясняет простоту и кратность отношений в составе химических соединений.
Следовательно, признание химической и механической неделимости простых атомов и их способности связываться друг с другом в различных соотношениях перекидывает еще один мостик
между макро- и микровеличинами в химии.
Наконец, четвертым, и самым важным, свойством, которым наделяет свои атомы Дальтон и которое объясняет стехиометрию химических соединений, является атомный вес*.
* В начале сентября 1803 г. Дж. Дальтону пришла идея исходить при определении веса частиц не из абсолютного веса атома, а из его относительного веса. Но для этого следовало принять за единицу вес атома одного какого-нибудь элемента. За такую единицу Дальтон принял вес атома водорода, равный 1.
6 сентября 1803 г. Дальтон составляет первую «таблицу относительных
весов первичных частиц»:
Первичный атом водорода I
кислорода 5,66
азота 4
углерода 4,5
воды 6,66
аммиака 5
селитренного газа 9,66
селитренной окиси 13,66
селитренной кислоты 15,32
серы 17
сернистой кислоты 22,66
серной кислоты 28,32
угольной кислоты 15,8
окиси углерода 10,2
Атомный вес кислорода Дальтон нашел на основании данных анализа воды (85% кислорода и 15% водорода), полученных А. Лавуазье. Относительный вес атома кислорода был вычислен из соотношения 85:15= =5,66. Атомный вес атома азота Дальтоп установил, пользуясь данными
247
ГЛАВА ШЕСТАЯ
Как мы уже видели, понятие «пай» имеет смысл в примец иии не к отдельному, изолированному, веществу, а только к рЯг/ веществ, вступающих в однотипные реакции. В связи с этщ находится и то, что абсолютный размер пая является величиной неопределенной. В рамках закона эквивалентов речь могла идТ11 лишь об относительном определении величины пая. Переход От эмпирического понятия «пай», или «эквивалент», к более общему и кардинальному понятию «атомный вес» предполагал ряд сту пеней.
Прежде всего необходимо было распространить понятие «пай» от сложных веществ (кислот, оснований, солей и окпслов) На отдельные элементы, входящие в состав этих сложных тел. Так например, Дальтон устанавливает, что в случае воды, для которой он принимает состав 87,4 весовой части кислорода и 12,6 части водорода, эквивалент кислорода в 7 раз больше эквивалента водорода. Теперь, чтобы определить атомный вес, нужно было еще знать, сколько же атомов кислорода и водорода образуют один «атом» воды.
Дальтон дает общий метод решения вопроса. Он допускает, что если имеется только одно соединение веществ Л и В, то этому соединению нужен атом водорода второго порядка, состоящий из одного атома А и одного атома В.
«Следующие правила,— пишет Дальтон,— могут быть применены как руководящие во всех наших исследованиях в области химического синтеза.
1. Если можно получить только одно соединение из двух тел, то нужно предполагать, что оно двойное, если против этого не выставлено каких-либо дополнительных данных.
2. Если имеются два соединения, то следует допустить наличие двойного и тройного» [5, с. 214] и т. д.
Следовательно, у Дальтона каждый ряд соединений двух элементов обязательно начинается с простейшего соединения. Эмпирических данных, доказывающих правомерность такого допущения в качестве общего закона, у Дальтона не было, да и быть не могло, так как действительные отношения вовсе не подчиняются подобного рода закономерности. Но вместе с тем у Дальтона
анализа аммиака NH (80% азота и 20% водорода), 80:20=4. Атомный вес углерода он вычислил из данных анализа углекислого газа (72% Уг' лерода), полученных Лавуазье; атомный вес серы был получен в результате анализа серной кислоты (61,5% серы), при этом учитывался уже и3" вестный атомный вес кислорода и предполагалось, что формула серной кислоты SO2. Дальтон приписывал низшему окислу формулу RO, а высшему RO2 (например, СпО и СиО2). Это правило он распространил 03 все окислы. Например, для цинка и серебра формулы Дальтона (в совре" менпых знаках) были ZnO2 и AgO2, так как он считал, что у всех металлов (и в том числе у цинка или серебра) имеются низшие окислы Znv и AgO, которые еще не открыты. Для окислов железа, кобальта, никслй и других металлов он предполагает формулы RO2 и RO3.— Прим. отв. реС>'
248
ХИМИЧЕСКАЯ АТОМИСТИКА
было в то время объективного критерия для того, чтобы правильно определить число элементарных атомов, образующих частиЦУ данного сложного вещества *.
Поэтому волей-неволей он вынужден был стать на путь произвольных допущений и ad hoc выбирать из общего числа возможных комбинаций наиболее простые.
На это правильно указывают Г. Роско и А. Гарден в работе о аропсхождении атомистической теории Дальтона. «Стремясь определить вес последней частицы, или атома, вещества,— пишут они,— Дальтон скоро заметил, что одно только знание пропорций, в которых одно вещество соединяется с определенным весовым количеством другого вещества, выбранного в качестве мерила, недостаточно; необходимо определить также число атомов, между которыми получается соединение. Для решения этого важного вопроса он не был в состоянии наметить более определенного пути, чем допущение «закона наибольшей простоты», по которому наиболее простая формула обладает и наибольшей вероятностью быть точной» [7, с. 81].
Как мы увидим далее3, на пути подобного допущения Дальтон пришел в непримиримое противоречие с открытием Ж.-Л. Гей-Люссаком закона кратных объемных отношений и этим сильно ослабил свои научные позиции **. Но вместе с тем принцип наибольшей простоты так или иначе позволил Дальтону определить
* В 1806—1810 гг. Дж. Дальтон использовал несколько способов для вычисления относительных атомных весов элементов на основании имевшихся в то время химических данных. Он применял такие методы анализа, как сжигание известного количества металла и определение веса получившегося окисла; растворение определенного количества металла в кислоте, осаждение и взвешивание прокаленного окисла; определение объема водорода, выделяющегося при растворении известного количества металла в кислоте; окисление низших окислов действием раствора хлорной извести и осаждение получившегося высшего окисла; определение объема окисла азота, выделяемого при растворении металла в азотной кислоте.— Прим. отв. ред.
** В 1808 г. во Франции появился перевод книги Т. Томсона «Система химии неорганических веществ» с предисловием К. Бертолле. Нельзя согласиться с А. Вюрцем, что Бертолле в этом предисловии «нападает весьма ожесточенно на атомистическую систему» [8, с. 17]. На самом деле Бертолле писал: «В этом сочинении находится элегантное изложение гениальной гипотезы Дальтона, с помощью которой он объясняет постоянные пропорции, наблюдаемые между элементами некоторых сложных тел... Эта гипотеза дает объяснение явления, причина которого до сих пор оставалась непонятной; но чем она привлекательнее, тем более нужно подвергнуть ее внимательному рассмотрению» (цит. по [9, с. 18]).
Как видно, сомнения К. Бертолле относились не столько к самой идее Дальтона, сколько к общности закона кратных отношений, а также к способу определения атомных весов исходя из постулата Дальтона о преимущественном соединении атомов в простейшей пропорции lj 1. Бертолле в этом ошибочном предположении Дальтона уловил самый слабый и уязвимый пункт его теории.— Прим. отв. ред.
См. главу седьмую.
249
ГЛАВА ШЕСТАЯ
первые значения относительных атомных весов в наиболее пых из известных в то время элементов.
Теперь, когда известен пай элемента и определено число ато мов, входящих в одну сложную частицу, остается сделать ц0' следний шаг для установления атомного веса. Для этого пуи^ только выбрать единицу, при помощи которой можно измеряв и численно выражать веса атомов. В качестве таковой Дальтоц выбрал эквивалент водорода как наименьший. Тогда автомата чески отношения всех паев остальных элементов к паю водород (с поправкой на число атомов, образующих данную сложную стицу) оказались равными как раз атомному весу соответствую, щих элементов. Короче говоря, атомный вес элементов выступил как отношение веса атома этого элемента к весу атома водорода Именно таков был путь самого Дальтона. Выбрав в качестве ру_ ководящего правила принцип наибольшей простоты и прилагая его к химическим фактам, Дальтон приходит к выводу, «что вода есть двойное соединение водорода и кислорода и что относительные веса двух элементарных атомов составляют почти 1:7» [5, с. 214-215].
Благодаря установлению атомных весов не только закон паев но и все количественные взаимоотношения в химии получили свою рациональную расшифровку. Именно в этом видел Дальтон главную задачу всей своей работы. «Основная цель этого произведения,—пишет он в «Новой системе»,—показать важность и пользу установления относительных весов первичных частиц как простых, так и сложных тел, числа простых элементарных частиц, образующих сложную частицу, и число менее сложных частиц, которые участвуют в образовании более сложных частиц» [5, с. 213]. ‘ I
Этой цели Дальтон в основном достиг. Правда, его атомные веса могли быть приняты лишь в качестве первого приближения; но самый метод их определения — сравнением весов составных частей химического соединения — был полностью оправдан всем последующим развитием химии.
Для определения же числа атомов в частице сложного тела метод Дальтона, опиравшийся на произвольный «закон наибольшей простоты», оказался несостоятельным и в дальнейшем был неправлен на основании молекулярного учения. Что же мы имеем в итоге работ Дальтона?
Прежде всего, благодаря тому, что Дальтон указал путь определения атомных весов, он установил вполне конкретную связь между теоретическими представлениями об атомах и опытными данными. Установить подобную связь обычно означает «объяснить» данное явление. Поэтому можно сказать, что учение об атомистическом строении тел «объяснило» правила и соотношения в составе тел. Достиг же этого Дальтон тем, что мысленно перенес па микровеличины все те соотношения и свойства, кото-
250 '
ХИМИЧЕСКАЯ АТОМИСТИКА
pALTON DALTON DALTON DALTON THOMSON
6 Sept. 1803 1807 1808 1807
ф HyJrogen (H) Hydrogen Q 0 Hydrogen 0 Hydrogen 0 Hydrogen
Q Oxygen О Oxygen Q О Oxygen Q Oxygen О Oxygen
ф Azote @ Azote 0 ) Azote 0 Azote © Azote
ф Carbon ф Carbon ф ф Carbon ф Carbon (J) Carbon
ф Sulphur © Sulphur 1 ) © Sulphur 0 Sulphur © Sulphur
Phosphorus 0 0 Phosphorus 0 Phosphorus © Phosphorus
© Magnesia @ Magnesia
@ Lime 1 ® Zircone ] © Lime
© Gold © Gold Muriatic Acid
Символы химических элементов по Дальтону и Томсону
рые чувственно были установлены для макровеличин. Совершенно правильно указывает на это биограф Дальтона Р. Смит, который следующим образом излагает подход Дальтона к установлению связи между макро- и микровеличинами в химии:
1. «Если мельчайшая часть имеет определенный состав, то же самое имеет место в самой большой части. Это основной закон определенного состава соединений».
2. «Раз большие куски построены так же, как и малые, кратные отношения становятся законом строения всех тел».
3. «Когда мы получаем относительные веса составных частей Тел, то одновременно мы получаем относительные веса атомов, тлк как мельчайшие части должны быть построены так же, как и самые большие» [6].
В результате этого атом из абстрактно-механической модели превратился в конкретное химическое понятие. У Дальтона элементарный атом наделен не фантастическими крючочками и за
251
ГЛаёа шестая
зубринками, а вполне конкретными физико-химическими своц ствами: тождественностью с другими атомами того же элеменД способностью соединяться с различным числом атомов друг^ ’ элементов, химической неразложимостью и механической неде лимостью, атомным весом.
Из всех этих свойств последнее является самым главным всеобъемлющим; именно оно превратило атом из чисто философ, ского понятия в естественнонаучное. Поэтому не случайно осо. был интерес приобретает вопрос о том, кто первый дал пде1о о различии атомов по их весу. Однако здесь не надо смешивать два вопроса: кто первый высказал (хотя бы в самой отвлеченной форме) эту идею и кто первый конкретно применил ее для объяснения определенных естественнонаучных фактов.
Было бы неверно утверждать, что Дальтон первый выдвинул самую идею различия атомов по весу, как это делают обычно многие химики и историки химии, не знающие историю философии 4. Не только представления о самих атомах, по и представления об их различии по величине и по весу были уже высказаны представителями античной философии. Так, еще Демокрит, по словам Аристотеля, говорил, «что каждое из неделимых (телец) бывает более тяжелым вследствие большего размера», и далее:! «Тем, которые утверждают, что первичные тела тверды (в противоположность Платону), более позволительно говорить, что большее из них более тяжело» [10, с. 49].
«...У Диогена Лаэрция (кн. X, §§ 43—44 и 61) можно прочесть, что уже Эпикур приписывал атомам не только различия по величине и форме, но также и различия по весу, т. е. что Эпикур по-своему уже знал атомный вес и атомный объем»5.
Наконец, и сам Дальтон в своих лекциях, прочитанных в 1810 г. в Лондонском Королевском институте, отнюдь не претендует на приоритет по вопросу об атомных весах, а прямо делает ссылку на Ньютона*, когда выставляет тезис, что «атомы различного рода неодинаковы по весу» (цит. по [7, с. 18]).
Однако если задолго до Дальтона была уже высказана мысль о различной весомости атомов, то это было сделано лишь в самой общей, самой абстрактной форме; Дальтон же впервые сделал эту мысль конкретной: с ее помощью он первый объяснил сте-
4 По мнению Смита, Дальтону «первому принадлежит идея атомных весов»-
5 Маркс К., Энгельс Ф. Соч. 2-е изд., т. 20, с. 367—368.
* -Труд И. Ньютона «Математические начала натуральной философии» («Philosophiae naturalis principia mathematica», 1687) был настольной книгой Дальтона. 23-я глава этой книги особенно привлекла внимание Дальтона. В ней Ньютон показал, что «упругий флюид» (т. е. воздух) состоит из маленьких корпускул, или атомов, вещества, которые отталкиваются друг от друга с силой, увеличивающейся пропорционально уменьшению расстояния между ними (более подробно см. [И]).— Прим, отв-ped.
252
ХИМИЧЕСКАЯ АТОМИСТИКА
.^метрические законы химии и первый на основании этих зако-^оВ указал эмпирический путь точного измерения атомных весов. «Для мира,—пишет Смит,—его (Дальтона.—Б. К.) заслуги со-сТОят в том, что он первый увидел огромное значение использования атомов для объяснения отношений и определенности состава» [6].
И если Смит ошибается, думая, что Дальтон первый выдвинул идею атомных весов, то в данном случае он прав: действительно, Дальтон первый до конца объяснил химическую стехиометрию при помощи своей атомистики.
Закон кратных отношений
Остановимся теперь на истории открытия Дальтоном важнейшего из стехиометрических законов — закона кратных отношений,— так как на этом примере отчетливо обнаруживается вся сила теоретического мышления Дальтона.
Обычная версия гласит, будто бы Дальтон сначала открыл закон кратных отношений, а затем, стремясь его объяснить, воспользовался атомистической гипотезой. Эта версия сложилась еще во времена Дальтона; сам Дальтон не описал истории возникновения своих идей; его современники восприняли атомистику прежде всего как объяснение простоты и кратности соотношений в химическом составе тел. Возможно, именно поэтому они приписали Дальтону тот же путь, какой проделали сами, воспринимая его идеи. Но опубликованные Г. Роско и А. Гарденом в 1896 г. рукописные записки Дальтона доказывают, что действительный ход развития идеи Дальтона был принципиально иным, нежели обычно изображают.
В своей 17-й лекции «О химических элементах» (см. [7, с. 13—18]) Дальтон рассказал, откуда, произошли и как развивались его атомистические идеи (эта лекция была опубликована лишь в самом конце XIX в.).
Исходным пунктом, положившим начало всему учению Дальтона, явились метеорологические наблюдения и попытки Дальтона выяснить природу и строение атмосферы. Основной факт, который поражал всегда химиков и требовал объяснения, состоял в следующем. Если привести в соприкосновение два различных газа (или, как их тогда называли, упругие, или эластичные, Жидкости), то эти газы диффундируют друг в друга и образуют Физически однородную смесь, но отнюдь не расслаиваются, подобно тому как это наблюдается у несмешивающихся жидкостей Различного удельного веса. При этом диффузия газов происходит сама собой, без участия внешней силы. Чтобы объяснить это, Дальтон должен был прежде всего выбрать определенную гипотезу строения газов вообще. В то время существовали две такие гипотезы.
253
ГЛАВА ШЕСТАЯ
Первая — химическая — была принята главным образом фра^ цузской школой химиков; в основу объяснения процесса дифф/ зии кладется химическое сродство газов; однородность газовой смеси объясняется тем, что газы растворяются друг в друге, обр^ зуя при этом своего рода химическое соединение.
Вторая гипотеза — механическая — была высказана И. Ньюто-ном; в ней исходят из признания атомов, которые взаимно от. талкиваются друг от друга. Однако эта гипотеза возникла еще тогда, когда не было известно, что атмосфера имеет сложный состав; поэтому ее нужно было развить применительно к явлению диффузии газов.
Дальтон, будучи горячим сторонником воззрений Ньютона становится на сторону атомистической гипотезы строения атмосферы. С точки зрения этой гипотезы он ищет объяснения, почему смесь различных газов является однородной. Прежде всего, он опровергает предположение, что при смешивании газов различные атомы соединяются друг с другом; он доказывает, что подобное предположение противоречит фактам. Затем он придумывает различные модели строения атмосферы исходя из атомистических представлений. После ряда неудачных попыток он приходит к следующему, более или менее оформленному, взгляду. Газ построен из атомов; между атомами действуют две противоположные силы — сила притяжения и сила отталкивания. Обе силы носят всеобщий характер; сила притяжения по своей природе тождественна ньютоновской силе тяготения и присуща самим атомам; силой же отталкивания обладает теплород, который является носителем всех тепловых явлений, происходящих в газе. В результате взаимного наложения этих сил происходят все процессы в газе: расширение, сжатие, нагревание, конденсация и т. д.
Модель однородного газа рисуется Дальтону в следующем виде. Каждый атом газа окружен атмосферой теплорода, плотность которого убывает с увеличением расстояния от центра атома; теплород мешает атомам вступить в непосредственное соприкосновение друг с другом; при этом объем теплородной оболочки может в 1000 и более раз превышать объем самого атома.
Независимо от формы твердого атома вся система, состоящая из атома и окружающей его атмосферы теплорода, имеет шаровидную форму. Но так как все эти шарики испытывают одно и то же давление, то они должны иметь равный объем и располагаться горизонтальными рядами, как это наблюдается в кучке дроби. В конце второй части «Повой системы...» Дальтон приводит ряд моделей подобного строения газов, причем он вводит особые обозначения атомов и их оболочек6. Например, для водо-рода эта модель будет иметь вид, изображенный на рис. 1.
6 Значком ® Дальтон обозначает атом водорода; ф — атом азота, О — атоМ кислорода; лучами изображается теплородная атмосфера.
254
ХИМИЧЕСКАЯ АТОМИСТИКА
Рис. 1.
Модель строения однородного газа (водорода) по Дальтону
Но как же теперь с этой точки зре-Н0Я объяснить процесс диффузии газов л образование однородной смеси?
Еще в 1801 г. Дальтон высказал предположение, что атомы одного рода отталкиваются от атомов того же рода, ио не от атомов другого рода. Раз это так, то, независимо от присутствия других газов, каждый газ равномерно должен распространяться на весь объем системы. Таким образом, Дальтон приходит к известному закону о парциальных давлениях, гласящему, что каждый газ в смеси оказывает такое давление на стенки сосуда, как если бы он один занимал все пространство7 [5, с. 191-192].
Хотя это предположение и объясняло, почему смесь получается однородной, но оно казалось неприемлемым, так как при этом выходило, что существует столько видов отталкивательных сил, сколько имеется газов, и что само тепло при этом не является отталкивательной силой, что противоречило исходному взгляду Дальтона на природу теплорода.
В этом противоречии вся суть вопроса. Чтобы разрешить это противоречие, Дальтону нужно было показать, что отталкивательная сила у всех атомов одна и та же — это теплород; но проявляется она так, что одинаковые частицы отталкиваются друг от друга, а различные — не взаимодействуют вовсе. И вот в 1805 г. Дальтону пришло в голову, как он сам писал, что он никогда не принимал во внимание различия в величине частиц, понимая под величиной твердую частицу в центре с окружающей ее атмосферой теплорода. Если же атомы различных родов обладают различными величинами, то исходя из этого можно допустить, что два разных газа, между которыми удалена разделяющая их перегородка, пе останутся в равновесии, а под влиянием отталкивательной силы теплорода начнут проникать друг в друга; произойдет это потому, что частицы, которые давят друг на друга, обладают различной величиной (подобно тому, как можно представить перемешивание крупных и мелких дробинок).
Дальтон ищет доказательство своему предположению; он находит, что один объем азота и один объем кислорода дают при —
7 Интересно отметить, что именно закон Дальтона о парциальных давлениях лежит в основе процесса диффузии газов. Игнорирование этого закона при рассмотрении диффузии газов привело в конце XIX в. к так называемому парадоксу Гиббса.
255
ГЛАВА ШЕСТАЯ
Рис. 2.
Модель строения атомов кислорода, азота и селитренного газа
Рис. 3.
Диаграмма, объясняющая диффузию одного газа в другой
химическом соединении примерно два объема селитренного газа (окиси азота), которому он приписывает двойной состав Ф О. Но, замечает Дальтон, два объема селитренного газа не могут содержать больше частиц этого газа, чем содержал один объем азота или один объем кислорода. А раз так, то отсюда неизбежно следует допустить, что у разных газов атомы обладают различной величиной, причем атом окиси азота должен быть примерно в два раза больше атома кислорода или атома азота.
Схематически это можно изобразить составленной нами диаграммой (рис. 2).
В том, что разные атомы обладают разной величиной, Дальтон видит причину, почему диффузия одного газа в другой происходит без помощи какой-либо иной отталкивательной силы, кроме хорошо известного тепла.
В «Новой системе...» он приводит диаграмму, которая иллюстрирует его мысль (рис. 3).
Здесь изображаются два газа — водород и азот. «Частица» водорода примерно раза в два больше «частицы» азота. Лучи, исходящие от атомов, изображают теплородные оболочки. В случае однородных атомов внутри азота или водорода эти лучи встречаются друг с другом и, как бы взаимно «упираясь», поддерживают общее равновесие между атомами; однородные атомы ие
256
ХИМИЧЕСКАЯ АТОМИСТИКА
рИближаются и не удаляются друг от друга, а находятся как во взвешенном состоянии. Но в плоскости Z соприкосновения Обоих газов лучи теплородных оболочек, исходящие от различ-gbix атомов, уже не «упираются» друг в друга. В результате этого равновесие сохраниться не может и начинается взаимное проникновение газов до тех пор, пока каждый из них не распространится на весь объем системы8.
Именно эти представления о разной величине атомов натолкнули Дальтона на мысль о различном их весе, а далее — на запои кратных отношений. «После того,— пишет он,— как я пришел п убеждению о различии в величине частиц упругих жидкостей при одинаковых условиях температуры и давления, передо мной встала задача определения относительных величин и весов, а вместе с тем и относительных чисел атомов в данном объеме. Это привело меня к исследованию соединений газов и чисел атомов, входящих в подобные соединения... Я подверг также исследованию и другие тела, а именно: жидкие и твердые, которые соединяются с упругими жидкостями. Так возник ряд исследований для определения числа и веса всех химически элементарных составных частей, которые соединяются друг с другом» (цит. по [7, с. 17]).
Здесь отчетливо видно, что идея об атомных весах и различном числе атомов, входящих в соединение, возникла у Дальтона не под влиянием открытия кратности отношений в составе тел, а как раз наоборот: самое исследование состава тел Дальтон предпринял исходя именно из допущения о различной величине и весомости атомов.
В другой, более ранней, работе Дальтона, относящейся к 1803 г., также выявляется связь атомистических идей Дальтона не с результатами химического анализа состава тел, а с изучением физических явлений в области газов, в данном случае — явлений абсорбции газов жидкостями. Так, пытаясь объяснить процесс абсорбции газов водой, Дальтон констатирует, что величайшее затруднение, которое встает перед механической (атомистической) гипотезой, состоит в том, что различные газы по-разному ведут себя в этом процессе. «Я почти убежден,— пишет Дальтон,— что это различие зависит от веса и числа мельчайших частиц у различных газов» [12]. И он поясняет, что те газы, У которых частиц меньше и сами частицы легче, поглощаются Жидкостью слабее; в случае же более тяжелых частиц и большего их количества абсорбция идет сильнее.
Таким образом, вполне правы Г. Роско и А. Гарден, которые возражают против взгляда, «что атомистическая теория якобы
8 Здесь мы видим, как ложное в своей основе представление о теплороде сыграло известную положительную роль, поскольку натолкнуло Дальтона на мысль о различии атомов по величине, а позднее — и по весу.
Всеобщая история химии
^7
ГЛАВА ШЕСТАЯ
была применена для объяснения фактов, уже установленных мическим анализом», :
кратных отношений. Они совершенно правильно считают,
и, прежде всего,—для объяснения закщ^
Y /- ТТ ’
чисто физические соображения навели Дальтона на мысль о том что атомы различных веществ должны иметь различный вес’ далее, на этом основании Дальтон сделал предположение, что химическое соединение происходит между различными количест-
вами атомов определенного веса и только позднее свою точку зрения ему удалось подтвердить результатами анализов. «Таким образом,—заключают Роско и Гарден,—действительные отношения были как раз обратные той картине, которая получила все-общее признание, а именно: теория существования атомов раз. личного веса привела Дальтона к открытию фактов соединения в кратных отношениях» [17, с. VII—IX].
К такому же заключению приходит и Р. Смит. По мнению Смита, теория у Дальтона опередила эксперимент и указала путь последнему. «Развивая свою (атомистическую.—Б. Я.) идею,-пишет Р. Смит,— он (Дальтон.— Б. К.) нашел в ней основной закон природы в его нынешнем виде (речь идет о законе кратных отношений и постоянства состава.—Б. Я.). Он понял важ
ность кратных отношений, или соединения атомов по два и по три, и с этой мыслью приступил к исследованию природы. Он
доказал, что тела следуют законам, соответствующим его гипотезе, которая с тех пор поднялась до уровня научной теории» [6].
Выше мы не случайно отметили, что закон кратных отношений явился логической, но не исторической предпосылкой атомистики Дальтона. Теперь мы видим, что действительное развитие химии шло так, что закон кратных отношений был теоретически предсказан, а затем экспериментально открыт Дальтоном как раз на основании атомистических представлений. Это означает, что атомистика Дальтона выступила не только как простое объяснение готовых фактов, но и, что особенно важно, как научное предвидение новых химических явлений. Тем самым она оправдала себя в качестве теории, освещающей путь экспериментальному
исследованию.
Именно эту сторону работы Дальтона игнорировали многие историки химии конца XIX и начала XX в. Они не понимали, что главная сила теоретического мышления состоит вовсе не в индуктивной обработке отдельных фактов, а в способности, опираясь на уже обобщенный фактический материал, научно прсД' видеть новые явления и закономерности. Это не поняли ни А. Ла-денбург [13, с. 55], ни Г. Герц [14, с. 59—60], ни Э. Мейер [15]. Например, Мейер считал, что при разработке химически11 атомистики Дальтон шел от отдельных опытных данных (от анализа метана и этилена) к открытию закона кратных отношен®11 и только после этого привлек для объяснения открытого закона
258
ХИМЙЧЕСКАЯ АТОМИСТИКА
гСмистические представления. По Мейеру, следовательно, получало ь, что теория Дальтона отнюдь не ориентировала опытного исследователя на новые открытия, а лишь подытоживала найден-цые уже результаты.
А между тем предвидение и последующее затем открытие Дальтоном закона кратных отношений на основании атомистической гипотезы представляют собой не меньший научный подвиг, чем, скажем, предсказание Менделеевым на основании периодического закона неизвестных еще химических элементов. Правда, Дальтон не сформулировал свое предсказание так же ясно и определенно, как это сделал Менделеев. Более того, возможно, что он вообще не отдавал себе отчета в том, что речь идет именно о научном предвидении. Однако это нисколько не умаляет заслуг его теоретического мышления.
Самое главное, что характеризует Дальтона, это то, что в период создания атомистики его научное мышление не было оторвано от практики лабораторного эксперимента и, в широком смысле, от практики тогдашней химической промышленности. По отношению к производству основных неорганических продуктов, как, например, кислоты и соли, атомистика Дальтона сыграла почти такую же роль, какую по отношению к анилинокрасоч-пой и химико-фармацевтической промышленности несколько десятилетий спустя сыграла теория строения органических соединений, выросшая как раз на основе дальтоновского учения.
Атомистика Дальтона сделала серьезный шаг к решению вопроса о сущности химической реакции, т. е. того основного вопроса, который выдвигала перед наукой практика того времени.
Благодаря этому удалось в первом приближении разъяснить «механизм»9 основных технологических процессов химического производства: а) образования сложных тел из элементов; б) разложения сложных тел на их более простые составные части и в) двойного обмена.
Далее, для правильного технического расчета производственного процесса необходимо было иметь в руках теоретически твердо обоснованные и экспериментально точно проверенные данные, которые давали бы возможность правильно ориентироваться при выборе оптимальных количественных соотношений исходных и конечных продуктов, т. е. тех соотношений, которые играют решающую роль в химических процессах. Эту задачу также реши-ла атомистика Дальтона.
Наконец, вся практика химического анализа, качественного 11 количественного, получила в атомистике Дальтона свое рациональное обоснование.
9 Здесь под «механизмом» мы понимаем объяснение химических процессов с точки зрения взаимодействия различных атомов.
259
11*
ГЛАВА ШЕСТАЯ
Carbon
Oxygen
Hydrogen Azote
Sulphur Phosphorus Gold
Platina
Silver
Mercury Copper Iron
Nickel
Tin Lead Zinc
Bismuth
Antimony
Arsenic Cobalt Manganese
Potash
Lime
Water
Uranium Tungsten Titanium
Magnesia Barytes Strontites Alumine Silex
Sulphurous oxide (Sulphur dioxide^
Sulphurous acid
Cerium
Yttria
Glucine
Carbonic oxide (Carbon monoxide)
Carbonic acid (Carbon dioxide)
Olefiant gas (Ethylene)
Carburetted hydrogen (Methane)
Muriatic acid
(Hydrochloric acid)
Sulphate of alumme
Химические символы Дальтона (1810)
Soda
Zircone
Sulphuric acid
Oxymuriatic acid (Chlorine)
Sugar (=Alcohol +
Hydrate of potash
260
ХИМИЧЕСКАЯ АТОМИСТИКА
Правда, связь с практикой химического производства у Даль-оЯа не выступает явно и непосредственно; сам Дальтон не решает задач, имеющих прямое технологическое значение, не разрабатывает рецептов получения новых химических продуктов, пе руководит теми или другими отраслями химической промышленности. Связь с практикой у него обнаруживается лишь в конечном счете и осуществляется через практику лабораторного эксперимента, через выявление тех общих проблем химической которых промышленность вынуждена
без разрешения
была бы вслепую, чисто эмпирически, нащупывать пути более рациональной постановки Однако если учение
тогда еще молодой химической промышленности тем, что подвело под нее общую научную базу, то в еще неизмеримо большей степени учение Дальтона было обязано и своим возникновением, и развитием, и торжеством именно практике химической промышленности, химического эксперимента.
пауки,
производства.
Дальтона оказало серьезную помощь
СТЕХИОМЕТРИЧЕСКИЕ АСПЕКТЫ ХИМИЧЕСКОЙ АТОМИСТИКИ В РАБОТАХ Я. БЕРЦЕЛИУСА*
Одной из главных заслуг Берцелиуса ** перед наукой является развитие им химической атомистики. Однако отношение шведского ученого к теории Дальтона было весьма сложным и со временем неоднократно изменялось. Чтобы понять позицию Берцелиуса, необходимо учесть следующее обстоятельство. Как известно, атомное учение Дальтона имело два аспекта — физи-
* Данный раздел написан И. С. Дмитриевым.
** Йенс Якоб Берцелиус родился 20 августа 1779 г. в селении Вэферсунда (юг Швеции) в семье учителя. В 1793 г. он поступил в гимназию Линчепинга, в 1797 г.— в Упсальскип университет, где начал заниматься химией. В 1802 г. Королевская медицинская коллегия назначила Берцелиуса адъюнктом медицины и фармации при Медико-хирургическом институте в Стокгольме. В 1808 г. Берцелиус избирается действительным членом, а в 1810 — президентом Шведской Академии наук. С 1818 г. он непременный секретарь Академии.
Я. Берцелиус воспитал таких шведских химиков, как И. А. Арфведсон, Н. Г. Сефстрём, К. Г. Мозандер. Его непосредственными учениками были известные немецкие ученые Генрих и Густав Розе, X. Г. Гмелин, Ф. Вёлер, Э. Митчерлих, Г. Магнус, швейцарский химик Ф. Плантамур. Из русских ученых в лаборатории Берцелиуса работали Г. И. Гесс, Й. ф. фрицше, Г. В. Струве. По словам Генриха Розе, Берцелиус был «в любознательном кругу учеников возвышенным примером практического и теоретического учителя».
Умер Я. Берцелиус в ночь с 6 на 7 августа 1848 г.
О жизни и деятельности Я. Берцелиуса см.: Soderbaum Н. G. Jacob Berzelius. Vol. I—III, Uppsala, 1929—1931. Соловьев Ю. И., Куринной В. И. Якоб Берцелиус. 2-е изд. М.: Наука, 1980.— Прим. отв. ред.
261
ГЛАВА ШЕСТАЯ g.AI -_ - — - '
ческпй и химический. Первый включал в себя представление веществе, состоящем из мельчайших, химически неделимых, териальных образований — атомов,— наделенных определенны^ размерами, формой и, что особенно важно, весом, а также сц0 собностью давать посредством соединения друг с другом частицы того или иного состава — молекулы, или, по терминологии Даль-тона, сложные атомы. Второй, химический, аспект атомной тео. рии включал определенные представления о составе сложных атомов, об отношении масс (или объемов, если речь шла о газах) реагирующих и образующихся веществ, методах эксперименталь-ного определения и расчета разнообразных стехиометрических величин, в том числе атомных весов и химических формул. Естественно, этот второй аспект атомистики в химическом плане был наиболее важен, так как именно соответствие или несоответствие теоретических представлений о составе химических соединений экспериментальным данным определяло отношение химика первой половины XIX в. к атомной теории в целом.
За полтора столетия существования научной химии недостач ка в корпускулярно-атомистических учениях абстрактно-метафизического характера не было. Поэтому получить признание у химиков могли лишь те идеи о дискретной структуре вещества, которые позволяли бы логически непротиворечивым образом однозначно связать микро- и макроуровни химической организации вещества. Путь к такому учению был открыт Дальтоном, однако конкретные представления английского ученого о составе химических соединений (например, его принцип наибольшей простоты) оказались далеко неадекватными, что и было отмечено Берцелиусом. «... Если отбросить атомистическую идею Дальтона,—писал он Д. Брюстеру в 1817 г.,— которая сделает его имя бессмертным, то остальное представляет собой лишь собрание довольно плохих и грубо поставленных экспериментов, а также математических выводов, противоречащих друг другу, но которые поначалу, в силу своего математического вида, убеждают» [16, с. 8].
К этому следует добавить, что шведский химик пришел к атомной теории путем, отличным от дальтоновского. Его отправной точкой послужило химико-аналитическое исследование, на первом этапе лишенное какой-либо атомистической трактовки-Разумеется, такой путь совершенно не обязательно должен был кончаться признанием атомистики, исследователь мог не пойти далее эмпирического определения процентного состава веществ и поиска элементарных стехиометрических закономерностей, как это и произошло со многими искуснейшими химиками начала прошлого века. Но Берцелиус был ученым иного склада. Е#У были одинаково чужды как необоснованные фактами теории, так и теоретически неосмысленное собирание фактов. Кроме того, 00 прекрасно понимал, что атомное учение в принципе могло послУ" жить основой для систематизации обширного аналитического
262
ХИМИЧЕСКАЯ АТОМИСТИКА
риала химии и его следует принять если пе как установленную истину, то, по крайней мере, как наилучшую гипотезу.
Конечно, понимание этого обстоятельства пришло к Берцелиусу не сразу. Более того, первоначальное знакомство с теорией Дальтона оттолкнуло от нее Берцелиуса именно вследствие неубедительной разработки английским исследователем химическо-г0 (точнее, стехиометрического) аспекта. Но в то же время целый ряд факторов: детальное изучение Берцелиусом дискретных химических отношений, влияние кристаллографической традиции, опиравшейся на идею дискретных материальных образований, составляющих твердые тела, неприязнь к чисто динамическому пониманию материи (материя — продукт действия сил), влияние творческих контактов с учеными-атомистами (Т. Томсоном, У. Волластоном и др.) — способствовал переходу Берцелиуса в середине 1810-х годов па атомистические позиции.
Немалую роль в формировании химических воззрений ученого сыграли электрохимические исследования, с которых он начал свой научный путь. Проведенное им совместно с В. Хизингером изучение разложения различных соединений в растворе и в расплаве под действием электрического тока показало, что «компоненты, собирающиеся около одного и того же полюса, имеют между собой некоторое сходство. Так, все горючие тела, щелочи и земли притягиваются к отрицательному полюсу батареи, тогда как кислород, кислоты и окисленные тела — к положительному» [17, с. 142]. Тем самым кислотность и основность химических соединений оказались тесно связанными с их электрохимическим характером, и принятая в химии ранее кислотно-основная классификация веществ была подчинена электрохимическому делению соединений на электроположительные и электроотрицательные. Но что является причиной, определяющей те или иные свойства данного сложного вещества? Какой фрагмент соединения «отвечает» за то, будет ли оно кислотой, основанием или нейтральным телом? Без ответа на эти вопросы дальнейшее развитие теоретической химии было бы крайне затруднено.
По мнению Берцелиуса, в кислородных соединениях свойствоопределяющим фрагментом служит радикал (или радикалы), а не кислород. Последний же, будучи абсолютно электроотрицательным элементом, создает, согласно концепции ученого, как бы общий отрицательный фоп, в то время как радикалы, в зависимости от своей электрической природы, частично, полностью или с избытком гасят отрицательный заряд кислорода (подробнее см. [18, с. 126-132]).
Однако природа радикала была не единственным фактором, определяющим свойства соединения, многое зависело от количест-ноипого состава последнего. Так, например, химические свойства °кпслов и солей во многом определялись степенью окисления соответствующего радикала. Для выяснения этой зависимости не-
ГЛАВА ШЕСТАЯ
обходимо было иметь надежные аналитические данные по состав широкого круга веществ. Таким образом, электрохимические Z стехиометрические исследования Берцелиуса оказались логическх неразрывно связанными друг с другом. Я
Остановимся на основных количественных закономерностях подмеченных Берцелиусом в процессе изучения составов неорга’ нических веществ.
Продолжая исследования И. Рихтера с целью определения состава различных окислов и солей, Берцелиус сделал ряд новых обобщений. Причем,— и это следует подчеркнуть особо,— основное внимание он сосредоточил на двух моментах: а) на стехиометрических отношениях между компонентами сложных веществ и б) на взаимосвязях между составом бинарных соединений и соединений более высокого порядка. Особое внимание Берцелиус уделял изучению состава солей. По словам Д. И. Менделеева «с историей солей долго были связаны судьбы теоретических воззрений химии. Яснейшее представление об этом предмете ведет свое начало от Лавуазье и было особенно строго развито Берцелиусом. Это представление называется дуализмом. Все сложные
соединения, а соли по преимуществу, представлялись состоящими из двух частей. Соли представляли себе как соединения основного окисла (основания) с кислотным (т. е. с ангидридом кислоты, его тогда называли кислотою), гидрат же представляли как соединение безводного окисла с водою. Такое выражение употребляли не только для того, чтобы выразить обыкновеннейший способ получения этих веществ (<что> было бы совершенно верно), но также и для выражения того внутреннего распределения элементов, которым предполагали объяснять все свойства названных соединений... Это есть гипотеза. Ее развитие совпало с электрохимической гипотезой, предполагающей, что две составные части удерживаются во взаимной связи потому, что одна часть (ангидрид кислоты) имеет электроотрицательные свойства, а другая (в солях основания) положительные» [19, с. 474]. 1
Таким образом, в исследованиях Берцелиуса, посвященных составу неорганических соединений, первоначально (в 1807" 1813 гг.) слились в единое целое три фундаментальных направления теоретической химии первой четверти XIX в.: учение о дуализме состава (восходящее к Г. Шталю, Г. Бургаве, Т. Бергману и, особенно, к Г. Руэлю и А. Лавуазье), электрохимически^ дуализм (основоположником которого был сам Берцелиус) и стехиометрия (созданная трудами К. Венцеля, Р. Кирваиа, Э. Ф0' шера, Ж. Пруста, И. Рихтера и других ученых). Впоследствии, в 1814—1816 гг., Берцелиус, после долгих колебаний, принял атомную теорию Дальтона, существенно, однако, уточнив и переработав ее стехиометрическую часть.
Рассмотрим теперь основные выводы, к которым пришел шве^-
ХИМИЧЕСКАЯ АТОМИСТИКА
гк0Й химик в первом цикле аналитических работ, законченных м в октябре 1809 г. и опубликованых в 1810—1812 гг. [20, 21].
Сюда входят как законы, открытые самим Берцелиусом, так и переосмысленные или обобщенные им результаты других исследователей.
1. В одной из статей 1810 г. Берцелиус писал: «... занимаясь эТими работами (т. е. изучением состава солей.—Я. Д.), я встретил в журнале Николсона (за ноябрь 1808 г.) статью г-на Волластона, посвященную кислым солям и связанную с гипотезой Дальтона» [20, а, с. 163] 10. В данном случае речь идет о даль-тоновском законе кратных отношений, к которому Берцелиус пришел, по-видимому, независимо и который он, опираясь на свои экспериментальные данные, поначалу сформулировал так: «... когда два тела, А и В, соединяются друг с другом различным образом, то их соединение всегда происходит в следующих твердо установленных пропорциях: 1А с 1В (минимальный состав); 1А с ВДВ (или, возможно, правильнее: 2А с ЗВ); 1А с 2В, 1А с 4В. В моих опытах, однако, не встретилось ни одного примера 1А с ЗВ» 11 [20, б, с. 251].
Хотя в цитированном отрывке Берцелиус приводит весовое отношение 1А: 1,5В как «твердо установленное» и даже дает его целочисленный вариант (2А: ЗВ), в последующих частях этой серии статей [21], а также в других своих работах он прямо указывает на трудности, связанные с открытием соединений «полуторного состава». Дело в том, что, по мнению Берцелиуса, если простые вещества А и В образуют несколько соединений, то одно из исходных тел, например А, непременно должно находиться в наименьшем весовом количестве, так что количество другого вещества (В) следует относить к этому, принятому за единицу, весу вещества А. При этом число весовых частей В, приходящихся на одну часть А, всегда целое (дробность химических отношений была чревата потерей их дискретного характера). Таким образом, могут существовать лишь бинарные соединения состава А+В, А+2В, А+ЗВ и т. д., но не 2А+2В, 2А+ЗВ, ЗА+5В и т. п. В противном случае «в другом месте надо будет принять 9А+10В и так далее до 999А+1000В. Каждому, однако, ясно, что тогда все учение об определенных пропорциях придет в упадок» [23, с. 189].
В другой статье ученый пояснял свою позицию следующим °бразом: «Представлять отдельный сложный атом первого порядка 12 состоящим из двух или более атомов А, соединенных
*° Берцелиус имеет в виду работу [22].
Впоследствии Берцелиус нашел соединения состава 1А+ЗВ, например SO3. Представление об атомах различных порядков было введено Берцелиусом в 1813 г.: «Атомы можно разделить на два класса: 1) элементарные и 2) сложные. Последние бывают трех типов: а) атомы, образованные из Двух элементарных частиц,— мы назовем сложными атомами первого по
265
ГЛАВА ШЕСТАЯ
с двумя или большим числом атомов В, например 2А+2В, 2Д-р +ЗВ, 7А+7В и т. д., значит противоречить здравой (sound) Ло гике; ибо в этом случае не существует ни механических, ни мических препятствий для чисто механического разделения тя' кого атома на два или более атомов, состав которых проще>> [24, с. 447].
К сожалению, Берцелиус не обосновал свои возражения про. тив полуторных составов более детально. По-видимому, причину неприятия им весовых отношений типа «1А: 1,5В» следует искать в его электрохимической теории сродства, согласно которой химическое соединение обусловлено притяжением противоположно заряженных атомов. С позиций этой теории один атом А мог координировать вокруг себя несколько (но не более девяти, см. [24, с. 443—454]) противоположно заряженных атомов В, тогда как удержать в молекуле два одинаково заряженных атома А мог только расположенный между ними атом В. Однако последнее предположение, во-первых, противоречило принятому дуалистическому распределению элементов в соединении, а во-вторых, представлялось Берцелиусу слишком умозрительным (по крайней мере, для бинарных соединений), так что в итоге он предпочел искать иное решение проблемы «1А: 1,5В» (см. далее).
Кроме того, увеличение коэффициентов при А и В приводило, по его мнению, к тому, что «по мере возрастания возможностей комбинирования < атомов) определенные пропорции в неорганических телах должны становиться все менее отчетливыми (dis-tinctes). Но так как эти определенные пропорции не вызывают никаких сомнений и были твердо установлены экспериментально, необходимо, чтобы существовали пределы, за которыми в неорганической природе не могут уже иметь место соединения элементарных атомов» [25, с. 336—337] (см. также [26]).
Другой проблемой, вставшей перед Берцелиусом в связи с законом кратных отношений, была проблема нецелочисленного отношения масс кислорода в ряду окислов одного и того же элемента, например:
^(O)l(so2):w(°)l(so8) = 1^’5i3, I
которая, по его мнению, также представляла существенную трудность для атомной теории.
рядка, б) атомы, составленные более чем из двух элементарных частил (атомов), встречаются в органических телах или в телах, полученных при их разложении, поэтому мы назовем эти атомы органическими» и в) атомы, образованные соединением двух или большего числа сложных атомов, как, например, в солях; в этих случаях мы будем говорить ° сложных атомах второго порядка» [24, с. 454].
13 Символ т(А)](дв) обозначает массу элемента А в соединении АВ, раС' считанную на определенную массу В или какого-либо другого тела. В круглых скобках приводятся современные формулы и названия. Следует отметить, что в работах Берцелиуса везде используется термин «вес», а не «масса».
266
ХИМИЧЕСКАЯ АТОМИСТИКА
На том, как эти трудности преодолевались ученым, мы остановимся далее, но уже из сказанного ясно, что закон кратных отношений Берцелиус понимал иначе, чем Дальтон* 14. Это дало основание некоторым современным историкам химии дать предложенной Берцелиусом трактовке закона кратных отношений специальные названия,— «закон единицы» (Enhetslagen) [27, с. 100], «закон единичного атома» (the law of unitary atom) [28, c. 403]
и т. Д.
2. Следующая стехиометрическая закономерность, отмеченная шведским химиком, состояла в том, что при образовании средней соли из двух окислов (основного и кислотного) содержание кислорода в любом основном окисле, рассчитанное на определенное количество данного кислотного окисла, постоянно [29, с. 259]:
^(°) Ir+по = const
(R+zzO — формула основного окисла).
Этот закон, открытый ранее Т. Бергманом и независимо от него И. Рихтером, Э. Мельхадо назвал законом постоянства основного кислорода (the law of the constancy of basic oxygen) [28, c. 260—268], а сам Берцелиус характеризовал его как «Meine Hauptsatz» [29, с. 259], так как с его помощью можно было не только определять состав основных окислов, но и рассчитывать составы солей.
3. Расширяя круг исследуемых бинарных соединений, Берцелиус обратился к сопоставительному анализу составов сульфидов и окислов металлов, в результате которого он пришел к следующему выводу: массы кислорода в низшем и высшем окислах металла относятся друг к другу как массы серы в соответствующих сульфидах этого металла, например:
(8) l(PbS) гп(О) |(рЬ0) _ 1
m(s) l(Pbs2) 1(рьо2) 2
В общем виде это утверждение можно записать так:
m(S) l(M2Sn) те(°) l(Mao„) п
W(S) |(W - «(О) |(W - Л
Кроме того, как показал анализ, в большинстве случаев окис-ЛУ металла можно сопоставить сульфид, состав которого удовлетворял бы такому отношению:
т<°) 1(М Оп) I
m(S) l(M2s ) 2
14 Последний, как известно, хотя и не указал в перечне возможных комбинаций атомов в соединениях соотношений типа 2А: ЗВ, ЗА: 5В и т. п., °граиичившись весьма неопределенным «и так далее» [3, с. 91], в то же время и не накладывал на эти комбинации каких-либо ограничений. Бо
267
ГЛАВА ШЕСТАЯ
Эти результаты, указывающие на связь степени окислецц (Oxydations-Stufen) и степени сульфирования (Schwefelungs-Stu* fen) элемента, уже сами по себе имели большое аналитическое значение, так как позволяли, зная, скажем, состав низшего суЛь фида, определить состав низшего окисла и наоборот. Характер ным примером могут служить исследования ученика БерцелиуСа Н. Сефстрёма, который показал, что киноварь (HgS) отвечает не низшему окислу ртути, а высшему (HgO), и затем получил неизвестный до тех пор низший окисел (Hg2S)15 [30, а, с. 186]
Однако для Берцелиуса эти наблюдения представляли интерес прежде всего потому, что позволяли перейти к решению глав-
ной задачи — установлению взаимосвязи между составами бинар-; ных и более сложных соединений (второго и высших порядков). Особый интерес представляют две подмеченные им закономерности: сохранение бинарных отношений и правило окислов.
Первую из них можно проиллюстрировать, сопоставляя составы сульфидов и сульфатов свинца, в которых отношение масс РЬ и S одинаково:
тп(РЬ) тп(РЬ)
т(&) (PbS) m(s)
(PbSO4) ’
Аналогичная картина наблюдалась и во многих других случаях (ср., к примеру, Na2S, Na2SO3, Na2SO4; BaS, BaSO3, BaSO4 и т. д.), что позволило Берцелиусу сделать следующее обобщение: отношение масс элементов А и В в их бинарном соединении (А+&В) остается неизменным и в средних солях, образованных окислами этих элементов (A+ZO) и (В+тО)16 [21, с. 246—320].
Эта закономерность (далеко, однако, не универсальная) широко использовалась Берцелиусом в различных стехиометрических расчетах.
4. Другим важным обобщением, связывающим составы соединений разных порядков, явилось так называемое правило окис-
лее того, в пятой главе II части «Новой системы...» он принял для «се-литроватой кислоты» состав 2N+3O [3, с. 128] (см. также графические формулы Дальтона на стр. 104—107, там же).
15 Следует, однако, заметить, что сульфид диртути (Hg2S) разлагается при температуре выше 0° С на HgS и металлическую ртуть. Поэтому труд00 сказать, что же Сефстрём получил в действительности. Правда, аналогия «окисел—сульфид» часто приводила к трудностям, причина которых, как теперь известно, заключалась в образовании полисульфидов. «Можно сравнить степень сульфирования тела с его степенью окисления, которое, однако, не всегда отвечают друг другу,—подчеркивал Берцелиус.—Йз; вестно, например, что мышьяк может соединяться с кислородом и серой в двух отношениях. Кислород в обоих окислах содержится в отношений 3 к 5 (As2O3 и As2O5 — И. Д.), тогда как сера в сульфидах — в отношений 2 к 3 (As4S4 (реальгар) и As2S3 (аурипигмент), другие сульфиды, Аз4Ьз и As2Ss, были тогда неизвестны.— И. Д.}» [20, в, с. 12]. Д
16 Мы несколько модернизировали формулировку Берцелиуса, сохранив, оД* цако, ее смысл,
268
ХИМИЧЕСКАЯ АТОМИСТИКА
лОв. Оно касалось распределения кислорода между основным и кислотным фрагментами в средних солях и было сформулировано Берцелиусом так: «В нейтральных солях количество кислорода, содержащееся в кислоте (т. е. в кислотном окисле.— И. Д.) является целым кратным количеству кислорода в основании (в основном окисле.—Я. Д.)» [21, с. 161]. Иными словами, для солей состава (А+/О) + (В+тпО) выполняется следующее условие:
^(°) Ia+zo I
-—_ m-----— — — целое число.
^(°) 1в+тпО т
— И, Д.)» [21, с. 161]. Иными словами, для солей
Таким образом, в согласии с правилом окислов, возможны лишь составы типа: (А+О) + (В+2О), (А+О) + (В+ЗО) и т. д., но не (А+ЗО) + (В+5О) и т. п.
Для солей, состоящих из трех и более окислов, в том числе и для кристаллогидратов, Берцелиус предложил обобщенное правило окислов17 [21, с. 246—320], в соответствии с которым количество кислорода в окислах, образующих такие соли, кратно его количеству в одном из них, например в алюмокалиевых квасцах:
l(K2o): I(A12o»): l(4so3): 1(24H2o) = 1: 3 :12 : 24.
Перечисленные выше четыре стехиометрических обобщения были объединены Берцелиусом в общий закон определенных пропорций: «Если два тела соединяются друг с другом в нескольких отношениях, то эти отношения являются целыми кратными (весового количества.— И. Д.) одного из тел. Если соединяются кислородные тела, то количество кислорода в том соединении, в котором его содержится меньше всего, будет общим делителем по отношению к его количеству в прочих компонентах или же последнее количество будет кратно первому. Наконец, если соединяются горючие тела, то их соединение происходит в таком отношении, что при их окислении кислород полученных окислов находится в приведенных выше кратных отношениях» [23, с. 171].
По существу эти утверждения явились эмпирическими правилами, полученными путем экстраполяции на все неорганические соединения результатов, установленных для весьма ограниченного круга веществ, главным образом соединений серы, кислорода и шести-семи металлов. Неудивительно поэтому, что, пе-Роходя к соединениям других элементов, Берцелиус столкнулся с «аномалиями». Так, срстац окисдов щедезд не удовлетворял закону единицы:
-m(°) l(FeO) ,
«(°) 1(ГегОа) ”
7 По терминологии Э. Мельуадо — закон единичного кислорода [28, с. 409].
§69
ГЛАВА ШЕСТАЯ
Аналогичные трудности возникали с окислами азота, фосфору мышьяка и сурьмы. Кроме того, соли названных элементов подчинялись правилу окислов (о чем подробнее см. [31, с. 74 75]).
Далеко не всегда удавалось согласовать результаты анализов с законом сохранения бинарных отношений. Например, неизменность отношения m(Fe):m(S) при переходе от низшего сульфида FeS к сульфату низшего окисла железа (FeSO4) нарушу лась в сульфате высшего окисла, так что
m(Fe) m(S)
3 m(Fe)
(FeSO4) 2
zn(Fe) (FeS) m(S)
[Fe2(SO4)3]
Заметим, что проблема «1А+1,5В», т. е. проблема несоизмеримости в целочисленном отношении весовых количеств реагирующих простых тел, равно как и другие «аномалии», долгое время удерживали Берцелиуса от принятия атомистической интерпретации стехиометрических обобщений. «Пусть О — кислород, а А и В — два горючих тела,— писал он.— Закон (окислов,— И. Д.) допускает соединение А+ЗО с 1,5(В+О), так как 1,5-2=3. И мы можем непосредственно убедиться, что такие соединения действительно существуют, хотя, согласно корпускулярной теории, их появление абсурдно. С другой стороны, закон не допускает соединения А+ЗО с В+2О, хотя существование таких соединений совместимо с атомной теорией» [24, с. 448].
Таким образом, будучи убежденным сторонником концепции дискретности химических отношений, Берцелиус не мог принять ни появление «дробных пропорций» типа 1А+1,5В, ни атомистического способа их устранения. И то, и другое было неприемлемым для него по одной общей причине: как «полуторные» составы, так и атомистическое приведение их к целочисленному виду грозило «упадком» всего учения об определенности химических пропорций, так как могло привести к переходу от дискретных стехиометрических отношений к континууму составов. Отсюда проистекает и крайне осторожное, чтобы не сказать скептическое, отношение Берцелиуса к атомной теории: «... было бы опрометчивым делать заключение, что в будущем мы не сможем объяснить эти кажущиеся аномалии удовлетворительным образом. Но до тех пор, пока это время не настало, гипотеза об атомах не может быть ни принята, ни рассматриваться как истинная» [24, с. 450]. Я
Понятие об атоме, начатое систематически употребляться .Берцелиусом примерно со второй половины 1812 г., поначалу соединяло в себе три группы значений: структурно-физическое (атом как мельчайшая, механически и химически неделимая частица вещества), стехиометрическое (атом как наименьшее количество вещества, фигурирующее в химических пропорциях, как «абсолютный минимум соединения тел друг с другом, которому
270
ХИМИЧЕСКАЯ АТОМИСТИКА
л)Кны быть кратны все другие соединения» [21, с. 297], и «объемное» (атом как наименьший объем (Raumtheile) простого вещества, находящийся в сложном теле). При этом последнее зна-чеНие в работах Берцелиуса 1813—1814 гг. оказалось превалирующим18: «...то, что в одной теории (Дальтона.— И. Д.) Называют атомом, в другой (Берцелиуса.—Я. Д.) —объемом. При настоящем состоянии наших знаний теория объемов имеет то преимущество, что она основана на хорошо установленном факте (имеется в виду закон Гей-Люссака.—Я. Д.), между тем как теория атомов зиждется на предположении. В теории объемов мы можем оперировать с половиной объема, тогда как в теории атомов половина атома — абсурд» [24, с. 450].
Из этой цитаты хорошо видно, что противопоставлять понятие «объем» понятию «атом» заставляли Берцелиуса трудности, связанные с проблемой дробных отношений, т. е. с упомянутыми выше «кажущимися аномалиями». Устранить последние ученый надеялся с помощью гипотезы о существовании неизвестных низших окислов (субокислов, по его терминологии) элементов, состав которых обеспечивал бы соблюдение всех четырех стехиометри
ческих законов.
«...В ряду окисленных тел,—писал Берцелиус,—все отношения, содержащие < число) 17г, скорее всего —только кажущиеся, тогда как истинные пропорции являются целыми кратными некоторой неизвестной или неисследованной низшей ступени окисления» [30, б, с. 73].
В качестве примера укажем на исследования Берцелиуса, посвященные кислородным соединениям серы, выполненные им в 1812—1814 гг. Для обнаружения субокислов этого элемента Берцелиус обратился к его соединениям с муриевой (соляной) кислотой, которую он в то время рассматривал как оксикислоту. Одно из этих соединений было открыто Т. Томсоном в 1803 г. [32, с. 104—109] и затем изучено X. Бухольцем в 1810 г. [33, с. 186—188], его современная формула S2C12; другое (SC12) исследовалось А. Бертолле-сыном в 1807 г. [34, с. 170—175]. Оба соединения Берцелиус рассматривал как муриаты низших окислов серы [20, б, с. 470—472]. «Мы знаем,—писал он, подытоживая результаты своих анализов серосодержащих веществ,—что сера соединяется с оксимуриевым газом (т. е. с С12, который тоже считался кислородным соединением.— И. Д.) в двух отношениях, дающих два муриата с разными основаниями. В одном из них сера присоединяет в 2 раза меньше кислорода, чем в сернистой кислоте (SO.— И. Д.), а в другом — в 4 раза меньше», откуда следовало, что «четыре степени окисления серы должны быть выражены таким образом: 2S+O, S+O, S+2O, S+3O» [24,
18 Заметим, что атомные веса в таблице 1814 г. [24, с. 363—364] трактовались Берцелиусом как удельные веса единичных объемов элементов.
271
ГЛАВА ШЕСТАЯ
с. 53], т. е. отношение весовых количеств кислорода в этих окис л ах оказалось равным 1: 2 : 4 : 6.
Другой пример — кислородные соединения азота. По мнению Берцелиуса, которого он придерживался в 1810—1813 гг., азот (подобно хлору) не был элементарным телом, а представлял собой очень прочный низший окисел радикала, названного им Nitricum (Nt)19 [35]. Этому радикалу Берцелиус сопоставил следующий ряд окислов: NtO — азот (N), NtO2 и NtO3—окислы азота, отве-чающие современным формулам NO и NO2, NtO4— азотистая кислота (т. е. N2O3) и NtO6— азотная кислота (N2O5).
Много сил потратил Берцелиус на выяснение состава кислородных соединений мышьяка и сурьмы [21; 24, с. 93—101, 248— 250]. В этих работах отчетливо просматривается его стремление подогнать составы окислов и солей этих (и всех остальных) элементов под общий стехиометрический эталон, в качестве которого была выбрана приведенная выше последовательность окислов серы. Так, после ряда неточных анализов20 и весьма произвольных допущений Берцелиус пришел, наконец, к желанному, но, как впоследствии выяснилось, неправильному выводу: «Известные степени окисления мышьяка таковы: 1. Субокисел, или черный порошок, который образуется из металлического мышьяка. В своих первых экспериментах я обнаружил, что 100 частей мышьяка соединяются с 8,5 частями кислорода. Это, разумеется, либо слишком много, либо слишком мало, так как субокисел должен иметь состав 2As+O или As+O. В будущем я проделаю соответствующие анализы. 2. Солеобразующий окисел As+3O. 3. Мышьяковистая кислота, As+4O. 4. Мышьяковая кислота,. As+бО. Но существует ли окисел состава As+2O?» [21, с. 178].
Еще более показателен пример с окислами сурьмы. По данным анализа, их состав должен быть следующим: Sb+50 и Sb+ЗО. Однако Берцелиус категорически отверг собственные результаты как «противоречащие всему опыту» [24, с. 249—250]. «Я полагаю,— писал он,— что аналогия (с принятыми им ранее составами окислов серы, мышьяка и других элементов.—17. Д.)
19 Этому радикалу Берцелиус приписывал атомный вес 79,64 (при О = 100) или 12,74 (при О = 16) [35, с. 284]. Заметим также, что эта работа была первой, где он начал широко использовать атомистическую терминологию.
20 К примеру, отношение масс
m(9) I(As2o6) : !(As«O8)’
по Берцелиусу, равно 1,5:1 (—3:2), а не 5:3. Кроме того, для арсенатов им было получено следующее отношение:
Wi(O) |в КИСЛОТНОЙ • 1в ОСНОВНОЙ 2.1,
части соли части соли
(по-видимому, это — неточный результат анализа оксоарсената), тогда как для арсенитов (точнее, метаарсенитов — MTAsO2) это отношение полула"
272
ХИМИЧЕСКАЯ АТОМИСТИКА
в столь сильной степени говорит в нашу пользу, что мы не сможем избежать принятия состава Sb+бО в качестве истинного. 0 особенно потому, что в настоящее время нам неизвестен ни один пример радикала, соединяющегося с пятью объемами кис-лОрода» [24, с. 250].
Итак, подавляющему большинству изученных в начале 1810-х гОдов химических элементов — сере, азоту (точнее радикалу Nt), фосфору, мышьяку, сурьме, олову, хрому, молибдену, вольфраму, железу и др.— Берцелиус сопоставил следующие две последовательности окислов: либо 2Э+О, Э+О, Э+2О, Э+30, либо Э+О, Э+20, Э+30, Э+4О, Э+6О.
В обоих случаях отношение масс кислорода, рассчитанных на определенную массу элемента, было примерно одинаковым — 1:2: (3) : 4:6. Зачем же понадобилось Берцелиусу столь упорно (иногда вопреки очевидным фактам) доказывать, что составы окислов и солей названных элементов отвечают единому стехиометрическому эталону? По-видимому, дело здесь не только в желании «спасти» установленные ранее стехиометрические закономерности. В задачи своей исследовательской программы Берцелиус включал не только поиски количественных соотношений, но и количественную конкретизацию некоторых важных качественных наблюдений. При этом особое внимание он уделял подмеченной еще Лавуазье корреляции между кислотно-основными свойствами окислов данного элемента и степенью окисления последнего в этих окислах.
Согласно Лавуазье, каждый радикал мог (по крайней мере, потенциально) находиться в любой из четырех возможных степеней окисления (les degres d’oxygenation) и кислотные свойства окислов менялись симбатно изменению степени окисления образующего их радикала21 [36, с. 60—61, 82—83, 203].
Берцелиус, как известно связывал кислотность окисла не только со степенью окисления соответствующего радикала (химического элемента), но и с положением последнего в электрохи
лось равным 3:1. В итоге, имело место явное несоответствие: в кислотной части соли высшего окисла кислорода оказывалось меньше, чем в такой же части соли низшего окисла, тогда как данные по составу самих окислов говорили об обратном. Еще более удивительным казалось то, что в ряде опытов
О) 1в КИСЛОТНОЙ части соли
2 Видимо, это было связано с образованием диполиарсенатов M2ITAs2O7.
проявилось и в номенклатуре. Окислы, отвечающие трем последним (высшим) степеням окисления радикала, Лавуазье относил к кислотам, названия которых отличались окончаниями (-еих — для второй степени окисления радикала, -ique — для третьей и -ique oxy gene — для четвертой). И только соединения радикалов с кислородом первой степени окисления он называл окислами, отмечая их основные или же слабые кислотные свойства.
1в ОСНОВНОЙ2* части соли
273
ГЛАВА ШЕСТАЯ
мическом ряду, т. е. с его электроотрицательностью. Это, в свою очередь, вело к важному (особенно в исторической перспективе^ выводу: элементы, различные по своей природе и потому заН11 мавшие в указанном ряду разные положения, в высших степеням окисления образовывали соединения, имевшие между собой известное сходство, например высшие окислы обладали ярко выра. женными кислотными свойствами. Разумеется, не для всех элементов, известных в первой четверти XIX в., удалось получить высшие (кислотные) окислы, но важно другое — эти соединения (а вслед за ними высшие сульфиды, галогениды и т. д.) получили в химии особый статус: с увеличением степени окисления (сульфирования, галоидирования) присущие элементарным телам индивидуальные различия несколько сглаживались — тезис, с наибольшей глубиной и детальностью развитый впоследствии Д. И. Менделеевым в период разработки им учения о периодичности 22.
Вместе с тем, развивая идеи Лавуазье в количественном аспекте23, Берцелиус абсолютизировал зависимость «степень окисления—свойство», полагая, что для всех элементов существует один и тот же набор степеней окисления и потому отношение
1высший • m (О) |низший окисел окисел
должно быть постоянным (равным, как правило, 6:1) и не зависеть от природы элемента. Именно здесь берет начало стремление ученого подогнать составы неорганических соединений под единый стехиометрический эталон, а затем рассмотреть характерный для каждого элемента и обусловленный его внутренней природой темп нарастания кислотных свойств окислов по мере увеличения в них содержания кислорода. И поначалу, как ему казалось, цель эта была почти достигнута. Было даже предложено следующее теоретическое обоснование найденных зависимостей: «Число простых атомов в сложном атоме с необходимостью должно влиять на форму последнего, а следовательно, и на его свойства. Это дает основание полагать, что окислы, содержащие одно и то же число атомов кислорода, имеют, по крайней мере, несколько общих свойств, которые отличают их от тех окислов, в которых кислорода больше или меньше» [37, с. 115—116]-Конкретизация этого положения приводит, по Берцелиусу, к следующим выводам:
22 См. главу восьмую настоящей монографии.
23 Заметим, что французский химик не связывал понятие степени окисления с какой-либо количественной мерой, и предложенные им четыре ст^ пепи окисления были по сути дела четырьмя качественно различным состояниями окисления. Далее утверждения о том, что «металлически вещества в процессе их окисления увеличивают вес пропорциопальи кислороду, который они поглощают» [36, с. 82—83], Лавуазье не шел.
274
ХИМИЧЕСКАЯ АТОМИСТИКА
а) для большинства элементов характерны четыре степени оКисления (см. выше);
б) окислы, содержащие один атом кислорода, как правило, 00Дифферентны;
в) окислы, включающие в свой состав два атома кислорода, имеют свойства солеобразующих оснований;
г) два высших окисла — всегда кислотные, причем отношение Jiacc кислорода в них равно 3 : 2=1,5 :1; например,
l(so3): I(so2) = 3:2;
д) отношение масс кислорода в высшем и низшем окислах чаще всего равно 6:1.
Однако вся эта красивая теория рухнула, как только весной 1816 г. Берцелиус обратился к изучению составов окислов и кислородсодержащих солей фосфора [25]. Это было не первое его знакомство с этими соединениями. Ранее, в статье 1811 г., он предложил следующую формулу для фосфатов бария и свинца (М+О) + (Р+2О)24 [21, с. 174—223], что полностью согласовывалось с законом единицы и правилом окислов.
Однако впоследствии, когда Берцелиус, по его словам, «приобрел больший опыт в этой части химии и лучше его продумал», он понял, что, во-первых, «фосфорная кислота имеет сходный состав с другой кислотой, так что кислород основания соединяется с этими кислотами в двух различных отношениях», и, во-вторых, что «кислород фосфористой кислоты относится к кислороду фосфорной кислоты, как 2:3, откуда следует, что фосфорная кислота содержит больше двух атомов или объемов (Raumtheile) кислорода» 25 [26, с. 394—395].
Это обстоятельство побудило Берцелиуса начать новые исследования, в ходе которых состав солей фосфорных кислот показался ему еще более запутанным, чем он представлял себе это ранее26. «Мне снится,—писал он А. Марсэ 9 мая 1816 г.,—что я оказался в лабиринте позабытый друзьями, и вся душа моя заполнена фосфорной кислотой» [38, с. 137—138].
В итоге, после долгих колебаний и трудоемких, изнурительных опытов Берцелиус пришел к следующему выводу: «... в раз
24 По-видимому, у Берцелиуса образовалась эквимолярная смесь фосфата и диполифосфата: BasfPCkh+BazPzO? (или в «окисной» записи: 5ВаО+ 2Р2О5), где отношение масс кислорода в кислотном и основном окислах равно 2:1.
Правильное отношение масс кислорода в РЮв и Р40ю равно 3:5, а не 2 : 3, как это поначалу принимал Берцелиус.
Дело в том, что Берцелиус рассматривал нейтральные фосфаты как основные соли, в качестве же нейтральных у него фигурировали только Дпполифосфаты, но не фосфаты. Отсюда — неоднозначность получаемых Результатов.
ГЛАВА ШЕСТАЯ
личных фосфорнокислых солях кислород основания составляет 3/ 2/б, 1,5/s и кислорода кислоты» [26, с. 434]. Этот результат’ однако, не согласовывался с правилом окислов. Чтобы «спасти»’ это правило, Берцелиус пошел сначала по проторенному путп предположив, что фосфор не является элементарным веществом’ а, подобно азоту, состоит из одного атома кислорода и неизвестного радикала. Тем самым в кислотных частях фосфорных солей число атомов кислорода возрастало на единицу, и тогда «кислород основания составлял бы 7г, 7з, 74 и 7б кислорода кислоты и фосфор в этом случае удовлетворял бы правилу, справедливому для всех остальных известных ныне тел» [там же], т. е. правилу окислов.
Из этой гипотезы следовало, что если она правильна, то при взаимодействии с металлами фосфор должен терять кислород и потому для получения окисла фосфора из фосфида потребуется больше кислорода, чем при его получении из фосфора. Однако тщательный химический анализ не выявил никаких различий — в обоих случаях количество присоединяемого кислорода было одним и тем же [26, с. 37]. Это заставило Берцелиуса отказаться от гипотезы о неэлементарной природе фосфора, что, в свою очередь, сильно подрывало его веру и в справедливость сделанных им ранее выводов о составе окислов и солей азота, мышьяка и сурьмы.
В работах 1817-1818 гг. [39, с. 179-180; 40, а, с. 455-475; 40, б, с. 170—186], сообщая о результатах повторных анализов соединений этих элементов, Берцелиус признает, что «во всех этих случаях (т. е. в окислах N, Р и As.—Я. Д.) кислород в высшей (vollkommenen) кислоте относится к кислороду в невысшей (unvollkomnienen), как 5:3» [40, б, с. 179—180].
Вместе с тем, придя к выводу о несоответствии составов окислов фосфора выбранному стехиометрическому эталону, Берцелиус изменил свой взгляд и на проблему «1А+1,5В»: «исключения (из закона единицы.—Я. Д.), где имеет место отношение 1:1,5, возможно проистекают не оттого, что принимали неправильный атомный вес для того или иного элемента, но скорее оттого, что в действительности в этих соединениях 2А может соединяться с ЗВ, хотя такие комбинации встречаются не часто» [25, с. 335—336]. Это снимало возражения, которые Берцелиус выдвигал против атомной теории, и открывало путь к атомисти^ ческой интерпретации стехиометрических законов, путь, который «наилучшим образом согласуется с нашей обычной манерой рассмотрения и понимания тел и их составов» [41, с. 81].
Не случайно поэтому статья, посвященная анализу фосфорный соединений, заканчивалась разделом: «Состав фосфорной и Ф°7 фористой кислот и их солей с точки зрения корпускулярной теории» [25, с. 329], где в первом же абзаце Берцелиус ясно сформулирордд спор отношение к атомистике; «Я убежден^ ИТО
О
ХИМИЧЕСКАЯ АТОМИСТИКА
обоснованная корпускулярная теория, отнюдь не пренебрегающая с0лами, от которых зависит соединение молекул, станет отныне основой химической и физической теории» 27 [там же].
Другим важным результатом исследования соединений азота, фосфора, мышьяка и сурьмы явилась идея стехиометрического рЯда, которая впоследствии, вместе с детально рассмотренной зависимостью свойств кислородных соединений элемента от его степени окисления в них, составила ядро учения о периодичности. Именно в работах Берцелиуса берет свое начало объединение элементов в отдельные совокупности по стехиометрическому признаку, по «форме сложного атома», хотя состав высшего окисла еще не играл toii важной классифицирующей роли, какую он стал играть у Менделеева, но тем не менее начало было положено.
В аналитических работах Берцелиуса, написанных после 1816 г., отчетливо выделяются три группы элементов: группа I включала элементы, окислы и соли которых подчиняются закону единицы, правилу окислов и соответствуют стехиометрическому эталону (S, Те, Cr, Mo, W, впоследствии Se, Sn, Pb, Си и др.), группа II объединяла такие элементы, как N, Р, As и Sb, в двух высших окислах которых массы кислорода относились, как 5 : 3, и к группе III принадлежали все остальные элементы, соединения которых удовлетворяли правилу окислов и закону единицы, но по составу отличались от аналогичных соединений первых двух групп.
Таким образом, в своих стехиометрических исследованиях Берцелиус не только развил и углубил атомное учение, но и создал предпосылки для последующих фундаментальных обобщений в химии.
СИСТЕМА АТОМНЫХ ВЕСОВ БЕРЦЕЛИУСА*
В работе «Опыт теории химических пропорций»28, появившейся в 1818 г. на шведском языке и переведенной в 1819 г. на Французский, а в 1820 г. на немецкий язык [42а], Я. Берцелиус Дает более развитое изложение своих атомистических взглядов (обобщив также свои исследования, сделанные им после 1814 г.) и связывает химические знаки с понятием об атоме и атомном Весе, а не с понятием об объеме.
Необходимо отметить, что таблица атомных всесов, предложения в 1818 г. (см. таблицу на стр. 281), принципиально мало отличается от таблицы «весов элементарных объемов» Берце-
27 Термины «корпускула», «атом», «молекула» Берцелиус употреблял как 28 синонимы.
См. Приложение VII.
* ,апный ц следующий разделы написаны М. Г, Фаерштейном [42].
?77
ГЛАВА ШЕСТАЯ
лиуса 1814 г. Обе таблицы построены на одной основе. Различая связаны с более точными аналитическими данными, получении ми за четыре года. Кроме того, имеются немногочисленные су щественные расхождения, которые касаются атомных весов ие^ которых неметаллов. В частности, атомный вес кремния в табли це 1814 г. равен 216,66, в то время как в таблице 1818 г.— 296,42. Это связано с тем, что в 1814 г. Берцелиус принимал для кремнезема формулу SiO2, а в 1818 г.— SiO3. Л
Для утверждения атомистики имели большое значение иссле-дования Э. Митчерлиха, подтвердившие мнение о связи между атомным составом и кристаллической формой соединения, высказанное еще У. Волластоном в 1808 г. и в дальнейшем развитое Берцелиусом (1818). Наряду с законами Гей-Люссака, давшими физическое обоснование атомистики для газов, закон Митчерлиха помог объяснить и применить атомистическую гипотезу для твердых тел.
В окончательном, общем, виде закон Митчерлиха гласит: «Одинаковое число атомов, если они связаны одинаковым образом, дает одинаковые кристаллические формы, и эти формы не зависят от природы атомов, а только от их числа и способа связи между ними» [42а, с. 17].
Берцелиус сразу оценил огромное значение этого закона для дальнейшего развития химии [43, с. 41]. Он указывал, что «это соотношение может дать такие же положительные результаты, как и измерение объема составных частей веществ в газообразной форме» [44, т. 7, с. 403]. Закон изоморфизма подтверждал аналогичность химических формул, установленных ранее Берцелиусом для окислов большинства металлов (Ba, Sr, Са, Pb, Ni, Zn и др.); с другой стороны, изоморфизм окислов алюминия, хрома и марганца подтвердил правильность вывода Берцелиуса о наличии в них одинакового числа (трех) атомов кислорода. Также подтвердилась аналогичность атомного состава фосфорной и мышьяковой кислот и т. д.
Закон изоморфизма, однако, не мог играть решающей роли при изменении системы атомных весов и при переходе к новой системе. Тем не менее некоторые историки химии, и в частности А. Ладенбург, считают, что Берцелиус отбросил свой постулат о наличии только одного атома элемента во всех окислах под влиянием закона Дюлонга и Пти, с которым не согласовывались атомные веса металлов таблицы 1818 г. [45, с. 74]. Однако более глубокое знакомство с работами Берцелиуса показывает, что ов не подходил односторонне и упрощенно к своей системе и что, принимая во внимание одновременно много соображений при ре' шении вопроса об атомном составе, он все же отделял главное от второстепенного.
Я. Берцелиус указывает, что поводом к пересмотру атомнЫ* весов послужила переработка его учебника для второго (немеН'
278
ХИМИЧЕСКАЯ АТОМИСТИКА
шли»!
® и е а в
von
Лдеоь Ведение.
Aus dem S с h w ed i scl) e n ubersetzt
i *
K. A. BlUde und K. Palmstedt.
Zweiie verbesterie duflage.
Erster Band, mit Tier К и j> f e r t a f e 1 n.
I ..it ; J -- .._ .
DRESDEN, in der Arnoldischen Bucbhandlung.
| 1823.
Титульный лист книги Я. Берцелиуса «Учебник химии» (1823)
коГо) издания (1821—1832) [46, с g7]. Он отмечал свое расхождение с другими химиками, считавшими, «что при образовании веществ происходит преимущественно соединение одного атома (элемента. — М. Ф.) с одним атомом (другого элемента,—71/. Ф.)» [47, с. 67]. Отвергая этот произвольный постулат, Берцелиус ссылается на то, что соображения, которыми он руководствовался еще при составлении первой таблицы, учитывая все аналитические данные и аналогии и, в частности правило о соотношении количества кислорода кислоты и кислорода основания, вполне оправдались.
Но один вопрос остался тогда нерешенным — вопрос о числе атомов радикала (элемента), соединяющегося с кислородом в кислотах и основаниях. Из двух возможных предположений Берцелиус в 1818 г. выбрал простейшее, т. е. признал существование только одного атома радика-
ла в соединениях. Но он вынужден был пересмотреть этот вопрос в связи с новыми обстоятельствами: открытие закона Дю-лонга и Пти, открытие закона Митчерлиха и, наконец, «...сравнение окислительных рядов азота и хлора, с одной стороны, и марганца и хрома — с другой, из которого стало весьма вероятным, что окислительный ряд обоих классов является одним и тем же, только в ряду марганца отсутствует первый член, а в ряду хрома — Два первых члена» [47, с. 69].
Хотя Берцелиус и не выделял, какое из трех обстоятельств было для него определяющим, но в 1826 г. впервые сообщил 0 новой системе атомных весов. Он писал: «Я откровенно признаюсь, что отношения хрома и марганца в первую очередь побудили меня избрать ряд (окисления — М, Ф.) азота в качестве более верного по всем данным и отказаться от мнения о простейшем ряде, которого я ранее придерживался» [44, т. 7, с. 416]. ^пдом окисления азота Берцелиус называл ряд: 2R+O; R+O; 2R+3O; 2R+5O, соответствующий кислородным соединениям азо-та и установленный на основе соображений объемного по-Рядка.
279
ГЛАВА ШЕСТАЯ
Здесь уместно отметить еще одно обстоятельство, на котор0е Берцелиус не указывает в своих статьях, а именно: признание ученым элементарности хлора и азота. При создании системы 1818 г. Берцелиус, хотя и признавал значение объемной теорц^ для развития атомно-молекулярного учения, но считал хлор ц азот сложными веществами, применяя свою объемную теорию фактически ограничивался только водородом и кислородов И лишь после 1820 г., Берцелиус вынужден был под давлением неоспоримых фактов признать элементарность хлора и азота.
Таким образом, первый толчок к признанию окислов, содержащих два атома элемента, связан с признанием элементарности хлора и азота и с применением вследствие этого объемной теории в более широком масштабе.
Убедительным доказательством того факта, что в основе новой системы атомных весов как неметаллов, так и металлов лежат соображения, прямо или косвенно связанные с объемным методом, служат следующие слова Берцелиуса: «Известно, что в окислительном ряду марганца кратные числа кислорода относятся, как 2, 3, 4, 5, и что последнее из них является кислотой; мы видели ранее, что кислоты, содержащие 5 атомов кислорода, по всей вероятности, всегда содержат 2 атома радикала, если, однако, марганцовая кислота является 2R+5O, то тогда окись марганца будет также 2R+3O, и, таким образом, все обстоятельства ведут... к принятию этого ряда (азотного.— М. Ф.)» [44, т. 7, с. 415]. И далее он писал: «...соотношения хрома и марганца побудили меня прежде всего избрать «ряд азота» в качестве наиболее правильного» [там же].
Таким образом, «мостом» между металлами и неметаллами был у Берцелиуса марганец. Изоморфизм окислов марганца и хрома укрепил эту связь. Переход к формулам Сг2О3 и СгО3 и к Fe2O3 и FeO (вместо FeO3 и FeO2) поставил вопрос о формулах окислов большинства металлов. Изоморфизм дал решение этого вопроса уже не только на основе априорного принципа аналогии, но и на прочной основе кристаллохимических данных.
При обосновании новой системы атомных весов немалую роль играли и чисто химические соображения. Так, касаясь свойств закисей ртути и меди, Берцелиус указывает, что «более вероятно признание того, что 2R+O выделяет один R и что большим химическим сродством обладает R+O, чем допущение того, что из двух «атомов» R+O один передает свой кислород другому, чтобы образовать один R и R+2O» [44, т. 7, с. 412]. I
При определении атомных весов Берцелиус отмечал, что одним из наиболее простых методов для определения относительного веса атомов является метод, основанный на взвешивании тел в газообразном виде с достаточной точностью и сравнении удель-ных весов [44, т. 7, с. 1]. Однако он указывал на ограниченность данного метода в связи с тем, что число газообразных вещесть
280
ХИМИЧЕСКАЯ АТОМИСТИКА
Таблица атомных весов по Берцелиусу (1818 и 1826 гг.)
Элемент 1818 г.* 1826 г. Элемент 1818 г.* 1826 г.
—— " Водород 1,06 1 Цинк 65 65
Кислород 16 16 Сурьма 122 129
Азот 14 14,2 Мышьяк 75 75
Углерод 12 12,2 Марганец 55 55
Сера 32 32,2 Калий 39 79
Фосфор 31 31 Натрий 23 47
Хлор 35 35 Кальций 40 41
Золото 187 199 Магний 24 25
Платина 195 197,10 Барий 137 137
Серебро 108 217 Стронций —• 88
Ртуть 201 203 Алюминий — 27
Медь 64 63,3 Кремний —— 44
Железо 56 54,2 Фтор 19
Олово 118 118 Бор 22
Свинец 208 2G7 1
* В данной таблице значения атомных весов 1818 г. для большинства элементов в два раза меньше, чем у Берцелиуса.
невелико, а также в связи с большими практическими трудностями при измерении объема и определении веса газообразных веществ. Берцелиус предложил общий метод определения атомных весов — на основе точных анализов атомного состава кислородных соединений данного элемента. Но, как мы уже видели, установление атомного состава веществ прямо или косвенно связано с применением объемных законов Гей-Люссака в их атомистической интерпретации. В конечном счете можно сказать, что в основе определения атомных весов всех элементов, согласно системе Берцелиуса 1826 г., лежит объемная теория. В частности, большое влияние при установлении Берцелиусом атомных весов и химических формул оказало признание для воды формулы Н2О. Это привело к тому, что, несмотря на то, что его формулы окислов большинства металлов совпадали с формулами Дальтона и Других ученых, атомные веса этих металлов оказались более близкими к современным, ибо он исходил из 0=16 (если взять Н=1,— М. Ф. ), а не 0=8. «Если считать, что молекула воды состоит из одного атома кислорода и двух атомов водорода, тогда °бе теории (атомистическая и теория объемов.— М. Ф.) становятся тождественными, отличаясь друг от друга только в отношении Представлений об агрегатном состоянии»,—писал Берцелиус И2а, с. 55].
Все изложенное выше указывает на то, что, несмотря на все косвенные соображения, применявшиеся Берцелиусом при уста
281
ГЛАВА ШЕСТАЯ
новлении атомного состава и атомного веса, он руководствовал^ принципом согласования весовых и объемных данных. Поэтом** нельзя согласиться с Г. Коппом, считавшим, что объемный метоп применялся Берцелиусом только для газообразных веществ и для твердых веществ он уже исходил только из закона изомоть физма и закона Дюлонга и Пти [48, с. 421], так же как нельзя согласиться с А. Ладенбургом, считавшим главной причиной перехода к новым атомным весам (системы 1826 г.) необходим мость согласовать атомные веса металлов с законом Дюлонга и Пти [49, с. 91]. Наконец, мы считаем, что мнение французского историка химии М. Делакра, будто Берцелиус при изменении своей системы 1818 г. исходил из «общей и неопределенной точки зрения» и что в его статьях, посвященных этому вопросу, нельзя «найти никакого соображения экспериментального характера в обоснование этого изменения» [50, с. 326], не отражает вполне точно и объективно историко-химической стороны этого вопроса. Делакр считал, что объемная теория не сыграла решающей роли при создании системы Берцелиуса, ибо, говоря о выводах объемных законов, Берцелиус указывал, «что они хороши, если дают простые объяснения». Далее Делакр указывал на то, что Берцелиус руководствовался главным образом выводами закона изоморфизма, а не законом Дюлонга и Пти. Таким образом, согласно Делакру, объемная теория, вытекавшая из непосредственных опытных данных, не сыграла будто бы никакой роли в изменении системы 1818 г. С этим мнением никак нельзя согласиться.
Закон изоморфизма и закон Дюлонга и Пти сыграли подчиненную роль. Они содействовали (в большой степени) более широкому и более последовательному применению Берцелиусом всех выводов объемной теории. Из всех историков химии более правильный взгляд на этот вопрос высказал, по нашему мнению, англичанин Мельдрум, считавший, что «объемная теория является краеугольным камнем системы Берцелиуса 1826 г.» [45, с. 75].
СИСТЕМА
ХИМИЧЕСКИХ ФОРМУЛ БЕРЦЕЛИУСА 1826 г. | И «ДВОЙНЫЕ АТОМЫ»
В символике Берцелиуса 1826 г. появляется нечто новое, что связано с новыми химическими формулами, а именно с призна-' нием окислов, содержащих два атома элемента. В 1818 г., когда единственным представителем таких соединений была только вода, Берцелиус употреблял для нее формулу Н2О.
Здесь необходимо отметить, что вообще он предпочитал пользоваться точками над символами для изображения кислорода 13 соединениях. Только в случае воды он вынужден был употреблЯт1,
282
ХИМИЧЕСКАЯ АТОМИСТИКА
(для ясности) символ кислорода О. Так, приводя формулу квасцов (KS2+2ALS3) +48Н2О, Берцелиус сделал следующее примеча-
«В тех случаях, когда два атома горючего радикала соеди-цяются с одним атомом кислорода, уже нельзя пользоваться точками, а надо применять символ кислорода О. Однако я ста-раюсь при обозначении атома воды пользоваться лучше знаком Aq» [42а, с. 119]. Таким образом, даже в 1818 г. Берцелиус избегал, по возможности, пользоваться формулой Н2О. Этого требовало единство обозначения формул, в которых атомы кислорода всегда бы обозначались точками над символами.
В 1826 г. в связи с изменением системы химических формул встал вопрос об обозначении двух атомов металла или неметалла в соединении при сохранении системы точек. С этой целью Берцелиус предложил следующее: «Во всех тех случаях, когда два атома радикала соединяются с 1, 3 или 5 атомами кислорода... ясность формул будет увеличена, если иметь специальное обозначение для двойного атома. Наиболее естественным знаком было бы удвоение начальных букв, но таким образом, чтоб они остались связанными вместе и выражали бы не два знака, а один. Для выражения этих формул я, однако, счел, что это можно осуществить легче и так же четко, пользуясь начальными буквами, но для изображения двух атомов проводя прямую черточку на расстоянии одной трети от нижней части буквы, так, например, Р обозначает один атом, а В — двойной атом фосфора, As — простой атом мышьяка, a As — двойной» [44, т. 8, с. 9].
Таким образом, происхождение перечеркнутых формул связано с чисто техническими, формальными соображениями. Но, с другой стороны, следует отметить, что применение двойных атомов вызвано не только признанием «ряда азота» для окислов металлов и неметаллов. Оно связано с новыми формулами солей, вытекавшими из новых формул оснований и из изменений формул некоторых кислот (например, хромовой). Дело в том, что система 1826 г. привела к тому, что большинство сильных оснований должно было быть сведено к более простому составу —из одного атома радикала и одного атома кислорода, а их соли — в одному атому основания и одному атому кислоты. Система химических формул солей Берцелиуса, таким образом, сблизилась с системами Дальтона, Томсона, Волластона и др.
В 1835 г. в «Учебнике химии» Берцелиус дает таблицу атомных весов и химических формул, принимая, с одной стороны, 0=100, а с другой — Н=1, что сближает его атомные веса с атомными весами Дальтона [51, с. 187]. Полагая, что в случае неметаллов вступают в реакцию вообще двойные атомы (кроме Нислорода), Берцелиус тем самым указал на то, что фактически эти элементы имеют удвоенный атомный вес по сравнению с другими, что приблизило его систему к системе химических аввпвалептов Волластона и других ученых, хотя сам Берцелиус
283
ГЛАВА ШЕСТАЯ:
предпочитал пользоваться системой атомных весов по кислорода которая сохраняла все достоинства системы 1826 г. ’
Определения Ж. Б. Дюма и Э. Митчерлихом плотности серы фосфора и мышьяка в некоторой степени подорвали доверие Бер’ целиуса к объемному методу как методу для непосредственного определения атомного веса по плотности элементов в газообра3 ном состоянии. Однако от своей системы он не отказался, ее подтверждали все соображения как чисто химического, так д физико-химического порядка (закон Дюлонга и Пти, закон изоморфизма). Сам Берцелиус не считал, что его система основана только на объемном методе, и всегда подчеркивал, что он против одностороннего, однозначного подхода к вопросу об атомных весах. Он это особо подчеркнул после неудачных попыток Дюма использовать объемный метод для непосредственного определения атомных весов: «...не существует никакого абсолютного метода для определения атомного веса; надо все принимать во внимание...» [52, с. 63].
Отождествление понятий «молекула» («сложный атом») и «эквивалент» вытекало у Берцелиуса из его дуалистической системы. В результате оказалось, что атомные веса щелочных металлов и серебра были удвоены, хотя это не вытекало ни из закона изоморфизма, ни из закона Дюлонга и Пти, а диктовалось только соображениями химической аналогии. Таким образом, дуалистическая система Берцелиуса, независимо от результатов исследования Дюма, уже оторвала химические формулы газообразных сложных веществ от их объемной основы.
Если, с одной стороны, дуалистическая система привела Берцелиуса к отождествлению для многих химических веществ их химических эквивалентов с молекулярным весом, то, с другой стороны, именно благодаря этой системе Берцелиус пришел к идее, что в реакциях многих элементов (неметаллов) принимают участие только двойные атомы. В результате начиная с 1833 г. он согласился принять удвоенный атом водорода за единицу (И =1), хотя и не отрицал существования простых атомов как водорода, так и других неметаллов.
Таким образом, система атомных весов Берцелиуса, введенная им с 1833 г., привела к еще большей путанице в понятиях «атомный вес» и «эквивалент».
Система атомных весов Берцелиуса 1826 г., отличавшаяся большой точностью и проверенная сопоставлением различных числовых данных, пользовалась авторитетом до середины 30-х годов прошлого века. Дюма во Франции и Митчерлих в Германии пытались укрепить эту систему путем распространения метода непосредственного определения плотности в газообразном состоя-нии и на нелетучие элементы. Однако, как известно, эти поныт-ки привели к обратным результатам: они подорвали доверие й объемному методу. Закон Дюлонга и Пти из-за многих отклоне-
284
ХИМИЧЕСКАЯ АТОМИСТИКА
ли# также не внушал доверия в качестве объективного метода определения атомных весов. Открытие диморфизма уменьшило значение закона изоморфизма. С другой стороны, открытие в 1834 г. электрохимических законов Фарадея, опровергших идею Берцелиуса о зависимости силы химического сродства от величины заряда атома, одновременно указало на то, что количество выделяющихся на электродах элементарных веществ пропорционально нх химическим эквивалентам. Этот факт привел Фарадея к отождествлению химических эквивалентов с атомными весами. Он считал метод электрохимического разложения верньш средством контроля при определении атомных весов [48, с. 435].
Все эти обстоятельства привели к тому, что химики все меньше начали доверять системе атомных весов и все больше стали применять химические эквиваленты.
ГЛАВА СЁДЬМАЙ
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
ОТКРЫТИЕ ЗАКОНА
ПРОСТЫХ ОБЪЕМНЫХ ОТНОШЕНИЙ |
И ЕГО РОЛЬ
В РАЗВИТИИ ХИМИЧЕСКОЙ АТОМИСТИКИ * *
Важнейшим открытием, оказавшимся исторически и логически связанным с химической атомистикой Дальтона, стало открытие в декабре 1808 г. французским ученым Жозефом Луи Гей-Люссаком ** закона простых объемных отношений реагирующих газов. Исследования газообразного состояния вещества занимают в творчестве Гей-Люссака особое место. При этом ученый «был совершенно равнодушен к возможностям альтернативной, гравиметрической, точки зрения» [1, с. 92] на состав химических соединений. Фактически Гей-Люссак противопоставил две формы закона кратных отношений — «весовую» и «объемную».
Если Дальтон, разрабатывая основы атомного учения, следовал ныотонианской традиции, ставившей во главу угла понятия о весе мельчайших частиц вещества и силах, действующих между ними, то Гей-Люссак опирался на картезианское представление о материи, согласно которому ее фундаментальным свойством служит протяженность, противопоставляемая «hard massy atoms» Ньютона. Об этом свидетельствуют и его двухтомные «Legons de physique», где о протяженности говорится, как о первом общем свойстве тел [2, с. 29], и работы по газам, и материалы личной библиотеки и архива французского исследователя [3].
Примечание. Литературу см. стр. 410—413.
* Разделы до стр. 309 написаны И. С. Дмитриевым.
** Жозеф Луи Гей-Люссак родился 6 декабря 1778 г. в Сен Леонаре (фран~ цузское графство Лимузен) в семье врача. С 1793 г. жил в Париже, где в 1797—1800 гг. учился в Политехнической школе. Здесь слушал лекции по химии К. Л. Бертолле, с которым в дальнейшем его связывала большая дружба. С 1809 г. Гей-Люссак почти одновременно стал профессоров физики в Сорбонне и профессором химии Политехнической школы. С 1806 г.— член Парижской Академии наук. 9 декабря 1829 г. избран п°' четным членом Петербургской Академии наук. Диапазон его химических исследований простирался на неорганическую и органическую, аналитическую и прикладную химию. Своими трудами и консультациями актиИ' но содействовал развитию химической промышленности. Долгие гоДь занимал руководящие должности в государственной пробирной слу^у и Монетном дворе. В 1839 г. получил звание пэра Франции. Умер Геи Люссак в Париже 9 мая 1850 г.
О жизни п деятельности Гей-Люссака см.: Crosland М. Р. Gay-Lussa ’ Scientists and Bourgeois. Cambridge: Univ, press, 1978.— Прим. отв. Peu'
286
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
Отсюда берет начало поиск Гей-Люссаком закономерностей, связанных в первую очередь с объемами газов и паров, а не с весовыми отношениями веществ. «Мы будем сопоставлять тела,— идеал Гей-Люссак в 1815 г.,— по их объемам в упругом (т. е. газообразном.— И. Д.) состоянии, а не по их весовым количествам, которые оказывают значительно меньшее влияние на соединение» 4 с. 188]. Не случайно, по-видимому, в знаменитой работе / g09 г., «Мемуар о соединении газообразных веществ друг с другом» [6], посвященной объемным отношениям реагирующих газов, Гей-Люссак не заметил, что, например, массы кислорода в окислах азота, рассчитанные па одну и ту же массу азота, относятся друг к другу, как небольшие целые числа [5, с. 64—65], п вместо этого подчеркнул прямо противоположную мысль — «крайне важно отметить, что при рассмотрении масс не наблюдаются никакие простые и конечные отношения между элементами» [6, с. 218].
Большое влияние на Гей-Люссака оказали также работы К. Л. Бертолле (1748—1822), ассистентом которого он стал в ноябре 1800 г. Многочисленные дискуссии в Аркёе по поводу химического сродства, химических пропорций, а также исследование К. Бертолле и А. Гумбольдтом (1769—1859) состава атмосферного воздуха способствовали пробуждению у Гей-Люссака глубокого интереса к проблеме взаимодействия частиц в газах. Поэтому его работы, посвященные изучению особенностей химических реакций в газовой фазе, были далеко не случайными. Однако, прежде чем переходить к истории открытия Гей-Люссаком закона простых объемных отношений, следует остановиться на теоретическом контексте, в котором этот закон был сформулирован.
Одним из наиболее острых вопросов теоретической химии начала XIX столетия был вопрос о составе химических соединений, что нашло свое отражение в известной полемике между Ж. Прустом и К. Бертолле, в которую так или иначе оказались втянутыми многие крупные химики того времени. Понимая, что обе спорящие стороны имеют свои веские аргументы, Гей-Люссак пошел по пути компромисса: «Прежде всего вместе с Бертолле следует принять, что химическое взаимодействие между молекулами тел протекает непрерывным образом и неопределенно, какими бы ни были число молекул и отношения между ними, так Чт°> вообще говоря, можно получать соединения в самых раз-личных отношениях. Но затем нужно в то же время допустить, Чт°, кроме нерастворимости, сцепления и упругости, т. е. фактору? способствующих образованию соединений в определенных отрешениях, химическое действие также оказывается более сильным, когда элементы находятся в простых или кратных друг ^Pyry отношениях, и тогда оно (химическое действие.—Я. Д.) составы, которые легче разделяются» [6, с. 232].
?87
ГЛАВА СЕДЬМАЯ
Таким образом, если реагирующие вещества оказываюТе «в сфере действия друг друга, они дают соединения самых ра* нообразных отношений, если только эти отношения не буД¥' определяться особыми условиями» [6, с. 232—233], к числу торых Гей-Люссак относил и условия протекания реакций в зовой фазе.
В чем же конкретно, по мнению французского ученого, заклю, чались отличительные свойства газов?
«Вещества,— читаем в той же статье 1809 г.,— как в твердом так в жидком и в газообразном состоянии обладают свойствами не зависящими от силы сцепления (la force de cohesion), но вместе с тем они наделены и другими свойствами, которые, по-види^ мому, подвержены действию этой силы (очень изменчивой ц0 своей интенсивности) и которые уже не подчиняются какому-либо закону. Так, одинаковое давление, приложенное ко всем твердым и жидким веществам, вызывает различное уменьшение их объема, тогда как для всех упругих флюидов (т. е. газов.-И. Д.) это уменьшение одинаково. Хотя теплота расширяет все тела, но до сего времени еще не найден определенный закон
для этого расширения; и только для газов оно одинаково и не зависит от природы тела. Поэтому притяжение молекул в твердых и жидких телах является причиной, изменяющей их специфические свойства, и, по-видимому, только когда притяжение полностью устранено, как это имеет место в газах, тела при одинаковых условиях удовлетворяют простым и правильным (regulieres) законам» [6, с. 207—208]. Иными словами, «только в газообразном состоянии тела находятся в одинаковых (внутренних.— И. Д.} условиях» [6, с. 233]. Отсюда — наличие у газов целого ряда характерных свойств, не зависящих от их химической природы и являющихся, по словам Ж. Б. Дюма, «следствием одинакового строения, присущего всем газообразным веществам» (цит. по [7, с. 99]).
Таким образом, первый тезис, который отстаивал Гей-Люссак и который важен для понимания ранней истории атомно-молекулярного учения, состоял в признании особой природы газообразного состояния, выражающейся в: а) отсутствии межчастич-ного взаимодействия и б) независимости некоторых свойств газов от их химической природы, вследствие чего реагирующие вещест' ва соединяются (в газовой фазе) в определенных отношениях (весовых и объемных) 1“2.
1-2 Последний вывод представляется, однако, не совсем правомерным Да?1л с позиций начала XIX в., так как определенность химических отношен11 как раз связана с природой реагирующих веществ. В газообразном состоянии (особенно, когда газы близки к идеальным) дискретность эТ11„ отношений проявляется с наибольшей отчетливостью, причем как н совых, так и объемных измерениях.
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
Далее, опираясь на данные объемного анализа, Гей-Люссак использовал «волюметрический» способ выражения количественного состава соединений, а не атомистический (дальтоновский). faK, например, аммиак, по его терминологии, состоит из одного объема азота и трех объемов водорода (NH3) 3. Сторонниками объемной теории были также У. Праут, Я. Берцелиус, Г. Дэви н некоторые другие химики.
Итак, второй тезис, сформулированный Гей-Люссаком, состоял н выражении состава соединения посредством указания объемов входящих в него элементов.
Из сказанного следует, что закон объемных отношений явился не только результатом эмпирического поиска, но и оказался включенным в определенный теоретический контекст, причем в такой, что кажется удивительным, почему Гей-Люссак не пришел в 1808—1809 гг. к идее, составляющей содержание закона Аво-гадро.
Во всяком случае, если обратиться к более позднему периоду творчества ученого (1820—1830), то обращает на себя внимание одно обстоятельство, отмеченное А. Роуком [8, с. 597— 598],—составы, приводимые Гей-Люссаком в 1820-х годах, отличаются от принятых им ранее. Например, вместо СО (1809) он предлагает для углекислого газа правильную формулу СО2 (1827), тогда как для воды вместо Н2О (1805—1809) дает состав ОН (1833). По мнению Роука, все становится на свои места, если допустить, что «Гей-Люссак рассматривал молекулы таких элементов, как водород, углерод, азот и хлор, состоящими из двух атомов, а молекулу кислорода — из четырех и, кроме того, придерживался мнения, что в равных объемах различных газов, при одинаковых температуре и давлении, находится равное число частиц» [8, с. 598]. Тогда некоторые хорошо известные в то время реакции можно записать так:
П2 + С122НС1, 2Н2 + О4->2(ОН)2 или 2Н2О2. 2С2 -J- 2Н2 С4Н4,
С2 + О4 —> С2О4 или (СО2)2.
По свидетельству весьма осведомленного в данном вопросе Дюма, предположение о делимости молекул простых газов во Франции «не было принято практически еще ни одним химиком, кроме Гей-Люссака» [9, с. 338]. С другой стороны, в 1832 г, Гей-Люссак совместно с А. Беккерелем доложили на заседании Парижской Академии наук свой одобрительный отзыв о работе
Гей-Люссак практически не пользовался химическими формулами, давая словеспое описание состава. Однако мы в дальнейшем будем для краткости использовать современную химическую символику, подразумевая чод индексами не число атомов, но объемы соответствующих элементов, как того требовала теория французского ученого,
И Всеобщая история химии
289
ГЛАВА СЕДЬМАЯ
М. А. Годепа «Исследования относительно группировки атомо [10, а], в котором отмечалось, что это исследование «основу на столь простых принципах, что ваши уполномоченные (т. е. р ° цензенты, представляющие работу Академии,— И. Д. полагаю необходимым доложить ее вам детально» [10, а, с. 142]. Идея Годена, который, в свою очередь, ссылался на Ампера как предшественника, состояла в том, что «молекулы простых тел^. кислорода, азота, водорода и т. д.— образуются соединением скольких атомов», но, в отличие от Ампера, Годен полагал, что
число их должно удовлетворять следующим двум условиям-«1) результатам анализа всех соединений, в которые эти тела входят, и 2) закону симметрии, который определяет все груп_ пировки атомов» [10, с. 144—145]. II далее, рецензенты останавливаются на некоторых примерах, рассмотренных Годеном, ци-
тируя его выводы о двухатомиости молекул водорода, хлора, азота и кислорода. При этом Гей-Люссак и Беккерель отмечают, что, «следуя тем же путем, можно показать, что молекулы азота пары брома и иода по крайней мере двухатомны, как и пары хлора; пары же ртути — одноатомны и т. д. Фосфор в парах по меньшей мере четырехатомен, а сера — шестиатомна» [10, с. 145].
Отсюда следует, что идеи Авогадро и Ампера (о которых см. далее) были близки Гей-Люссаку, хотя данных, позволяющих доказать, что последний пришел к ним самостоятельно, а тем более ранее Авогадро и Ампера, пока явно недостаточно.
Вернемся, однако, к статье [6]. Еще в 1805 г. А. Гумбольдт и Гей-Люссак начали изучать зависимость состава атмосферного воздуха от географической широты местности. Так как содержание
кислорода в воздухе определялось пми по количеству водяных паров, образующихся при сжигании смеси воздуха с водородом, то возникла необходимость в точном определении состава воды. Этим вопросом, по просьбе Гумбольдта, и занялся Гей-Люссак. В результате им было найдено, что 100 объемов кислорода соединяются с 200 объемами водорода. Однако до 1808 г. это наблюдение оставалось единичным фактом. Обратиться к систематическому изучению объемных отношений реагирующих газов заставили ученого его исследования трифторида бора4. Гей-Люссак установил, что это соединение легко реагирует с водой и аммиаком, причем последняя реакция напоминала взаимодействие с аммиаком сухого хлористого водорода, а именно: во всех случаях отношение объемов реагирующих газов равно 1:1.
Другим стимулом к поиску объемных закономерностей, по-видимому, послужили для Гей-Люссака работы У. Волластона
4 Ж. Л. Гей-Люссак и Л. Тепар надеялись получить фтор из плавпково® кислоты, разлагая ее затем калием. Одновременно они проделали ряд Д0-полнительных экспериментов, в одном из которых был получен ВГз(гяз) путем нагревания фторида кальция с борной кислотой, а из негр зртеН получили плавиковую кислоту [11].
290
МОЛЕКУЛЯРНАЯ ТЕОРИЙ
Основные результаты объемнометрических исследований Гей-Люссака
уравнение реакции (в современной записи)
Отношение объемов реагирующих и образующихся газов по данным Гей-Люссака
—-—' ~
^Нз(газ) + НС1(газ) = NH4C1(Tb) 1:1
2?Шз(газ) + Н20(ж) + СО2(газ) = (ЫШ^СО^р-р) 2:1
^тНз(газ) + Н20(ж) + СО2(газ) = NH4HCO3(p-p) 1:1
рШз(газ) + BF3(ras) = NH3«BF3(tb) 1:1
2Н2(газ) + О2(газ) = 2Н2О(Газ) 2:1:2
2N2(ras) + О2(газ) = 2Р42О(газ) 2:1:2
Nj(ra3) + О2(газ) = 2РЮ(Газ) 1:1:2
И2(газ) + 2О2(газ) = 2NO2(ra3) 1:2:2
2NO(ra3) + О2(газ) = 2NO2(ra3) 2:1:2
4NO(ras) + ОгСгаз) = 2N2O3(tb) 3:1
N2(ras) + ЗН2(газ) — 2МН3(Газ) 1:3:2
2йО2(газ) + О2(газ) = 2SO3(tb) 2:1
2СО(газ) + О2(газ) = 2СО2(газ) 2:1:2
4НС1(газ) О2(газ) = 2Н2О(газ) ф- 2С12(газ) 3:1:?:?
[12] и Т. Томсона [13], посвященные исследованию весовых отношений реагирующих тел5.
Теперь обратимся к рассмотрению выводов и рассуждений французского ученого, к которым он пришел на исходе 1808 г., изучая газофазные реакции.
Вывод первый: «Взаимодействие газообразных веществ происходит всегда в наиболее простых отношениях, так что с одним объемом газообразного вещества всегда соединяется такой же, или двойной, или, самое большее, тройной объем другого газа» [6, с. 233].
Вывод второй-. «Наблюдаемое сжатие объема, которое претер-иевают газы в результате соединения друг с другом, находится также в простом отношении к объему одного из них» [6, с. 234].
В таблице представлены некоторые газофазные реакции, изученные либо самим Гей-Люссаком, либо другими исследователя-А1п, на данные которых он опирался. Из этой таблицы видно, что в подавляющем большинстве случаев реакция протекала с Уменьшением объема,— отсюда приведенная формулировка вто-
Ж. Л. Гей-Люссак упоминает об этих работах, отмечая, что они, «кажется, подтверждают эту теорию (т. е. атомистику Дальтона.— И. Д.)» [6, с. 231]. Кроме того, ученый приводит в статье [6], наряду с объемными, также и весовые отношения между реагирующими газами, которые, хотя 11 не выражались им в целочисленном виде (как это делал Берцелиус,— см. главу шестую), по тем пе менее указывали на «определенность» (точнее — дискретность) химических отношений.
291
12*
ГЛАВА СЕДЬМАЯ
МО-
рого вывода. Кроме того, как будет показано далее, Гей-Люс интересовал вопрос о связи между изменением объемов г в ходе реакции и изменением объемов самих реагирующих лекул — вопрос, обсуждавшийся ранее Дж. Дальтоном [14].
Обращает на себя внимание и первый вывод, где фактич постулируется только отношение объемов типа 1: п (где 2, 3), а не отношения более общего вида к: п (где к и п — большие целые числа). Из таблицы также видно, что к такой формулировке толкали Гей-Люссака имеющиеся в его распоря
не’
жении экспериментальные данные, хотя вполне возможно, что и здесь имели место определенные теоретические соображения о «механизме» протекания химических реакций (одна молекула присоединяет — как бы «координирует» вокруг себя — несколько других молекул) 6 7.
Впоследствии, в 1811 г., А. Авогадро дал более общую формулировку обоих выводов, которая отвечала его представлениям о газах и газофазных реакциях и которая почти без изменений приводится в современных учебниках: «..соединение газов происходит всегда в весьма простых объемных отношениях, и если в результате соединения образуется газ, то его объем также на-
ходится в весьма простом отношении к объемам своих компонентов» [15, с. 58].
Полученные результаты позволили Гей-Люссаку рассчитать плотность любого из участвующих в реакции газов, если плотность остальных газов известна. Например, так как при взаимодействии окиси углерода и кислорода с образованием углекислого газа отношение объемов реагирующих веществ и продукта равно 2:1:2, то из закона сохранения массы следует, что плотности (р) этих газов удовлетворяют следующему равенству:
2р (окиси углерода)р (кислорода) = 2р (углекислого газа),
откуда Гей-Люссак нашел: плотность окиси углерода равна 0,9578 при плотности воздуха, принятой за единицу, что хорошо согласовывалось с данными У. Круйкшанка (0,9569) 7.
Кроме того, по мнению Гей-Люссака, открытые им газовые законы позволяют определять плотность паров простых тел, находящихся при обычных условиях в твердом или жидком состоянии, если при реакции они дают газообразные соединения. «Наблюдение, показывающее, что горючие газы соединяются с кислородом в простых соотношениях 1:1, или 1 : 2, или 1 : 1,5, моЖсГ дать средство для определения плотности паров горючих тел или» по крайней мере, заметно приблизить нас к этому» [6, с. 229] • Однако для реализации подобных расчетов требовалось устано-
6 Ср. со взглядами Берцелиуса по этому вопросу (глава шестая иастояШе' го издания).
7 Современное значение: 0,9670 при 0° С.
292
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
нлть аналогии в составе соединений, что далеко не всегда уда-ваЛ°сь сДелать корректно. Так, неправильная аналогия между составами соединений ртути и азота, проведеная Гей-Люссаком, повлекла за собой ошибку в определении плотности паров ртути [6, 0. 23].
II, наконец, крайне важными представляются также следующие две идеи, высказанные в работе [6].
Первая состояла в предположении, что отношение объемов реагирующих простых газов соответствовало отношению элементов в образующемся сложном газе, или, говоря словами Гей-Люссака, «элементы соединяются в простых отношениях, которые должны рассматриваться как пределы их отношений (des limites de lour proportions)» [6, c. 213]. Символически эту мысль можно выразить следующим образом:
аА ЬВ —* cAaB&t
где а, b и с — объемы газов А, В и АаВь соответственно8. Отсюда получались следующие простейшие (или «предельные») составы, которые Гей-Люссак принимал за истинные:
Н2О — для воды, поскольку два объема водорода реагируют с одним объемом кислорода;
С2О —для окиси углерода (угарного газа), поскольку Гей-Люссак принял, что при его образовании из простых тел углерод предварительно переходит в газообразное состояние и отношение объемов углерода и кислорода равно 2:1;
C2N — для циана — из расчета, что два объема газообразного углерода реагируют с одним объемом азота и т. д.
Другая идея, рассмотренная Гей-Люссаком в работе [6], была связана с тем парадоксальным, по его мнению, обстоятельством, что плотность кислорода оказывалась больше плотности окиси углерода: «...вспоминая великий закон химического сродства, гласящий, что каждое соединение приводит к сближению элементарных молекул, трудно понять, почему окись углерода (современная формула: СО.—И. Д.) легче кислорода» [6, с. 227]. Причина недоумения состояла в следующем. Масса молекулы угарного газа должна была быть больше массы молекулы кислорода, так как реакция образования окиси углерода — это реакция присоединения. Кроме того, в ходе этой реакции происходит уменьшение объема. Казалось бы, оба указанных обстоятельства свидетельствовали о том, что р (угарного газа) > р (кислорода), 0Днако эксперимент говорил об обратном. По мнению Гей-Люсса-Ка, «эта трудность происходит, оттого что полагают, будто сближение молекул в газе сопровождается уменьшением объема,
Еще раз подчеркнем,— в соответствии с теорией Гей-Люссака, индексы а и Ъ в формуле АаВь указывают не числа атомов А и В, но отношение объемов веществ А п В при образовании ими соединения, т. е. характеризуют, так сказать, «волюметрическую генеалогию» данного соединения.
293
frtfABA СЕДЬМАЯ
которое они испытывают при соединении. Однако это предпод жепие не всегда оправданно, п можно назвать множество газ^' образных соединений, молекулы (molecules const ituantes) которы" сильно сближены, хотя пет никакого уменьшения объема и даже наоборот, может иметь место некоторое его увеличение» [g с. 227—228]. В качестве примеров Гей-Люссак приводит две pear’ цпи, которые в современной записи имеют вид:
N2 + О2 = 2NO — не происходит изменения объема,
2N2O + О2 = 4NO — происходит увеличение объема.
При этом ученый совершенно определенно высказался пр0_ тив того мнения, что «сжатие (частиц.— //. Д.) элементов находится в непосредственной связи с уменьшением объема» [6, с. 228] в ходе реакции.
Здесь Гей-Люссак особенно близко подходит к тому кругу проблем, которые привели затем Авогадро к его открытию, ибо устранение зависимости «объем молекулы—объем газа» (на которой настаивал Дальтон) открывало, в конечном счете, путь к идее о равенстве числа частиц в равных объемах различных газов.
Завершая рассмотрение открытия Гей-Люссаком закона объемных отношений, следует отметить, что французский исследователь не только дал экспериментальный материал, послуживший источником концепций Авогадро, но и в известной мере подготовил появление этих концепций в теоретическом плане, а именно:
— им была ясно сформулирована модель, называемая ныне моделью идеального газа;
— взаимодействие газов рассматривалось в терминах молекулярных представлений;
— изменение объемов молекул в ходе реакции не связывалось с изменением объемов газов, образованных этими молекулами 9.
ОТНОШЕНИЕ ДАЛЬТОНА К ОТКРЫТИЮ ЗАКОНА ПРОСТЫХ ОБЪЕМНЫХ ОТНОШЕНИЙ
К открытию Гей-Люссака Дж. Дальтон отнесся критически: «Его (Гей-Люссака.— //. Д.) представление об объемах аналогично моему представлению об атомах, и если бы можно было казать, -что все упругие флюиды имеют в одинаковых объемах . равное число атомов или числа, относящиеся, как 1, 2, 3 и т. Д-» то обе гипотезы стали бы одной, с тем лишь различием, что моя
9 Термин «молекула» (molecule elementaire, molecule constituante) использовался Гей-Люссаком столь же широко, как термины «атом» и «сложная атом» Дальтоном.
294
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
гипотеза универсальна, а его применима только к упругим флюи-тгаМ. Гей-Люссак не мог не видеть, что подобная гипотеза была развита мною и отброшена как невыдержавшая критики» [14, с 61]. Дальтон полагал, что Геп-Люссак допустил экспериментальную ошибку, и приводил в пример результаты опытов К. Бертолле, Г. Дэви, У. Генри и своих собственных, «произведенных с добросовестной внимательностью в отношении точности» [14, с 62]. В итоге английский ученый приходит к следующему выводу: «Истина, я полагаю, состоит в том, что газы в любом случае не соединяются в равных или точных объемах; когда же они, как кажется, ведут себя именно так, то это происходит вследствие неточности измерений» [там же] 10.
В письме к Берцелиусу от 20 сентября 1812 г., когда уже точность опытов Гей-Люссака была неоднократно перепроверена, Дальтон более определенно указывает на неприемлемость для пего закона объемных отношений прежде всего в теоретическом плане: «Французское учение о соединении равных объемов газов я не могу признать, понимая его в математическом смысле» [14, с. 102]. Поэтому следует специально остановиться на том, почему же Дальтон отвергал закон Гей-Люссака.
Прежде всего заметим, что английский ученый сразу схватил суть дела, связав выводы своего французского коллеги с проблемой числа частиц различных газов в равных объемах. Дальтон прекрасно понимал, что в создавшейся обстановке необходимо либо предположить, что в равных объемах различных газов содержится разное число частиц, и тем самым поставить под сомнение закон объемных отношений, либо признать закон и одновременно принять гипотезу о равенстве числа частиц.
Такая альтернатива вытекала из следующих соображений, которые мы рассмотрим на примере реакции
N2q_Q2_^2NO
(в современной записи).
Явно или неявно и Гей-Люссак, и Дальтон (а в известной мере, и Авогадро — см. далее) принимали тот «великий закон», согласно которому если при взаимодействии двух веществ А и В образуется вещество С, то эта реакция непременно должна рассматриваться как реакция простого присоединения (приводя к своего рода компаунд-образованию), т. е.
А -4- пВ —* АВ . 1 п
Иными словами, частицы одного вещества как бы „прилипают" к частице (частицам) другого, или, по терминологии «объемной
10 По-видимому, Дальтои не совсем точно выразился — речь идет о соединении газов не в равных объемах, а в целочисленном отношении (по объемам),
ГЛАВА СЕДЬМАЯ
Закон Гей-Люссака и гипотеза о равенстве числа частиц не выполняются, реакция идет по ’’механизму’”простого присс^единения
Закон Гей-Люссака и гипотеза о равенстве числа частиц выполнены; в ходе реакции исходные частицы делятся
теории», один или несколько объемов вещества А присоединяется к одному или нескольким объемам вещества В, например:
Азот
+ кислород
> окись азота (селитряный
1 объем 1 объем или 1 частица 1 частица
Простое присоединение частиц
1 объем
1 частица
Эксперимент
2 объема
Если теперь принять гипотезу о равенстве числа частиц, распространив ее как на простые газы, так и на сложные, то в двух образующихся объемах окиси азота должно находиться в два раза большее число частиц, чем это имело бы место в случае простого присоединения. Но откуда появятся эти „лишние“ частицы?
Следовательно, либо надо отказаться от гипотезы о равенстве числа частиц и закона Гей Люссака, либо от идеи протекания реакции по «механизму» простого присоединения. В последнем случае необходимо допустить наличие более сложных превращений, связанных с делением и перегруппировкой фрагментов исходных частиц (атомов, по Дальтону) азота и кислорода. Графически приведенную альтернативу можно условно изобразить так, как показано на схеме.
Дж. Дальтон пошел по первому пути, и па то у него были веские основания, для понимания которых следует остановиться на некоторых моментах в истории создания им атомной теории.
Как* уже было сказано выше, Дальтон, отвергая химическую • теорию смешанных газов Бертолле, опирался на модель газа, восходящую к И. Ньютону, согласно которой газ представляет собой совокупность мельчайших частиц, отталкивающихся ДРУГ от друга. При этом Дальтон обратился к заимствованному и3 арсенала метафизики XVIII в. представлению о тепловых обо-
МОЛЕКУЛЯРНАЯ: ТЕОРИЙ
пОчках атомов, которые и обусловливали отталкивание последних [14, с. 44-60].
Сначала Дальтон предположил, что «частицы одного газа упруги и обладают отталкивательной силой лишь в отношении частиц одинакового с ними рода, но не в отношениии частиц другого рода» [14, с. 48]. Это утверждение, называемое в историко-химической литературе первой теорией смешанных газов (1801), объясняло однородность атмосферы (т. е. отсутствие расслоения газов в зависимости от относительны^ весов их частиц) , а также подтверждалось рядом открытых впоследствии закономерностей (в частности, законом растворимости газов Генри, 1803).
Однако первая теория смешанных газов Дальтона не обладала в полной мере «внутренним совершенством». Так, было непонятно, почему теплородные оболочки, которые всегда отталкиваются, ведут себя столь избирательно по отношению к типу центральной части атома. «Приняв ееи,— вспоминал в 1810 г. Дж. Дальтон,— мы должны были бы предположить столько же различных видов отталкивательных сил, сколько имеется газов, п, более того,— допустить, что тепло ни в одном случае не является отталкивательной силой; такое предположение, естественно, не очень вероятно. Кроме того, я нашел... что диффузия одного газа в другой является процессом медленным, требующим значительных сил» [14, с. 142—143].
Это и ряд других обстоятельств привели Дальтона к новой идее — ко второй теории смешанных газов (1804—1805), согласно которой частицы различных газов при одинаковых температуре и давлении обладают оболочками разных размеров, что приводит к одинаковому отталкиванию частиц одного и того же газа, но к различному отталкиванию частиц разных газов [14, с. 58].
В нашем контексте здесь важны два момента:
1) обе теории объясняли широкий круг явлений только благодаря идее о существовании отталкивающихся теплородных оболочек атомов, и отказ от нее ставил под удар всю физическую атомистику в целом;
2) обе теории поднимали проблему определения относительных атомных весов, для решения которой необходимо было знать размеры (диаметры) теплородных оболочек атомов и число частиц в данном объеме газа (или же отношение этих чисел для разных газов, находящихся в одинаковых объемах и при одинаковых температуре и давлении) 11 12.
Кроме того, Дальтон полагал, что в газе сферические частицы касаются друг друга (расположение атомов «должно быть аналогично квадратной кучке дроби» [14, с. 59]), и потому пренебрегал
11 Т. е. первую теорию смешанных газов.— И. Д.
12 Интересные и важные соображения по поводу создания атомной теории были высказаны Т. Коул ем [16].
297
Глава седьмая
пустым межатомным пространством. Тогда диаметр атомных оболочек (d) оказывался пропорциональным величине Уv (где v Объем атома вместе с теплородной оболочкой) или
, з---- 1
где р — атомный вес, ар — плотность газа.
Отсюда следовало, что поскольку вес частиц п их объем полагались Дальтоном прямо пропорциональными друг другу (рсор), то в равных объемах различных газов, отличающихся, в соответствии со второй теорией смешанных газов, объемом и весом их частиц содержится разное число последних [14, с. 143].
Таким образом, в силу названных особенностей принятых Дальтоном моделей газа и «структуры» атомов (сердцевина + теплородная оболочка) центральное положение его атомной теории о различии весов и объемов атомов разных веществ оказалось неразрывно связанным с представлением о неодинаковом числе частиц в равных объемах различных газов.
Ж.-Л. Гей-Люссак же, как было показано выше, исходил из иной модели газа, и, кроме того, он вообще был не склонен переоценивать роль теплоты в химических явлениях [17, с. 32].
ГИПОТЕЗЫ АВОГАДРО
В 1811 г. в издаваемом Ж. К. Деламетри «Journal de physique, de chimie, et d’histoire naturelie» появилась статья итальянского физика А. Авогадро * «Очерк способа определения относительных масс элементарных молекул тел и пропорций, в соответствии с которыми они вступают в соединения» [15].
Среди историков химии уже более ста лет идет оживленная полемика по поводу этой работы, в ходе которой высказываются и доказываются подчас диаметрально противоположные мнения. Это обстоятельство делает задачу «объективного», внедискуссион-ного рассмотрения ранней истории атомно-молекулярного учения практически невыполнимой. Поэтому дальнейшее изложение будет
* Амедео Авогадро родился 9 августа 1776 г. в Турине в семье служащего судебного ведомства. Он получил юридическое образование, по работа в Бюро адвокатов не удовлетворяла молодого доктора права. Труды по физике и математике больше привлекали его внимание. От их изучения он переходит к выполнению самостоятельных работ. В 1803—1804 гг. представил в Туринскую Академию паук две работы по электричеству, выполненные совместно с братом. С 1809 — преподаватель физики лицея небольшого городка неподалеку от Турина (Верчелли). Здесь он написал свои знаменитые работы, которые легли в основу молекулярной теории. В 1820—1822 и 1834—1850 — профессор физики в Туринском университете. Умер Авогадро 9 июля 1856 г. в Турине.
О жизни и деятельности А. Авогадро см.: Guareschi I. Amedeo Avogad-ю е la teoria molecolare. Torino, 1901; Быков Г. В. Амедео Авогадро: Очерк жизни и деятельности. М.: Наука, 1970.— Прим. отв. ред.
298
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
достроено с учетом наиболее важных и убедительных результатов ксследованпй последних лет, в первую очередь работ [17—24], ос-110Бпая тенденция которых состоит в устранении чрезмерной модернизации взглядов итальянского ученого и в воссоздании реального контекста, в котором следует рассматривать его идеи.
Основные концепции были изложим Авогадро в первых двух: частях статьи [15], поэтому на них следует остановиться более детально. Первая часть начинается с приведенной нами выше
(точнее, переформулировки) выводов Гей-Люссака,,
после чего автор переходит к их теоретической интерпретации,, подчеркнув, что «отношения количеств веществ в соединениях,, по-видимому, могут зависеть только от относительного числа соединяющихся молекул и от числа сложных молекул, которые при этом образуются (курсив наш.—If. Д.)» [15, с. 58]. Это не столь тривиальное замечание, как может показаться на первый взгляд. Переведем его, для лучшего понимания, па язык математических формул.
Допустим, в реакцию вступают два газа — А и В, давая газообразный продукт С; обозначим далее через VA, Ув и VG объемы соответствующих газов, через vA, vB и vc — объемы их частиц, а через пА, пв и пс — число частиц этих газов в объемах VА, Ув п Ус соответственно.
Тогда, согласно второй теории смешанных газов Дальтона, имеет место следующее отношение:
^А : : Т С “ nAVA : ПВГВ : ПсУс
(1)
Приведенное же выше утверждение Авогадро фактически сво-
дит это отношение к другому:
“ пА * пВ : ПС’
(2)
что при учете законов Гей-Люссака (отвергнутых Дальтоном) означает признание зависимости между числом частиц газа, находящихся в единице объема, объемами участвующих в реакции газов
и составом соединений. При этом эффективные размеры реагирую-
щих и образующихся частиц оказываются несущественными — важны лишь расстояния между их центрами, которые, если принять формулу (1), должны полагаться неизменными13. И действительно, уже в следующем предложении Авогадро формулирует свою знаменитую гипотезу как следствие законов Гей-Люссака и приведенного допущения (2): «Следовательно, необходимо принять, что имеются также очень простые отношения между объемами газообразных веществ и числом простых или сложных молекул, из которых они состоят. Наиболее простая гипотеза, которая в этой связи может быть высказана и которая представляется
13 Кстати, в рукописи статьп Авогадро там, где обсуждается структура газов, говорится, что теплородные оболочки газовых частиц препятствуют изменению «расстояния между центрами молекул» [25, с. 155].
299
ГЛАВА СЕДЬМАЯ
даже единственно приемлемой (после того как принято предпОЛо жение (2).— И. Д.), состоит в допущении, что в равных объемах число интегральных молекул (des molecules integrantes) у любых газов всегда одинаково или всегда пропорционально объемам (курсив наш.—Я. Д.)» [15, с. 58].
Сформулировав свою гипотезу, итальянский физик переходцт к ее обоснованию: «Действительно, если предположить, что число молекул, содержащихся в данном объеме, различно для различных газов [что могло бы проистекать только вследствие различного сродства молекул к теплороду) 14, то было бы совершенно непонятно, каким образом закон, который определяет различные расстояния между молекулами, может во всех случаях давать те простые отношения между объемом и числом молекул, которые вынуждают нас принять указанные выше факты (т. е. законы Гей-Люссака.— И. Д.). Напротив, легко понять, что молекулы газа находятся па таком расстоянии, что их взаимное притяжение не может проявиться, а их различное притяжение к теплороду может ограничиваться его конденсацией в больших или в меньших количествах около молекул, так что теплородная атмосфера не будет для одних молекул иметь большую протяженность, чем для других, и, следовательно, расстояние между [центрами молекул] молекулами не будут изменяться, или, другими словами, число молекул, содержащихся в данном объеме, также не будет различным. Правда, Дальтон предложил в этой связи прямо противоположную гипотезу, а именно, что количество теплорода всегда одинаково у молекул любых тел в газообразном состоянии и большее или меньшее притяжение к теплороду приводит к конденсации большего или меньшего его количества около молекул и к изменению расстояния между ними. Но при имеющемся у нас незнании способа, коим это притяжение молекул к теплороду осуществляется [ни одна из этих гипотез не будет более обоснованной, чем], ничто не может a priori склонить нас к одной из этих гипотез в большей мере, чем к другой, и уж скорее мы придем к принятию смешанной гипотезы, по которой и расстояния между молекулами, и количество теплорода изменяются по неизвестным законам...» [15, с. 58—59].
Таким образом, обоснование своей гипотезы Авогадро связывает с теплородной теорией, разработке которой ученый посвятил едва ли не большую часть своих научных работ. При этом «покушение на авторитет Дальтона (в вопросе о характере теплородных оболочек.— И. Д.) вряд ли могло способствовать популярности взглядов Авогадро» [26, с. 29]. К этому следует добавить, что главную щель своих физических и химических работ Авогадро видел не в развитии химической атомистики, а в количественном
14 Слова приводимые в квадратных скобках, взяты из рукописи статьи [15], опубликованной М. Морселли [25], и не вошли в окончательный текст.— И. Д.
300
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
определении сил сродства, в том числе сродства различных веществ к теплороду, для чего необходимо было знать, во-первых, силу притяжения материи к теплороду, а во-вгорых, уметь определять молекулярные массы. Силу притяжения можно было, по мысли Двогадро, найти из данных по удельным теплоемкостям (подробнее см. [24, с. 198—200]), тогда как гипотеза Авогадро позволяла заменить относительные молекулярные массы тел их плотностями [15]. Однако это направление исследований, ведущее начало от Бертолле и Лапласа и уходящее своими корнями к динамическим концепциям вещества XVIII столетия, к 1810-м годам уже начало терять свой былой престиж и постепенно уходило на периферию исследовательской (особенно химической) деятельности.
Поэтому формулировка гипотезы Авогадро в контексте учения о теплороде и теплородных оболочках частиц не способствовала ее признанию среди химиков. По мнению Р. Фокса, правильнее было бы рассматривать Авогадро «не как химика, наделенного глубокой интуицией, чьи взгляды были неправильно поняты при его жизни, но как физика, придерживающегося дискредитированной теории тепла» [22, с. 196]. Конечно, в этой оценке есть свои преувеличения, но вместе с тем не следует забывать, что химику наших дней гораздо легче находить «зерна истины» (зная всю истину) в рассуждениях Авогадро, нежели современникам итальянского ученого. Вернемся, однако, к статье [15].
Из своей гипотезы Авогадро вывел два следствия:
1) «отношения молекулярных масс те же, что и отношения плотностей различных газов при одинаковых давлении и температуре» и
2) «относительное число молекул в соединении непосредственно задается отношением объемов тех газов, которые данное соединение образуют» [15, с. 59].
Первое следствие Авогадро иллюстрирует следующим примером: отношение плотностей кислорода и водорода равно 1,10359: : 0,07321=15:1, следовательно, «масса молекулы кислорода будет примерно в 15 раз больше массы молекулы водорода» [15, с. 59]. Никаких эпитетов к слову «молекула» Авогадро здесь не дает, что, вообще говоря, затрудняет ретроспективную интерпретацию его вывода, ибо отношение масс 15:1 (современное значение 16:1) имеет место как для молекул кислорода и водорода (в современном смысле слова), так и для атомов. Заголовок статьи [15], однако, подсказывает, что речь идет об «элементарных молекулах», но, как будет показано далее, это понятие у Авогадро весьма неопределенно по своему смыслу.
Кроме того, от химиков — современников Авогадро — не ускользнуло и то, что из-за отсутствия в начале 1810-х годов независимого от его гипотезы метода определения молекулярных масс приведенное следствие не могло служить доказа1ельством справедливости самой гипотезы.
801
ГЛАВА СЕДЬМАЯ
Принятие гипотезы итальянского физика открыло бы путь к определению отношений молекулярных масс, «если бы эта пь потеза могла быть обоснована, но, к сожалению, нет средств, зволяющих подтвердить ее реальность» [21, с. 305].
Что касается второго следствия, то оно принималось многими исследователями, которые придерживались теории объемов (Гей-Люссаком, Берцелиусом и др.), отождествлявшей понятия атома (молекулы) и объема [7, с. 48—52]. Сам Авогадро так комментировал это следствие в 1814 г.: «... если даже эти объемы и не представляют самих молекул, они, по крайней мере, хороши тем, что заменяют их при современном состоянии наших знаний...» [27, с. 132]. Поэтому словам Авогадро о том, что, скажем, «вода образуется путем соединения одной молекулы кислорода с двумя молекулами водорода», так как «отношение объемов водорода и кислорода при образовании воды равно 2:1» [15, с. 60], не следует придавать современного смысла, ибо, как будет показано далее, состав исходных молекул водорода и кислорода (а потому и состав воды) оставался весьма неопределенным.
Теперь обратимся ко второй гипотезе Авогадро, которую сам он называл «I'hypothese du partage» (т. е. «гипотеза о делении»). Ей посвящен второй раздел статьи [15], который мы процитируем почти полностью:
«Поначалу кажется, что некоторое размышление говорит пе в пользу этой (т. е. первой.— И. Д.) гипотезы, когда речь идет о сложных телах. Так, представляется, что молекула, состоящая из двух или нескольких элементарных молекул, должна обладать массой, равной сумме масс этих элементарных молекул, и что, в частности, если в соединении каждая молекула одного тела присоединяется к двум или нескольким молекулам другого тела, то число сложных молекул должно оставаться тем же самым, что п число молекул первого тела. В связи с этим, согласно нашей гипотезе, когда [один объем] некоторый газ соединяется с двойным или кратным объемом другого газа, то соединение, которое при этом получается, если оно газообразное, может иметь только объем, равный объему первого газа. Но это в действительности, вообще говоря, не так. Например, объем воды в газообразном состоянии оказывается, как показал Гей-Люссак, удвоенным по сравнению с объемом входящего в ее состав кислорода, или, что то же, равен объему водорода, вместо того чтобы быть равным объему кислорода. Но довольно естественно напрашивается способ объяснения фактов такого рода в соответствии с нашей (первой.— И, Д.} гипотезой, а именно: следует предположить, что [интегральные] молекулы, составляющие какой-либо простой газ, т. о. молекулы, находящиеся па таком расстоянии, что их взаимодействие не может проявиться, не образованы одной элементарной молекулой, но состоят из некоторого числа этих молекул, объединенных в одну притяжением [которое определяет число входящих
302
Молекулярная теорий
_ г. ., :_-.L .- - :_ . 22- -я.-.--., j ..и , -^еа
в не© элементарных молекул]. И когда к ним присоединяются мо-декулы другого вещества с образованием сложных молекул, тогда интегральная молекула, которая должна при этом образоваться, делится на две или большее число частей, пли интегральных молекул, состоящих из 7г, 74 и т. д. числа элементарных молекул, образующих молекулу (la molecule constituante) первого вещества, соединенных с 7г, 7i и т. д. числа молекул другого вещества (курсив наш.—Я. Д.)» [15, с. 60—61].
Иными словами, некоторое число полу-, четверть- и т. д. молекул одного простого газа должны соединяться с «равным числом полу-, четверть, и т. д. молекул другого вещества, так что число интегральных молекул соединения должно быть двойным, четверным и т. д. по сравнению с тем числом, которое было бы без этого деления» [15, с. 61], и, кроме того, число образующихся интегральных молекул должно отвечать объему получающегося при данной реакции газа. «Так, например,— поясняет Авогадро,— интегральная молекула воды будет состоять из полумолекулы кислорода и одной молекулы, или,что то же самое, двух полумолекул, водорода» [15, с. 61].
Как правило, в литературе этот фрагмент статьи [15] рассматривают как формулировку идеи о многоатомности (часто даже — двухатомности) молекул простых газов. При этом в заслугу Авогадро ставится четкое разграничение физического понятия о молекуле и химического — об атоме [7, с. 38] и «механизм» реакции, скажем водорода с кислородом, в современных обозначениях представляют так:
2Н2 + О2 Н4О2 2Н2О
(см., например, [26, с. 30]).
Однако такая интерпретация грешит чрезмерной модернизацией взглядов Авогадро, который, как видно из приведенной цитаты, нигде не уточняет предела делимости молекул простых газов. Более того, в разных случаях он допускает деление одних и тех же молекул на разное число частей в зависимости от условий. Так, его интерпретация газофазной реакции водорода с кислородом основана на допущении деления их молекул надвое, т. е. на две элементарные, или парциальные, молекулы, тогда как в другой, более поздней, работе Авогадро полагает, что «атом твердой или Жидкой воды образован из 74 атома водорода и 7в атома кислорода» 15 [28, с. 86].
Или другой пример, на этот раз из статьи 1811 г.: «... из опытов 1 ей-Люссака следует, что эта кислота (имеется в виду 1’acide nitreux — азотистая кислота; N2O3 — в современной записи.— И. Д.) образована из 1 объема кислорода и 3 объемов селитряного газа (gaz nitreux, т. е. NO.— //. Д.), или, что то же, из 3 частей азота
Подробнее об этом выводе см. [24, с. 205].
303
ГЛАСА СЕДЬМАЯ
и 5 кислорода16, тогда как по нашей гипотезе молекула этой кисло, ты должна состоять из 3 молекул азота и 5 кислорода, если принимать во внимание возможность деления. Но этот состав, может быть сведен к указанным ранее более простым формам если его рассматривать как результат соединения одной молекулы* кислорода с тремя молекулами селитряного газа, каждая из которых состоит из одной полумолекулы кислорода и одной полумолекулы азота, что, таким образом, включает деление молекул кислорода, входящих в азотистую кислоту. Если ие предполагать другого, деления, то молекулярная масса последней будет равна 57,542: (при массе водорода, принятой за единицу), а плотность селитряного газа 4,21267 (при плотности воздуха, равной 1). Но имеет место, по крайней мере, еще одно деление па два и, следовательно,, уменьшение плотности вдвое...» [15, с. 64].
Иными словами, молекула кислорода должна, по Авогадро,, делиться дважды: сначала при образовании «селитряного газа»,, а затем еще раз, при образовании двух объемов «азотистой кислоты» (детально этот пример рассмотрен Б. Манди [20]).
Из приведенных примеров (число которых можно было бы увеличить) следует, что у Авогадро речь шла не столько о много-атомности молекул простых газов, сколько о «степени их субмо-лекулярности», которую ученый, однако, не конкретизировал. Поэтому «механизм» реакции образования воды по Авогадро правильнее было бы представить так:
2НП + (Hn)2(OR) 2(Нп/2)2(Оа72), пли 2НО,/2> 1
а реакцию образования «азотистой кислоты» как
+ (NzW0^ пли
где п, к и I — степени субмолекулярности соответствующих молекул 17.
Первая «стадия» указанных реакций сводится к образованию интегральных молекул путем соединения нескольких молекул реагентов. Состав этой интегральной молекулы определяется, в соответствии со следствием 2 из первой гипотезы Авогадро (см. стр. 301), отношением объемов реагирующих газов. Вторая «стадия» представляет собой деление промежуточной интегральной молекулы на число частей, численно равное объему получающегося в реакции газа, как правило, на 2.
Таким образом, если у Дальтона, как принято считать, практически отсутствовало понятие о молекуле как специфической
1С Точнее, из 4 объемов NO и 1 объема кислорода или 2 объемов азота и 3 объемов кислорода.— И. Д.
17 Можно, правда, допустить, что Авогадро, для простоты, принимал степени субмолекулярности равными друг другу для всех простых газов В противном случае отношение плотностей газов не будет равно отноше нию масс элементарных молекул.
304
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
сТруктурной единице вещества (что, впрочем, весьма спорно), т0 у Авогадро не было понятия об атоме в дальтоновском смысле слова, его теория была именно молекулярной по существу, а не только терминологически. Те относительные массы, с которыми имел дело итальянский физик, были на деле массами химически делимых молекул, состав которых он не конкретизировал. Кроме того, Авогадро нигде не говорил о том, что составные молекулы газа и продукты их «дупликации» различны по своей природе, наоборот, он говорит о «делении слояшой молекулы на две (и более.— И. Д.) молекулы той же природы (курсив наш.—Л. Д.)» [15, с. 62].
Сказанное в известной мере объясняет причины долгого непризнания химиками взглядов Авогадро. В самом деле, какова должна была быть реакция химика начала прошлого века, внимательно прочитавшего статью [15]? Во-первых, он вынужден был бы признать, что автор принимает гипотезу ad hoc, выдвигая ее специально для объяснения выводов Гей-Люссака. Во-вторых, что более существенно, эта гипотеза основывалась на весьма неопределенных умозрительных рассуждениях,— что, кстати, признавал и сам Авогадро,— опирающихся на теплородную теорию, которая с годами все более утрачивала свой авторитет среди химиков18, как и вся исследовательская программа, направленная на сведение химических явлений к действию сил сродства. В-третьих, следствия из первой гипотезы Авогадро, хотя и казались привлекательными, однако не могли быть проверены ни путем определения молекулярных масс независимым способом, ни измерением межчастичных расстояний в газе, ни посредством экспериментального доказательства двух- или вообще А-атомности молекул простых газов. В-четвертых, вторая гипотеза Авогадро, выдвинутая им единственно с целью согласовать первую с выводами Гей-Люссака, представлялась искусственной и весьма неопределенной. И, наконец, в-пятых, теория Авогадро не решала коренную проблему химической атомистики, состоящую в поисках независимых методов определения атомных масс19 и химических формул.
Последнее обстоятельство особенно ярко проявилось в работах Ж. Б. Дюма, выполненных в 1826—1832 гг. и посвященных определению «атомного веса большого числа тел по их плотности в вазо- или парообразной форме» [9, с. 337]. При этом в основу своего метода Дюма кладет гипотезы, которые по существу совпа-
18 Так, статья Ж. Молле «О конституции газов и их сродстве к теплороду» [29], автор которой, опираясь на теплородную теорию, пришел к гипотезе, аналогичной первой гипотезе Авогадро, практически прошла незамеченной. Тем самым, надежда Авогадро, что работа [29] «будет принята всеми, кто пожелает обратить внимание на данный вопрос» [30, с. 10], 1q 11 е оправдалась.
Хотя подавляющее большинство ученых понимало разницу между понятиями «масса» и «вес», практически оба термина использовались как синонимы.
305
Глава седьмая
дают с обеими гипотезами Авогадро (хотя имя последнего упоминается им лишь мимоходом в конце статьи и главную заслугу в выдвижении этих гипотез французский химик приписывает Амперу и Гей-Люссаку).
«...Не остается ничего другого,— писал Ж. Б. Дюма в 1826 г.,-< как принять лишь одну гипотезу, и все физики согласны с пей: во всех упругих флюидах при одинаковых условиях молекулы находятся на одинаковом расстоянии друг от друга, т. е. они находятся в одинаковом числе.
Непосредственное следствие этого подхода... состоит в том, что надо считать молекулы простых газов способными к последующему делению; деление, которое происходит в момент соединения и которое изменяется в зависимости от природы соединения. Хотя это следствие еще до сих пор не общепризнано, его невозможно избежать, если считать верным предыдущее предположение о строении газообразных тел» [9, с. 337].
Однако, принимая обе гипотезы, Ж. Б. Дюма вместе с тем подчеркивал, что частицы, образующиеся при делении молекул простых газов, не следует рассматривать как некие ультиматы, т. е. предел деления вещества: «... при настоящем состоянии науки существуют непреодолимые трудности, связанные с определением истинных атомов посредством изучения газов и паров. Фактически если молекулы простого вещества, переходя в газообразное состояние, остаются, однако, объединенными в группы в определенных количествах, то мы легко можем сравнить эти вещества при таких условиях, когда они содержат одинаковое число таких групп; но в настоящее время мы не располагаем способом определения числа элементарных молекул в каждой из групп» [9, с. 338].
Здесь уместно остановиться на терминологии Дюма, так как она не совпадает ни с той, которую принимали его предшественники, ни с современной ему. «Мы будем называть атомами,— писал он в «Трактате по химии» (1828),—группы химических молекул, которые существуют изолированно в газах. Таким образом, атомы простых газов всегда содержат некоторое число молекул, нам неизвестное» [31-32, с. X]. Под терминами «атом», «физический атом» и «физическая молекула» Дюма, таким образом, понимал некие субъединицы «химических атомов (молекул)»-Современному читателю такая терминология может показаться неудачной и даже надуманной, но у Дюма были основания именно для такого словоупотребления. С одной стороны, поскольку «химия делит те атомы, которые физика разделить не может» [там же], • логично, по мнению французского химика, назвать продукты такого деления «химическими атомами (молекулами) », а то, что делится," «физическими атомами (молекулами)». С другой стороны, поскольку истинный предел деления (физического и химического) был неизвестен, то, учитывая этимологию слова «атом», логичнее вообще изъять его из научного лексикона («если бы это было я
306
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
^оей власти,—писал Дюма,—я бы вычеркнул слово «атом» из цауки, так как убежден, что это понятие лежит вне нашего опыта») (цит. по [7, с. 104]). Для того времени — доводы вполне разумные. Остановимся теперь на расчетах Дюма, выбрав в качестве примера еГо исследование летучих соединений фосфора.
С целью определения плотности паров фосфора р(Р(паР)) французский химик обратился к двум соединениям этого элемента — фосфину (современная формула РН3) и трихлориду фосфора (рС13). Полагая, что реакция образования фосфина аналогична реакции образования аммиака, т. е. 1 объем паров фосфора реагирует с 3 объемами водорода, давая 2 объема газообразного продукта, и найдя плотность последнего равной 1,213, Дюма следующим образом рассчитал плотность паров фосфора20:
«Рассматривая протофосфид водорода (т. е. фосфин.— И. Д.) как соединение, образованное 3 объемами водорода и 1 объемом фосфора, сжатыми до 2 объемов, находим:
1,213 х2 = 2,426 —2 объема протофосфида водорода 0,0687 x 3 = 0,2061—3 объема водорода
2,2199 — плотность паров фосфора.
Принимая в качестве атомного веса фосфора 200 (при 0=100, т. е. округляя значение этой величины, данное Берцелиусом: 196,155 при О = 100.— И, Д.) и 1,1026 для плотности кислорода, находим плотность паров фосфора равной 2,2052» [9, с. 348]. Оба значения р(Р(паР)) совпали, таким образом, с хорошей точностью.
Чтобы окончательно убедиться в правильности полученного результата, Дюма сопоставляет плотность паров трихлорида фосфора (4,8076), рассчитанную с помощью найденного значения21 р(Р(паР)), с ее экспериментальным значением (4,8750). Полученное неплохое совпадение указывало, по мнению Дюма, на правильность принятых им составов соединений фосфора и приведенных значений плотности паров и атомного веса этого элемента22.
20 Плотность водорода была принята равной 0,0687.
21 При этом Дюма полагал, что 2 объема РС13 образуются из 3 объемов хлора и 1 объема паров фосфора:
«2,470X3 = 7,410 —3 объема хлора
"Р 2,2052—1 объем фосфора
9,6152 4,8076 для плотности паров
2 ~ хлористого фосфора» [9, с. 348].
22 С ретроспективной точки зрения, однако, ясно, что без прямого определения р(Р<пар)) все эти совпадения совершенно ничего не доказывают, ибо в действительности в газовой фазе идет реакция
Р4 + 6Н2 4РН3, нто, с использованием приведенных выше данных по плотностям водорода и фосфина, должно дать р(Р4(паР)) = 4,4398. Кстати, примерно такое значение (4.35) и получил Дюма (к своему великому удивлению) в 1832 г. [33], после чего оп вынужден был признать, что «фосфор может
307
ГЛАВА СЕДЬМАЯ
Таким образом, расчет Дюма сводился к следующему: а) сца чала определялся (или допускался по тем или иным соображе' ниям) состав летучего соединения элемента А в газо- или пар0, образном состоянии, а затем б) экспериментально устанавливалась плотность этого газа или пара, после чего в) вычислялась плот, ность паров соответствующего элементу А простого вещества по уравнению
ар(А) + fep(B) = ср(АаВь)
(где а, b п с - объемы реагирующих и образующихся газов (паров), а р —их плотности) и, наконец, г) полученная величина р(А) проверялась либо путем ее соответствия принятому атомному весу А, либо путем расчета плотности паров другого летучего соединения А и сопоставления ее с экспериментально найденной.
Ж. Б. Дюма допускал при этом, что атомы простых газов (т. е. их молекулы, по современной терминологии) могут делиться в ходе рекции, хотя и не отражал этой делимости в формулах [8, с. 599—600]. J
Нетрудно заметить, что в расчетной процедуре Дюма было, по крайней мере, два уязвимых места — выбор состава соединения данного элемента А и выбор атомного веса А. Принятие теории Авогадро не столько упрощало, сколько усложняло ситуацию. В самом деле, любые стехиометрические расчеты предполагали знание трех характеристик: 1) относительного количественного элементного состава данного соединения (который в ряде случаев мог быть определен по данным весового анализа); 2) его молекулярного веса (для чего можно было воспользоваться плотностью паров вещества) и 3) атомных весов входящих в него элементов. Определение атомных весов до открытия закона Дюлонга—Пти представляло наибольшие трудности23, которые в случае простых газов усугублялись невозможностью определить их состав известными методами, так что три указанные стехиометрические характеристики образовывали замкнутый круг взаимной зависимости. В результате можно было одновременно варьировать, скажем, химическую формулу соединения (уменьшая или увеличивая в п раз атомный состав) и атомные веса, оставляя без изменения молекулярный вес и (или) процентный состав соединения и получать согласующиеся между собой стехиометрические данные. Примером может служить расчет Дюма состава и других характеристик хлорида кремния. Критикуя Берцелиуса,
войти одной четвертью объема в газообразное соединение» [33, с. 174]-Аналогичную «аномалию» он обнаружил и при определении плотности паров серы [33].
23 Заметим, что и закон Дюлонга — Пти не решал всех проблем, так как о выполняется для элементов с атомным весом более 35 и с его помощь*0 не удается определить атомные веса простых газов и паров (Пг, Ог, ^2’ Hg(nap) и др.).
308
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
приписывавшего кремнезему формулу SiO3, а хлориду кремния giCl6, французский химик писал: «Можно прийти к более про-cTbiM отношениям, если уменьшить до одной трети атомный вес кремния; он тогда станет равным 92,5. В этом случае для плотности его паров получается значение 1,0197, а для составов кремнезема и хлористого кремния: Si+O и Si+Cl2. Согласно этой гипотезе мы имеем:
2,470X2 = 4,94 —2 объема хлора
1,0197—1 объем кремния
5,9597—1 объем хлористого кремния» [9, с. 368р4.
Итак, путем уменьшения в три раза как атомного веса кремния, так и числа атомов хлора в его хлориде (что эквивалентно троекратному уменьшению его (хлорида) молекулярного веса) Дюма удалось согласовать «теорию» и эксперимент.
В историко-химической литературе принято упрекать Дюма за большую путаницу, внесенную им в атомно-молекулярную теорию, и даже за отход от взглядов Авогадро и Ампера [26, с. 65; 7, с. 79—86]. Между тем, эти упреки вряд ли справедливы, ибо Дюма при всем своеобразии используемой им терминологии прекрасно понимал, что «физические молекулы» простых газов состоят из N «химических молекул» и всегда принимал гипотезу о равенстве числа частиц [8].
Проблема состояла в другом,— вследствие неопределенности числа А, понятия «атом» (в дальтоновском смысле) и «молекула» (как частица, состоящая из атомов) оказались «несостыко-ванными», так как было неясно, на какой стадии дробления молекулы, допускаемого второй гипотезой Авогадро, получается дальтоновский атом, и тем самым атомные веса оказались отделенными от своего материального субстрата.
Разработка химической атомистики в начале XIX в. шла по Двум направлениям — «восходящему» — от атома к молекуле (сложному атому) (Дальтон) и «нисходящему» — от молекулы, путем ее дробления, к субмолекулярным единицам (Авогадро, Дюма). При этом было совершенно не очевидно, что предел химического деления молекул есть дальтоновский атом. Для того чтобы обе линии развития атомно-молекулярной теории оказались соотнесенными друг с другом, необходимо было, во-первых, разорить замкнутый круг взаимной зависимости в выборе формул, молекулярных и атомных весов, разграничив понятия «атомный всс» и «химический эквивалент»; во-вторых, дать химические доказательства многоатомпости молекул простых газов и паров и, в~третьих, привести все численные стехиометрические характеристики соединений в соответствие друг с другом.
24 Экспериментальное значение плотности паров хлорида кремния, по данным Дюма, составляет 5,9390.
309
ГЛАВА СЕДЬМАЯ f... . ,111,1,1м. „,П„ ПВ-Т1ЖГ л! ! ПД| !! | —- - _
М. ГОДЕН И ГИПОТЕЗА АВОГАДРО * I
Все неувязки и противоречия, которые не смог разрешу Ж. Б. Дюма, пытаясь применить гипотезу Авогадро, устранил французский ученый Марк Антуан Годен (1804—1880). В 1827 г он посещал курс лекций по физике в Коллеж де Франс, и лекции А. М. Ампера по атомной теории зажгли его желанием посвятить свою жизнь разработке теорий расположения атомов в молекуле и молекул в кристалле. В 1831 г. Годен представил в Академию паук статью* 25, в которой он поддерживал гипотезу Авогадро—Ампера, представив, по-видимому, наиболее ясное до работ Канниццаро ее толкование [10, а]. Исходя из предположения, что такие элементарные газы, как водород и кислород, являются двухатомными, он объяснил кажущиеся аномальными данные о парах других элементов, полагая, что их молекулы могут содержать отличное от молекул элементарных газов число атомов. Например, он предположил, что молекулы паров ртути являются одноатомными, а серы — гексаатомными.
М. Годен впервые указал на необходимость четко разграничить понятия «атом» и «молекула». Он ввел понятие об одно атомных, двухатомных и многоатомных молекулах простых веществ, правильно объяснив плотности фосфора, мышьяка, сурьмы, серы и ртути в парообразном состоянии.
Одновременно с Годеном французский химик А. Бодримон в книге «Введение в изучение химии на основе атомной теории» (1833) четко сформулировал молекулярную гипотезу и указал, что она принадлежит Авогадро [34]. В учебнике «Курс общей и практической химии» (1844) Бодримон привел следующее определение «молекулы»:
«Молекулы.— Несколько атомов, связанных друг с другом и расположенных симметрично в пространстве, составляют корпускулярную систему, которую Ампер назвал молекулой. До сих пор слово молекула использовалось безотносительно к атому: лишь этот естествоиспытатель замечательным образом связал смысл этих слов, сохранив их этимологическое значение.
Молекулы, как и атомы, невозможно наблюдать непосредственно. Однако нельзя сомневаться в симметрии, которая является определяющей при их образовании, так как, связываясь друг с другом, они в конечном счете составляют тела, доступные чувственному наблюдению и предпочитающие правильные и симметричные формы. Этими телами являются кристаллы» [35].
* Данный и последующие разделы написаны М. Г. Фаерштейпом.
25 Эта статья была благожелательно отмечена и кратко пересказана в отзыве на нее, представленном 5 ноября 1832 г. в Академию наук физиком А. Беккерелем и химиком Ж.-Л. Гей-Люссаком. В 1873 г. М. Годси опуо-ликовал обобщенный труд сорока лет своей работы — книгу «Архитектура мира атомов», в которой приведен также текст отзыва Беккереля и Гей-Люссака [10, б, с. 155—157].
310
МОЛеКуЛЯРНЛЙ Феорий
Однако идеи, высказанные в работах Годена и Бодримона, п привлекли к себе большого внимания26. В то время француз-cKIie химики относились с недоверием к атомистике в целом и придерживались системы эквивалентов. Кроме того, Годен вы-сТупил со своими работами в неблагоприятный период укрепления дуализма. А ведь именно дуалистические формулы приводили, как мы уже сказали, к различным молекулярным (грамм-молеку-•1ЯрнЫм) объемам сложных веществ в газообразном состоянии *.
Все же работа Годена оказала некоторое влияние на распространение атомистических идей. Некоторые ученые стали пользоваться более четкой терминологией. Так, например, А. Ампер, который называл в 1814 г. молекулы «частицами» (particules), а атомы «молекулами», начал применять терминологию Годена, соответствующую современной. Даже Берцелиус стал использовать термины «атом», «молекула». Более того, он признал, что «идея о группах атомов в газообразных простых веществах также имеет нечто заманчивое» и соглашался с тем, что у фосфора п серы имеются такие «группы». Но он не распространял это представление на простые газы, считая, что у них отсутствует сцепление и поэтому образование «групп атомов» (молекул) невозможно, в то время как в парах серы и фосфора это сцепление может быть еще не полностью разрушенным. Мнение о том, что будто бы Берцелиус не признавал двухатомности молекул простых газов (в связи с тем, что одинаковые атомы отталкиваются своими одноименными зарядами), таким образом, является неверным.
Ж. Дюма в 1836 г. в своих «Лекциях по химической философии» (теоретической химии) говорил: «Химические атомы, по-видимому, образуют группы: так, газообразные частицы фосфора или мышьяка содержат вдвое больше атомов, чем частицы азота;
6 Сам Годен впоследствии писал: «Вот уже сорок лет, как это началось. Если оставить в стороне очень лестное мне одобрение со стороны некоторых представителей научной элиты, то можно сказать, что никогда научная теория не встречала такого полного безразличия. Несмотря на очень большое число публикаций, никогда ни во Франции, ни за рубежом она не вызывала дискуссии, как будто бы мои появляющиеся статьи не означали даже и того, что я выдвигал новые идеи относительно атомного и молекулярного строения тел...
Я должен отметить, что во Франции я все же слышал столь драгоценные для меня слова одобрения. М. Дюма никогда не прекращал меня поддерживать. А Эли де Бомон, который является не только одним из известных геологов, но и талантливым кристаллографом, уже довольно давно мне прямо сказал: „Вы на верном пути, и все, что Вас смущает, впоследствии прояснится*4. Это предсказание, действительно, мало-помалу Реализовалось» [10, б, с. 215—217].
Статья Годена не прошла незамеченной. В начале 1850-х годов ее прочитал С. Канниццаро и тем самым впервые познакомился с взглядами Аво-ГаДро, развитию и подтверждению которых он посвятил всю свою ^изнь,— Прим. отв. ред.
311
ГЛАВА СЕДЬМАЯ
< , . . - — _
газообразные частицы серы включают втрое больше химически атомов, чем частицы кислорода [36]. Дюма, однако, не допуск^ двухатомности молекул элементарных газов и одноатомности pTv ти и в то же время считал, что «молекулярные объемы» газо образных веществ могут быть различными.
Таким образом, в 1830-х годах сложилось своеобразное поло-жение в области атомно-молекулярных представлений: идея 0 молекулах простых веществ (хотя и в ограниченном виде) начала постепенно утверждаться, в то время как идея о равенстве числа частиц в одинаковых объемах газообразных веществ оказалась дискредитированной.
В 1840-х годах, когда уже назрела необходимость обобщения огромного эмпирического материала об органических соединениях и их систематизации, перед химиками-органиками в первую очередь встал вопрос о нахождении объективного метода определения молекулярного веса и о четком определении понятия «молекула». Важное объективное условие, способствовавшее решению этой задачи, было связано с началом крушения дуалистической системы под влиянием новых накопленных экспериментальных данных. Это устранило препятствия для признания гипотезы Авогадро, ибо падение дуалистической системы привело к необходимости отличать молекулу и атом от эквивалента и в результате — к установлению равенства «молекулярных объемов» газообразных веществ.
СОСТАВ ХИМИЧЕСКИХ СОЕДИНЕНИЙ: ДУАЛИСТИЧЕСКАЯ И УНИТАРНАЯ КОНЦЕПЦИИ.
ИХ РОЛЬ В ИСТОРИИ
АТОМНО-МОЛЕКУЛЯРНОГО УЧЕНИЯ
Еще в конце XVIII в., после возникновения дуалистической системы Лавуазье, в химии распространяются дуалистические представления, которые находят свое оправдание в химических реакцих соединения и разложения, образования и разложения соли, а также в способности солей к реакциям вытеснения (вытеснение кислоты более сильной кислотой, основания — более сильным основанием) и реакциям двойного обмена. Все это позволяло считать, что «кислота» (ангидрид) и «основание» (окисел металла) образуют обособленные части (группы) в соединениях.
Идеи Лавуазье нашли свое отражение в символических формулах «сложных атомов» Дальтона, изображавших строение солей. Эти формулы носили дуалистический характер, ибо изображали графически обособленную «группировку атомов», образуй щих «кислоту», и обособленно «основание». Дуалистическая концепция Лавуазье ждала своего теоретического обоснования. Все это явилось важной предпосылкой для возникновения электрохИ'
312
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
л1цческого дуализма. Я. Берцелиусу принадлежит историческая заслуга объединения химического дуализма с электрохимической гипотезой [37, с. 139].
Возникновение электрохимических представлений (1807—1818) связано с расширением данных о механизме горения, с новым толкованием этого процесса (по аналогии с возникновением электрической искры) и, наконец, главным образом с открытием явления электролиза химических соединений. Электрохимическая гипотеза была предложена в 1807 г. Г. Дэви, который считал, что в результате контакта реагирующих веществ между ними возникает электрическая полярность, обусловливающая химическое взаимодействие веществ. Эти идеи были развиты в 1809 г. Д. Авогадро, а затем И. Швейгером, X. Эрстедом, Я. Берцелиусом и другими учеными. Авогадро выдвинул идею об относительности электрохимических свойств в зависимости от природы реагирующих веществ и пришел к важным научным обобщениям по таким кардинальным понятиям в химии, как кислотность, щелочность, основность, нейтральность, амфотерность, окисление и восстановление. В 1812 г. Я. Берцелиус выдвигает гипотезу о существовании «готовых» полярных атомов (диполей). Представление о диполыюсти атомов дает Берцелиусу возможность объяснить образование химических соединений различных порядков. Он пишет: «Когда А и В, являясь двумя полярными частицами, соприкасаются друг с друюм своими противоположными полюсами, то их химическое соединение состоит в нейтрализации электричеств этих соприкоснувшихся полюсов; однако этот сложный атом является еще и теперь полярным благодаря электричеству в двух других полюсах» [38, с. 100]. Исходя из этого положения Берцелиус объяснил образование «отрицательных» кислот (окислов неметаллов), «положительных» оснований (окислов металлов), обычных солей, двойных солей и кристаллогидратов. Вода играла роль основания в «гидратированных кислотах», кислоты — в «гидратированных основаниях», а в кристаллогидратах — роль соли со слабыми электрохимическими свойствами.
Чтобы объяснить преобладание у элементов преимущественно одной полярности, Берцелиус приписывает двум полюсам атома различные по величине заряды, что приводит к относительной «униполярности». А для объяснения большей склонности некоторых электроотрицательных элементов (например, серы) соединяться с кислородом, чем это наблюдается у некоторых электроположительных элементов (Си, Ag, Аи), Берцелиус выдвигает гипотезу о различной «полярной интенсивности» атомов, зависящей от абсолютной величины заряда полюсов. Берцелиус дает также атомистическую и электрохимическую интерпретации идей К. Бертолле об обратимости химических реакций и о влиянии мас-СЬ1 веществ на сдвиг равновесия. Он считает, что меньшая электрохимическая сила действующих атомов может компенсироваться
ГЛАВА СЕДЬМАЯ
большим числом атомов, т. с. большей массой. И Берцелиус удовлетворением констатирует, что «правильные наблюдения Вг толле, использованные последним как пример неопределенна пропорций, получают благодаря электрохимической теории д^ гое объяснение, не противоречащее закону постоянства состав' [38, с. 112].
Согласно дуалистической схеме, соли —это, как мы уже знаем1 сочетание двух обособленных частей: «кислоты» и «основания»’ (причем формулу кислоты находили, «отнимая» от формулы соли формулу основания). В системе Берцелиуса 1818 г. большинству окислов металлов приписывалась формула МО2 (низшая степень окисления); позже, в 1826 г., Берцелиус приписывал этим же окислам формулу МО (в том числе и окислам щелочных металлов). Если формулы некоторых неорганических «кислот» совпа-SO2, С(\ то формулы, предложенные Берцелиусом для орга-
С ер.
дали в системе 1826 г. с формулами ангидридов (SO3, N2O5 и т. д.), ' ~
нических «кислот», были гипотетическими и часто соответствовали удвоенным формулам солей. Например, формулу ацетата калия записывали так: С4Н6О3-КО; формулу уксусной кислоты — С4Н60з (ее называли «обезвоженной» уксусной кислотой). За «водную» уксусную кислоту принимали С4Н6О3-Н2О. Здесь вода играла роль положительной части, т. е. основания. Для щавелевой кислоты принимали формулу С2О3 (которая следовала из формулы С2О3-КО).
Электрохимическая теория Берцелиуса сыграла известную роль в конкретизации понятия «молекула сложных веществ» («сложные атомы») как в отношении строения, так и в отношении их состава. Подчинив объемный и весовой методы определения атомных весов элементов дуалистическому принципу, Берцелиус пришел к определенным химическим формулам. Эти два метода служили для установления относительного числа атомов в молекуле, в то время как абсолютное число атомов диктовалось дуалистическими соображениями и зависело, кроме этого, еще от системы атомных весов. Так, формула уксусной кислоты изображалась С4Н6О3. Если же, согласно Дюма, принять, что атомный вес углерода С=6, тогда формула уксусной кислоты записывалась так: С8Н6О3. Неправильные представления о внутренней «конституции» молекул приводили к искажению формул, отражающих их состав.
Искажение химических формул коснулось не только органических кислот и их производных, по и других органических соединений. То же самое произошло с формулами некоторых неорганических соединений. Галогеноводородные кислоты изображали удвоенными формулами (TI2C12, H2Br2, H2F2), сероводород-" одинарной H2S, аммиак — N2H6 и т. д. Все это привело к тому» что при измерении «молекулярных» объемов веществ в газообразном состоянии появились соединения с разными объемами:
314
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
«двухобъемные» и «четырехобъемные». Дело в том, что если Припять объем «атома» водорода (объем одной весовой части) за единицу, то молекулярный объем тех веществ, которые имели рбьтчные формулы (неудвоенные), например щавелевой кислоты С2О2Н2, сероводорода, соответствует двум объемам (т. е. объему молекулы водорода Н2), в то время как молекулярный объем химических соединений, имеющих удвоенные формулы, соответствует в случае газообразного состояния четырем объемам.
Дуалистическая концепция коснулась только вопроса о конституции органических кислот и их солей. Однако, когда речь шла о других органических соединениях, единого мнения уже не было. Некоторые ученые (Гей-Люссак, Деберейнер, Дюма и др.), исходя из реакции разложения и некоторых аналогий с неорганическими соединениями, расчленяли формулу органического соединения на более простые формулы (например, этиловый спирт изображался так: С2Н4+Н2О, а этиловый эфир: 2С2Н4+Н2О).
Необходимость установления объективного критерия для правильной классификации огромного эмпирического материала органической химии (чему мешало применение различных систем атомных весов и эквивалентов и различных химических формул для одного и того же соединения) требовала унификации в области химических обозначений, а это, в свою очередь, требовало четкого разграничения понятий «атом», «молекула» и «эквивалент». В такой обстановке возникла унитарная система Ш. Жерара [39].
Попытка создания рациональной классификации органических соединений привела Жерара к необходимости унификации химических формул, что, в свою очередь, привело его к признанию одинакового молекулярного объема для всех веществ в газообразном состоянии, т. е. к признанию объемного метода в интерпретации Авогадро. Практическое применение этого метода было несколько искусственным, так как Жерар исходил из предполагаемых химических формул и использовал относительные плотности составных элементов (в некоторых случаях гипотетические). Относительная плотность исследуемого газообразного вещества использовалась только для контроля, а не как исходная величина. Например, при установлении правильности химической формулы этилового спирта С2Н6О, предложенной Жераром на основе аналитических данных, он делает следующий расчет:
2 X плотность углерода
6 X плотность газа водорода
1Хплотность газа кислорода
2x0,826 = 1,652
6x0,068 = 0,408
1X1,105 = 1,105
Сумма = 3,165
В этом расчете цифра 0,826 — гипотетическая относительная плотность углерода; 3,165 — «плотность» двух объемов паров спирта.
315
?ЛАВА СЕДЬМАЙ
Величина 3’165/2=1,582 представляет собой теоретическую плОт ность спирта, и так как это число приблизительно совпадает реальной плотностью этого вещества, то предложенная формулС верна и по пей можно вычислить молекулярный вес. а
Переход Жерара от дуалистических формул к правильны^ формулам органических и многих неорганических соединений по ставил на повестку дня вопрос о пересмотре системы «эквивалентов» (атомных весов), применявшейся в то время большинством ученых. В связи с этим Жерар приходит к правильным значениям атомных весов неметаллов. Интересно отметить, что в 1843 г., когда Жерар впервые предложил свой метод установления химических формул и свою новую систему атомных весов он еще называл атомные и молекулярные веса «эквивалентами»’ Только в 1848 г. Жерар начинает пользоваться правильной терминологией, четко разграничивая понятия «атом», «молекула» «эквивалент».
Система атомных весов Жерара выгодно отличалась от системы эквивалентов Гмелина, ибо атомные веса неметаллов, щелоч ных металлов и серебра приближались к современным. Атомные веса других металлов были заниженными в связи с тем, что он произвольно приписывал низшим окислам формулу В2О.
Таблица Жерара была менее совершенной, чем система Берцелиуса 1826 г., однако преимущество системы Жерара в целом состояло в том, что опа опиралась на правильные эмпирические формулы химических соединений, а это позволило Жерару прийти к новым теоретическим обобщениям, которые способствовали искоренению дуалистических формул.
Ш. Жерар, под влиянием О. Лорана, распространяет свои представления об одинаковом «молекулярном» объеме не только на сложные вещества, но также и на простые вещества в газообразном состоянии. Считая, что и простые вещества в газообразном состоянии должны быть двухобъемпыми, Лоран приходит к выводу, что формулы этих газов должны быть: Н2, О2, N2, С12 и т. д. Следовательно, чисто химическим путем и Жерар, и Лоран приходят к основным положениям гипотезы Авогадро.
О. Лоран распространил вывод Жерара о механизме реакции между сложными веществами как реакции двойного обмена иа реакции между элементарными газами. Это привело его к выводу, что между понятиями «атом» и «эквивалент» существует принципиальная разница 27. Лоран нашел, что один атом кислорода эквивалентен двум атомам водорода или двум атомам галогена 28. Он доказал, что между атомами О, S, Se есть эквивалентность, так же как между Н, Cl, Br, J. Но каждый атом первого
27 Идею о различной эквивалентности атомов различных элементов впервые высказал А. Авогадро еще в 1821 г.
28 Большинство химиков считали водород эквивалентным кислороду, поэтому они записывали формулу воды как НО.
316
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
дда эквивалентен двум атомам второго ряда. Это вытекало из хех химических формул, которые начал применять Жерар.
Таким образом, важная заслуга и Жерара, и Лорана заключается в том, что они в своих исследованиях химическим путем пришли к разграничению понятий «атом, молекула, эквивалент».
Систематическое изложение взглядов Жерара и Лорана дано в учебнике Жерара, опубликованном в 1848 г. [40]. Жерар подчеркивал, что в противовес дуалистам он рассматривает молекулу как единую систему атомов, однородных или разнородных. Касаясь определения понятия «эквивалент», Жерар отмечал, что величина эквивалента сложного вещества непостоянна — один и тот же элемент может иметь два или несколько эквивалентов.
Ш. Жерар, рассматривая все реакции как реакции двойного обмена, в своих учебниках и научных статьях широко применял уравнения химических реакций, что не было характерно для дуалистов. Дело в том, что дуалистические формулы Берцелиуса сами представляли собой химические уравнения реакции присоединения. Последовательное применение химических уравнений при изучении химических превращений в свое время (1842—1843) помогло Жерару разобраться в том, что только химические формулы, которым соответствуют одинаковые («двухобъемные») молекулярные объемы, являются истинными, т. е. отвечают наименьшему количеству веществ, вступающих в химическую реакцию.
Еще при жизни Ш. Жерара его идеи получили признание среди наиболее передовых ученых мира.
Д. И. Менделеев в своей магистерской диссертации «Удельные объемы» (1856) дал историко-критический анализ проблемы молекулярных и атомных объемов и развивал дальше идеи и методы Жерара, связанные с использованием молекулярных объемов газообразных веществ [41]. Менделеев доказал универсальность гипотезы Авогадро. Он объяснил причины отклонения плотности паров некоторых веществ при высокой температуре термической диссоциацией этих веществ — это еще до того, как А. Сент-Клер Девиль открыл явление термической диссоциации (1857).
В отличие от Авогадро и Жерара, использовавших относительные плотности простых веществ в газообразном состоянии (реальные и гипотетические) и данные химического анализа для определения численного значения молекулярного веса газообразных веществ, Д. И. Менделеев, впервые в 1856 г., предложил формулу для определения молекулярного веса таких веществ непосредственно по их относительной плотности, независимо от того, известен их химический состав или нет (М=29ДВоздУх). Он писал: «Это дает легчайший способ по удельному весу (относительной плотности.— М, Ф.) узнавать приближенный вес частицы и обратно. Эта формула должна заменить существующий ныне аРНоп-ческий способ определения удельного веса (относительной
317
ГЛАВА СЕДЬМАЯ
плотности) судя по его составу (судя по удельному весу его с0 ставляющпх)» [42, с. 222].
Д. 11. Менделеев, последовательно применяя эту формулу уточнил состав молекул различных простых веществ, доказав* что существуют вещества, которые в газообразном состоянии содержат различное число атомов (Н2, Р4, S6).
Итальянский ученый С. Канниццаро сыграл большую роль в устранении непоследовательности системы атомных весов металлов Жерара [43]. Трудности, с которыми столкнулся Канниццаро в процессе преподавания неорганической химии, натолкнули его на мысль о необходимости пересмотра атомных весов металлов. Его исследования исторического характера в области развития атомистики в первой половине XIX в. привели к выводу «что он [Жерар] в химии металлов не применял правило равенства объемов частиц, приведшее его к столь плодотворным результатам в химии углерода и других металлоидов», и Канниццаро предлагает: «Сделаем же, сказал я, для жераровских формул металлических соединений то же самое, что он сделал для формул органической химии, бывших до него в употреблении, т. е. сведем их к объемам, одинаковым с объемами других соединений» [44 с. 161].
В 1858 г. С. Канниццаро, независимо от Д. И. Менделеева, предложил формулу М=2РИ2. Взяв за основу гипотезу Авогадро и ее следствия и применив эту формулу, Канниццаро установил правильные атомные веса различных металлов, доказав тем самым непоследовательность системы Жерара. Канниццаро указал на объективный метод определения атомных весов исходя из определения молекулярных весов летучих соединений этих элементов.
В «Конспекте лекции по теоретической химии» (1858) С. Канниццаро дал научное обоснование своей новой системы атомных весов. Пользуясь плотностью паров органических и металлоорганических соединений соответствующих металлов, он пришел к выводу о необходимости удвоить атомные веса большинства металлов в системе Жерара. Он подчеркивал, что эти новые атомные веса как раз и удовлетворяют совокупности факторов: теплоемкости, изоморфизма и химической аналогии.
С. Канниццаро предлагает свое общее правило определения атомных весов: «Различные количества одного и того же элемента, содержащиеся в различных молекулах, являются целыми кратными одной и той же величины, которая входит неделимо в эти соединения и по праву называется атомом» [44, с. 305].
Применяя этот метод, С. Канниццаро исходил из молекулярного веса газообразного вещества, определенного по его относительной плотности. Канниццаро считал определяющим критерием при установлении атомных весов применение следствий гипотезы Авогадро. Согласование результатов с законом Дюлонга и ПтИ
318
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
п изоморфизмом еще больше укрепляло доверие к данному ме-тОду. Пользуясь законом изоморфизма и законом теплоемкости, в случае когда нельзя было применять непосредственную гипотезу Авогадро, Канниццаро подчеркивал подчиненную роль этих законов.
Математическая разработка кинетической теории газов, начавшаяся в 1856—1857 гг., привела к физическому обоснованию гипотезы Авогадро — как постулата о равенстве числа молекул в равных объемах при одинаковых условиях, так и второго постулата о наличии различного числа атомов в молекулах элементарных газов.
Р. Клаузиус и Дж. Максвелл развили учение о теплоемкости газов, учитывая различные виды движения молекул (поступательное, вращательное, колебательное) и теоретически обосновав зависимость отношения молярных теплоемкостей, при одинаковом давлении и одинаковом объеме, от числа атомов в молекуле *. Сопоставление теоретических значений CP!CV с опытными дало возможность физический! путем доказать существование у простых веществ в газообразном состоянии молекул с различным числом атомов.
Важным событием в истории атомно-молекулярного учения был Первый Международный конгресс химиков в Карлсруэ в 1860 г. [45]. К этому времени уже была подготовлена почва для утверждения молекулярных представлений. Путаница и неразбериха, которые господствовали в то время в химии — различные' системы атомных весов и эквивалентов, разнобой в применении химических формул,— приносили большой вред как процессу преподавания химии, так и самой науке. Отставание теоретической мысли от беспрерывно накоплявшегося экспериментального материала тормозило дальнейшее развитие прикладной химии.
Центральным вопросом, стоявшим на повестке дня Конгресса, был вопрос о разграничении понятий «атом», «молекула», «эквивалент». Кроме этого, был рассмотрен вопрос о введении новой системы атомных весов и химических формул Жерара. Работа Конгресса показала, что большинство химиков согласны с разграничением понятий «атом» и «молекула» в духе унитарного учения, но в то же время выявилось, что дуалистическое направление еще сильно (особенно среди неоргаников). Выявилась некоторая непоследовательность ученых, допускавших различие между «физической» молекулой и «химической» — даже у газов. Они придерживались мнения, что молекулярный вес газообразных веществ надо определять по химическим данным, по пред
* Атомно-молекулярное учение обеспечило предметную связь химии с физикой. Эта связь приобретала различную конкретную форму в зависимости от объекта исследования (ион, молекула, электрон) и новых методов. • Прим. отв. ред.
319
ГЛАВА СЕДЬМАЯ
полагаемой химической формуле вещества и пользоваться Ве личиной относительной плотности «только для контроля».
На Конгрессе получило утверждение научное понятие «моле кула» как наименьшее количество простого или сложного щества, вступающего в реакцию, в отличие от понятия «атом» как наименьшего количества элемента, входящего в состав мо лекул. Это было исторически необходимым шагом в истории химии [7]. «Приняв различие атома и частицы (молекулы),— писал Д. И. Менделеев,— химики всех стран приняли начало унитарной системы; теперь было бы большой непоследовательностью: приз, нав начало, не признать его следствий» (цит. по [7]).
Главное отличие идей Жерара и Лорана от идей Дальтона Берцелиуса и их сторонников — это признание молекулы в качестве основной единицы всех веществ, как простых, так и сложных. Кроме однородности или неоднородности атомов, Жерар не допускал никакого принципиального различия между молекулами простых пли сложных веществ. Таким образом, был выдвинут тезис о том, что химические силы взаимодействия не зависят от того, являются атомы одинаковыми или разными.
В системе Жерара атом был провозглашен наименьшей частицей, входящей в состав молекулы. Однако только после того, как Канниццаро устранил недостатки системы Жерара, касающиеся металлов, и предложил практические методы определения атомных весов, атом стал действительно наименьшей частицей в молекуле, причем однородные молекулы простых веществ, согласно Канниццаро, могут быть одноатомными, двухатомными тт многоатомными. В системе Жерара эквивалент приобретает своп истинный смысл, по только для неметаллов и их производных. Для металлов же эквиваленты совпадали с атомными весами. Канниццаро внес ясность и в этот вопрос. Установление правильных атомных весов для металлов привело к правильной системе эквивалентов для всех веществ. Таким образом, можно сказать, что в системе Канниццаро разграничение понятий «молекула», «атом», «эквивалент» достигло своего завершения. Однако теоретическое обоснование понятия «эквивалент» было признано только в 1860-х годах, после утверждения учения о валентности.
ИСТОКИ УЧЕНИЯ О ВАЛЕНТНОСТИ
Первые зародыши понятия валентности можно найти в работах* А. Авогадро, опубликованных в 1814—1821 гг. Он впервые пришел к выводу об эквивалентности двух атомов галогена одному атому кислорода или серы. Авогадро установил аналогию и эквивалентность фосфора, мышьяка и сурьмы исходя из правильных химических формул их соединений с галогенами и кислородом. Таким же образом он установил аналогию между углеро-
320
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
пом, кремнием, титаном, цирконием и оловом задолго до того, как это стало общепризнанным.
В 1842—1843 гг., когда Ш. Жерар предложил правильные эм-лирические формулы для органических соединений, стала возможной правильная интерпретация их атомного состава. Теория типов Жерара, развитая А. Вильямсоном, У. Одлингом, А. Вюр-цем, А. Кекуле и др., способствовала возникновению понятия «валентность». Была установлена эквивалентность радикалов СН3, C2Hs, С2Н3О одному атому водорода. Вильямсон и Жерар высказали затем предположение об удвоенных, утроенных или многоатомных типах и в связи с этим о многоатомных (многовалентных) радикалах. Под влиянием этих идей ученик Вильямсона Одлинг высказал идею о существовании многовалентных атомов (1851-1852).
В историко-химической литературе до сих пор вызывает много споров вопрос о том, кто первый ввел понятие «валентность» и заложил основы учения о валентности. Существует мнение, что это понятие впервые ввел английский химик Э. Франкланд в 1852 г. Как же в действительности обстояло дело?
В 1852 г. Э. Франкланд опубликовал статью [46], в которой на основе своих исследований металлоорганических соединений сделал некоторые выводы, имеющие отношение к понятию валентности. Используя типические представления Ж. Дюма, а также выводы А. Вюрца и А. Гофмана об аминах как продуктах замещения аммиака и А. Вильямсона о спиртах и эфирах как продуктах замещения воды, Франкланд пришел к выводу, что полученные им металлоорганические соединения являются производными неорганических «типов». В качестве «типов» он принимал окислы металлов (RO, RO2, RO3, RO5), в которых органический радикал замещает один эквивалент кислорода29.
Из своих формул Э. Франкланд делает следующий вывод: «При рассмотрении формул неорганических химических соединений даже поверхностному наблюдателю бросается в глаза господствующая в этих формулах симметрия. Особенно соединения азота, фосфора, сурьмы и мышьяка обнаруживают тенденцию этих элементов образовывать соединения, в которых содержатся три или пять эквивалентов других элементов; именно при этих отношениях имеет место наиболее полное насыщение сродства этих тел. С отношением эквивалентов 1 : 3 мы имеем соединения N03, NH3, NJ3, РО3, РН3, РС13, SbO3, SbH3, SbCl3, AsO3, AsH3, AsCl3 и др., а с отношением эквивалентов 1:5: NO6, NH4O, NH4J, ₽O5, РНД и др. He предполагая создавать гипотезу (подчеркнуто нами.— М. Ф.) о причине этой однородности в группировке атомов, я все же считаю совершенно ясным из приведенных приме-
В своих формулах Э. Франкланд пользовался эквивалентами С = 6, О — 8, а эквиваленты металлов соответствуют половине их атомных весов. 13
13 Всеобщая история химии 321
ГЛАВА СЕДЬМАЯ
ров, что такого рода тенденция или закономерность (подчеркнуто нами.— М. Ф.) является господствующей и что сродство атомов вышеуказанных элементов всегда удовлетворяется одним и тем же числом присоединяющихся атомов, независимо от их химической природы (подчеркнуто нами.— М. Ф.)» (цит. по [47]).
Приведенная цитата часто используется в качестве доказательства приоритета Фраикланда в создании теории валентности Первое, что бросается в глаза,— это то, что Франкланд пользуется эквивалентами и соответствующими формулами. Отсюда он п приходит к неправильному выводу, что атомы перечисленных выше элементов (N, Р, Sb, As) присоединяют одно и то же число других атомов «независимо от их химической природы» (т. е. что водород и хлор имеют ту же валентность, что и кислород) 30.
Таким образом, можно говорить, что Э. Франкланд подошел близко к понятию о валентности (которую он называл «соединительной силой»). Его заслуга состоит в том, что он подтвердил идеи О. Лорана, А. Вильямсона и У. Одлинга об эквивалентности радикалов и атомов на примере металлоорганических соединений.
Но, как признает К. Рассел, «применение Франкландом неправильных атомных весов мешало ему прийти к правильным значениям валентности» [49, с. 116]. Он даже не сумел установить единицу валентности.
Тот факт, что выводы Э. Фраикланда о «соединительной силе» занимают очень незначительное место в его большой работе о металлоорганических соединениях (1852), а также то, что он после этого не развивал свои идеи, показывает, что он не придавал им в 1850-х годах должного значения. Кроме того, хотя Франкланд в своей работе о металлоорганических соединениях отступил от некоторых положений теории радикалов, он все же в глазах химиков-органиков, в большинстве своем сторонников унитарной системы, считался приверженцем электрохимического дуализма и системы эквивалентов, и поэтому его идеи не могли оказать большого влияния.
Вот почему, по нашему мнению, идеи Фраикланда не вызвали до 1857—1858 гг., т. е. до работ А. Кекуле, никаких новых обобщений в этой области.
Истинная теория валентности могла возникнуть только тогда, когда начали применять правильные эмпирические формулы как для неорганических, так и для органических соединений, когда начали разграничивать понятия «атом», «молекула», «эквивалент» Этому в большой степени способствовала унитарная система Ш. Жерара и О. Лорана.
30 Отметим, что В. Палмер в своей кпиге «История концепции о валентности» [48], а также К. Рассел в своей «Истории валентности» [49, с. 116L цитируя Фраикланда. не приводят последние слова: «независимо от ИХ химической пр нроды ».
322
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
СОЗДАНИЕ УЧЕНИЯ О ВАЛЕНТНОСТИ
Истинным основателем учения о валентности следует считать д Кекуле [50]. Предпосылками для создания Кекуле теории валентности послужили теории типов Дюма и в особенности теория типов Жерара. В статье, опубликованной в 1857 г., Кекуле раскрывает теоретический смысл трех типов Жерара (П2, Н2О, дНз) и приходит к выводу о существовании трех групп элементов до отношению к водороду: одноатомной (одновалентной), двухатомной и трехатомной [51]. В этой же статье Кекуле также устанавливает, что «один атом углерода... эквивалентен четырем атомам И». Эти выводы Кекуле заложили основы теории валентности.
Почти одновременно с Кекуле в создание и утверждение учения о валентности большой вклад внес С. Канниццаро. Заслуга Канниццаро состоит в том, что, опираясь на объективный метод определения значений атомных весов элементов, как неметаллов, так и металлов, он смог установить правильную валентность металлов. Дело в том, что Кекуле, следуя за Жераром, приписывал окислам большинства металлов формулу К2О (т. е. он считал металлы одновалентными). На основе экспериментальных данных Канниццаро опроверг это неверное положение, доказав исходя из правильных химических формул соединений металлов (пользуясь для этого как определением плотности паров этих соединений, так и данными о теплоемкости металлов), что есть многовалентные металлы и что один и тот же металл может иметь переменную валентность.
Таким образом, можно сказать, что в 1858 г. уже существовала определенная система представлений о валентности элементов, опиравшаяся на правильные формулы соединений неметаллов (Кекуле) и металлов (Канниццаро). Эти закономерности исторически и логически связаны с новой системой атомных весов и химических формул Жерара—Канниццаро.
Однако понятие валентности до Первого Международного конгресса химиков в Карлсруэ (1860) и на самом Конгрессе еще не было в центре внимания химиков, хотя в обращении, разослан-, ном Инициативным комитетом Конгресса до его созыва, наряду с другими вопросами, фигурировал вопрос об «атомности» (валентности) . Конгресс, однако, этот вопрос не обсуждал. Р. Аншюц в связи с этим писал: «Многих может удивить, что вопрос о валентности даже не обсуждался на Конгрессе. Но это само собой понятно, если подумать, что основу теории валентности составляет определение атомных весов и что как раз этот вопрос и обсуждался на Конгрессе до конца» [52, с. 201]. Очевидно, что причина здесь была более серьезная, чем та, которую выдвигает Аншюц. Это, по-видимому, объясняется тем, что большинство Участников Конгресса, в том числе и сам А. Кекуле, еще не при
323
13*
ГЛАВА СЕДЬМАЯ
давали тогда понятию валентности того огромного значения, к торое было показано в 1861 г. А. М. Бутлеровым в его стать' «О химическом строении веществ» (см. [53, с. 46—54]).
Это подтверждается и тем, что на Конгрессе было принято решение считать, что «понятие „эквивалент44 является эмпирц ческим и независимым (подчеркнуто нами.— М. Ф.) от понятий „атом44 и „молекула44» [54, с. 985]. Это решение показывает, что химики тогда еще не понимали связи между понятиями «валентность» и «эквивалент», что величину эквивалента можно определять не только эмпирически, по и теоретически.
Известный интерес представляет также вопрос о возникновении термина «валентность». В научной и историко-научной литературе этот вопрос освещен или мало, или не совсем точно.
А. Кекуле вначале (1857) пользовался для обозначения понятия «валентность» терминами «основность» и «атомность» (одноатомный, двухатомный и т. д.). А. Купер в своей статье «О новой химической теории», опубликованной в 1858 г. (см. [53 с. 31—45]), применяет термин «степень сродства» (degree of affinity). Однако этот термин, как и термин «соединительная сила», предложенный Э. Франкландом (1852), не привился. Не привилась и терминология У. Одлинга (1864): «монад», «диад», «триад» (одновалентный, двухвалентный, трехвалентный). В 1860-х годах утверждается термин «атомность», но одновременно начинает применяться термин «эквивалентность», предложенный Од-лингом в одной из его работ.
В 1865 г. А. Гофман в своем учебнике «Введение в современную химию» назвал термин «атомность» двусмысленным, ибо он обозначает также число атомов в элементарной молекуле, и предложил заменить этот термин термином «квантивалентность» (quantivalency), обозначая соответствующие элементы терминами «унивалентный», «бивалентный», «тривалентный» и т. д. Термин «квантивалентность» вошел в английскую учебную и журнальную литературу (в журнале Лондонского химического общества он применялся вплоть до 1885 г.). Однако в Германии в конце 1860-х годов начинает применяться термин «валентность» (Va-lenz). Впервые этот термин применил А. Кекуле в 1867 г. в одной из научных статей [55, с. 217], а в 1868 г. его ученик Г. Вихель-хауз использовал этот термин в двух своих статьях. Во второй статье Вихельхауз дал определение этого термина: «„валентность (Valenz) применяется как более краткое слово вместо термина „квантивалентность44, введенного Гофманом, имея тот же смысл» [56, с. 257]. 4
Как видно из изложенного выше, термин «валентность» я® был взят непосредственно из латинского языка. В средневековой латыни «Valentia» обозначало «значение», «стоимость», «ценность». Но в химию этот термин пришел путем своеобразной метаморфозы эквивалентность -> квантивалентность -> валентность.
324
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
В английской научной литературе термин «валентность» стал общепринятым после 1876 г.; в немецкой и французской литературе—в 1870-х годах. В русской учебной и научной литературе Б XIX в. и даже в начале XX в. применялись термины «атомность» и «эквивалентность». Только в переводной литературе, появившейся в начале XX в., уже применяется термин «валентность». В работах Л. А. Чугаева начиная с 1913 г. используется только термин «валентность», и можно утверждать, что благодаря Чугаеву этот термин вошел в русскую учебную и научную литературу. Первым учебником химии, в котором последовательно применялся термин «валентность», был учебник Б. Н. Менптуткина, изданный в 1924 г. [57].
ВАЛЕНТНОСТЬ И ХИМИЧЕСКАЯ СВЯЗЬ
В связи с возникновением понятия «валентность» и учения о валентности родилось и понятие о химической связи атомов в молекуле. Еще А. Кекуле в 1857 г. в той же статье [51], в которой он излагал основные положения учения о валентности, писал, что «число атомов, связанное (verbundenen) с атомом другого элемента, зависит от основности (валентности) составных частей (атомов,— М. Ф.)». В 1858 г. и чуть позже Кекуле применяет для выражения образования химической связи чаще всего слова «прилегание», «прилегать» (Anlagerung, lagern) (см. [53, с. 26-28]).
Идея о том, что валентность атома указывает на число химических связей, которое имеет данный атом, как видим, впервые была высказана Кекуле. Но сам термин «химическая связь» был впервые введен А. М. Бутлеровым в 1863 г. В работе «О различных способах объяснения некоторых случаев изомерии» Бутлеров говорит о «способе химической связи атомов», о «химической связи отдельных атомов» [53, с. 57].
Э. Франкланд в 1866 г. применил термин «связь» (bond), отождествляя его с термином «атомность» (валентность). Многие английские ученые в конце XIX в. применяли термин «валентность» для обозначения понятия «химическая связь». Это привело к тому, что Дж. Фринд предложил ввести два термина: valency — в смысле валентности и valence — химическая связь между атомами [58]. Некоторые ученые поддержали это предложение и применяли его.
Терминологическое смешение понятий связано с тем, что многие ученые Запада их четко не разграничивали. Дело в том, что идея о наличии особой силы — химического сродства,— способствующей образованию химического соединения из различных элементов, существовала давно, еще до возникновения химической атомистики. Пройдя период «гравитационной» интерпретации (И. Ньютон), а затем «электрохимической» (Я. Берцелиус),
325
ГЛАВА СЕДЬМАЯ
это понятие после создания унитарной системы сохранилось, ц0 получило особый, специфический, необъяснимый с точки зрения макроскопической физики, смысл. По после формулирования понятия «валентность» возникла возможность связать между С(Я бой эмпирические данные об атомном составе с гипотетическим понятием о химическом сродстве.
А. Кекуле понятие о химическом сродстве связал с понятием валентности. Он подчеркивал, что валентность является численным выражением величины сродства (Verwandscliaftsgrosse) и числа химических связей атома. Кекуле говорил о единице «сродства» (Verwandschafeinlieit) как о единице валентности, так и о единице химической связи [59, с. 251]. Такое смешение вытекало из того, что Кекуле не мыслил себе атом вне молекулы и поэтому считал, что валентность проявляется в виде определенного числа химических связей. Кекуле смотрел на свои химические формулы, изображавшие графические связи между атомами (в том числе и между атомами углерода), как на условные модели, не отражающие реальную физическую структуру молекулы в пространстве (т. е. считал их функциональными, а не структурными моделями). Такой взгляд вытекал из концепции Кекуле, разграничивавшей «физический атом» и «химический», «физическую молекулу» и «химическую».
Большим достижением в развитии идей Кекуле о валентности и химической связи является его представление о способности атомов углерода (и вообще атомов одного и того же элемента) образовывать химические связи за счет единицы валентности каждого атома. Это новое представление возникло не случайно. Его корни можно найти в идеях Жерара о молекулах элементарных газов, в теории типов французского ученого, и в частности /нп
типа Н2 I тт I , где каждый атом может быть замещен углеводе-\ n J /
родными радикалами.
Таким образом, возрождение идей Авогадро о молекулах элементарных газов сыграло известную роль в развитии новых представлений. Такое предположение может быть подтверждено тем, что Кекуле в 1859 г. в своем «Учебнике органической химии» изобразил молекулу кислорода графически, как состоящую из двух атомов кислорода, соединенных двумя химическими связями, а именно . С другой стороны, Кекуле считал необходимым обосновать свой вывод о цепеобразных связях между атомами углерода экспериментальными данными о сохранении углеродного скелета органического соединения при различных его химических превращениях [53, с. 27—28]. Эта идея дала Кекуле возможность теоретически обосновать общую формулу гомологического ряда метана СпН2эт+2.
326
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
Вывод Кекуле о возможности образования химической связи меЖДУ атомами одного и того же элемента является наиболее оригинальным и наиболее важным из его учения о валентности, ибо он противоречил всем предшествовавшим представлениям о специфичности и полярности химического сродства, которые исключали такую возможность. Только признание идеи Авогадро и унитарной системы Жерара логически привело к этому выводу.
Важный вклад в развитие учения о валентности внес д. М. Бутлеров. В своей работе «О химическом строении веществ» (1861) Бутлеров высказал идею, что «каждому элементарному паю (атому,— М. Ф.) прирождено определенное количество силы, производящей химические явления (сродства). При химическом соединении потребляется (связывается, переходит в новую форму) часть этой силы или все се количество» (цит. по [53, с. 51]). Дальше он подчеркивал дискретный характер этой силы по сравнению со сродством (валентностью) водорода, принятым за единицу. Как видим, Бутлеров допускал переменную валентность у одного и того же атома. Кроме того, из приведенной цитаты вытекает, что Бутлеров видел различие между свободным (изолированным) атомом и атомом, вступившим в соединение с другим, когда он «связывается, переходит в новую форму (подчеркнуто нами.— М. Ф.)».
Далее русский химик подчеркивал: «От количества сродства необходимо отличать его напряжение, большую или меньшую энергию, с которой оно связывает вещества между собой. Напряжение это изменяется смотря по натуре действующих веществ и по условиям, при которых это действие происходит» [там же]. Он правильно отличал величину единицы валентности от величины энергии связи.
А. М. Бутлеров делил все элементы на две группы: с четной и нечетной валентностью, подчеркивая еще раз ее переменный характер: «Число единиц свободного (подчеркнуто нами.— М, Ф.) сродства всегда бывает четное (=0, =2, =4 и т. д.), так что атом, одаренный 2 единицами свободного сродства, может существовать самостоятельно, т. е. представлять частицу» (цит. по [53, с. 52]). И дальше он продолжал: «...прямое соединение может иметь место только между веществами, обладающими четным числом единиц сродства и притом, даже и в том случае, когда количество сродства одной стороны = 2, а в другой = 0. При этом можно себе представить, что частица, не имеющая свободного сродства, по необходимо обладающая по крайней мере 2 единицами сродства скрытого (потребленного), претерпевает в момент реакции распадение, так что 2 единицы, бывшие в ней соединенными между собой, теперь соединяются с 2 единицами сродства, принадлежащего двуатомпой (двухвалентной.— М. Ф.) частице» [там же].
А. М. Бутлеров более четко различает химическую связь как
^7
ГЛАВА СЕДЬМАЯ
проявление валентности атома. Оп считает валентность природ-денным свойством атома, которое «переходит в новую форму» при образовании химической связи, и, в отличие от Кекуле, д0, пускает существование у атомов, вступающих в соединение с дру„ гими атомами, неиспользованных «спаренных» частиц сродства [60, с. 33].
Выяснилось, что энергия химической связи бывает различной и зависит не только от валентности данного элемента, но и от природы «партнера» и от других факторов. Стало очевидно, что нельзя связывать валентность с понятием химического сродства с распределением действия этой силы, и начали больше склоняться к мысли, что валентность носит чисто формальный характер и не служит количественной характеристикой пи величины химического сродства, ни величины энергии связи, хотя между ними есть определенная взаимосвязь. Гофман подчеркивал, что эта взаимосвязь довольно сложная и даже противоречивая.
Вопрос о природе химического сродства, несмотря на большие успехи унитарного направления во второй половине XIX в., нашедшего свое воплощение в учении о валентности и теории химического строения, не был снят с повестки дня.
Интересной попыткой объяснить некоторые закономерности валентности и вникнуть в природу валентных связей является гипотеза французского ученого Ш. Делаво, высказанная им впервые, очевидно, не позднее 1863 г., ибо А. Вюрц ссылается на нее в 1864 г. [61, с. 75]. Подробное изложение своих взглядов Делаво опубликовал в 1865 г. Он высказал оригинальную мысль о сложном строении атомов. «Почему мы должны считать эту атомность абстрактным свойством атомов, в то время как мы можем приписать это свойство «составным атомам» (atomes constitutifs) низшего порядка...» Далее он называет эти составные атомы «субатомами» (sous-atomes), считая их носителями единицы валентности [62, с. 424].
Говоря об изменчивости валентности одного и того же элемента, Ш. Делаво писал: «Изменчивость атомности химического элемента... объясняется соединением его составных атомов или единиц сродства» [там же]. Он указывал, что валентность одного и того же элемента всегда увеличивается на две единицы, так что четная валентность всегда остается четной, а нечетная — нечетной. Это привело Делаво к важному заключению: «Почему „присоединяются41 только по две единицы сродства? Потому, что атомные частички (субатомы), которым принадлежат эти силы (сродства), соединены между собою по две. В присутствии тела, • которое может вступить с ними в реакцию, эти соединения тождественных частичек (субатомов) размыкаются (se defout). В действительности в молекуле пет свободных сродств; иначе нельзя было бы понять, почему молекулы с нечетной атомностью не существуют в свободном виде» [62, с. 426].
328
ЭДоЛёкуЛярйай теорий
Исходя из своей гипотезы Делаво строит модели различных угонов и молекул. Атом азота он изображает как систему пяти субатомов, связанных между собой «сцеплением». В молекуле аммиака три из этих единиц сродства насыщены сродствами атомов водорода, а остальные (а и Ь) «нейтрализуют» друг друга:
Далее Делаво пишет: «Если мы попытаемся установить аналогию между химическим сродством и физическими явлениями полярного характера, в частности магнетизмом, то надо будет сравнивать каждый составной атом (т. е. субатом.— М. Ф.) с магнитом и каждую пару (таких субатомов) — с системой двух противоположных магнитов, что согласуется с теорией Ампера, развитой Видеманом» [62, с. 428].
Таким образом, согласно Делаво, спаривание единиц сродства субатомов в атоме любого элемента имеет магнитное происхождение. Он считал, что магнитные и электрические свойства — проявление более общей причины. Общность магнитных и электрических сил Делаво видел не только в их взаимозависимости, по и в том, что и те, и другие полярны. Он выражал полярность химических сил сродства знаками (+) и (—). Но Делаво полагал, что эта полярность относительна и может меняться (по знаку) в зависимости от взаимодействующих атомов. Так, реакцию между окисью углерода (СО) и кислородом он изображал следующим образом:
329
tЛАВА СЕДЬМАЯ
Как видим, Делаво в своей гипотезе интуитивно предугадал некоторые закономерности, которые были окончательно раскрьь ты и обоснованы в современной теории сшш-валептности. Подтвердилась также идея о роли магнитного взаимодействия в процессе спаривания единиц сродства (электронов).
Известный интерес представляет отношение современников к идеям Ш. Делаво. А. Кекуле не признавал «спаривания» единиц сродства у одного и того же атома. А. М. Бутлеров, напротив был первым ученым, который до Делаво высказал подобную мысль. Французский химик А. Наке считал эту гипотезу умозрительной и поэтому не заслуживающей внимания. Шведский химик К. Бломстранд отверг гипотезу Делаво по нескольким соображениям. Он в принципе не признавал существование субатомов как противоречившее химическим данным. Но в то же время он понимал, что правило об элементах с четными и нечетными валентностями имеет какое-то объяснение, связанное с внутренними свойствами атомов. Бломстранд приписывал атомам не только электрохимическую полярность, но и магнитную, и, используя графические модели наподобие моделей Кекуле (слитые шарики), пытался объяснить, почему одни элементы (атомы) имеют только четную валентность, а другие — нечетную. Его надуманные схемы о симметричности и несимметричности магнитного флюида атомов различных элементов при взаимодействии с одновалентными атомами других элементов содержат в скрытом виде те же идеи, которые высказал Делаво, но без признания делимости атома на субатомы [63, с. 395, 397].
Интересно отметить, что до 1966 г. гипотеза Делаво не нашла своего отражения в историко-химической литературе не только у нас, но и за рубежом. Английский историк химии К. Рассел в своей «Истории валентности» [49] не излагает эту гипотезу, но зато приводит высказывания Э. Франкланда (1866), которые имеют косвенное отношение к гипотезе Делаво. Не признавая существования «субатомов», Франкланд для объяснения переменной, четной или нечетной, валентности предположил, что «одна или несколько пар связей (bonds), принадлежащих одному и тому же атому какого-либо элемента, могут соединиться и, насыщая друг друга, стать, если можно так выразиться, скрытыми. Так, пятиатомный азот становится трехатомным, когда одна его пара оказывается скрытой» [64, с. 378].
О причинах непризнания в свое время гипотезы Делаво можно высказать разные предположения. Мы думаем, что главной причиной было то, что в конце 1860-х годов атомистика в целом многими химиками оспаривалась и в лучшем случае принималась как рабочая гипотеза. Вот почему гипотеза Делаво, которая признавала, помимо атомов, еще и «субатомы», не могла найти благоприятную почву.
Заслуживает, однако, внимания тот факт, что в связи с пер-
330
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
А.Вюрц (1865)
К. Бломстранд (1869)
Таблица графических моделей молекул, отражающих эволюцию учения о валентности
выми попытками вникнуть в сущность химической связи, познать ее природу возникли первые, хотя и наивные, представления о сложности атома, первые модели строения атома.
Обобщая сказанное, мы можем констатировать, что в работах Э. Франкланда, А. Кекуле, А. М. Бутлерова, Ш. Делаво и других ученых валентность рассматривается в первую очередь как сродство свободного, изолированного атома, но это свойство практически изучается в его проявлениях, когда валентные возможности
331
ГЛАВА СЕДЬМАЯ
становятся реализованными. Соотношение валентность — химическая связь рассматривается, таким образом, как причинно-следственное, наподобие той связи, которая существует между потенциальной энергией и работой.
ПРЕДСТАВЛЕНИЯ О ПОСТОЯННОЙ И ПЕРЕМЕННОЙ ВАЛЕНТНОСТИ
Основатель учения о валентности А. Кекуле полагал, что валентность элементов постоянна. Он считал, что, являясь прирожденным свойством атома, валентность должна быть такой же постоянной, как и атомный вес. Максимальную валентность, равную четырем, Кекуле приписывал углероду. Защищая свою концепцию, он объяснял наличие меньшей валентности у некоторых элементов образованием цепеобразных связей между их атомами. Для тех случаев, когда валентность по эмпирической формуле оказывалась больше признанной им для данного элемента, он допускал образование молекулярных соединений. Так, например, РС15, NH4C1, SeCL он считал молекулярными соединениями: РС13-С12, NH3-HC1, SeCl2-Cl2*.
Среди сторонников переменной валентности мы встречаем Э. Франкланда, А. М. Бутлерова, У. Одлинга, А. Вюрца и др. Они допускали существование у каждого элемента определенной максимальной валентности, признавая, однако, возможность неполного использования всех единиц валентности. Для объяснения «кажущейся» низшей валентности эти ученые прибегали к различным моделям. В одних случаях они объясняли ее полимеризацией, в других — образованием цепеобразных связей между одинаковыми атомами и, наконец, спариванием между собой неиспользованных валентностей. Они также допускали существование пятивалентных, шестивалентных и семивалентных атомов.
Так, например, А. Вюрц приписывал атому железа валентность, равную четырем (исходя из формулы FeS2), и объяснял димеризацию хлорида железа следующим образом:
С1 С1
I I
Cl—Fe—Fe—Cl.
I I С1 С1
* Учение о постоянной валентности сыграло отрицательную роль в развитии представлений о строении комплексных соединений. Более тридцати лет продолжались попытки применить идею о цепеобразпом строении молекул к комплексным соединениям. В итоге оказалось, что эта идея, способствовавшая бурному и плодотворному развитию органической химии, не в состоянии была дать правильное представление о строений комплексных соединений.— Прим. отв. ред,
Я32
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
Экспериментальные данные по изучению плотности так называемых молекулярных соединений доказали, что речь идет об обычных «атомных соединениях», которые при высокой температуре могут подвергаться частичной или полной диссоциации алл не подвергаться такой диссоциации. Так, РС15 диссоциирует на РС13 и С12, в то время как PF5 не диссоциирует при высокой температуре.
При решении спора о постоянной или переменной валентности неметаллов известное значение имело изучение химического строения органических производных неметаллов. Дело в том, что существование различных окислов у этих элементов объяснялось сторонниками постоянной валентности цепеобразными связями между атомами кислорода. Например, SO2 и SO3 изображали так:
Изучение химического строения сульфоновых соединений с двумя ароматическими радикалами показало, что сера является максимально шестивалентной. Это вытекает из структурной формулы, обоснованной химически:
О
II
Ar-S-Ar.
II О
Таким образом, если после возникновения понятия валентности исходя из формул простейших водородных соединений это понятие послужило основой для установления химической связи и химического строения в соответствии с химическими свойствами веществ, то позднее, в 1870-х годах, спор о переменной валентности по отношению к кислороду решался чисто химическим путем — изучением строения веществ и их свойств.
Развитие учения о валентности в 1860-е и последующие годы пошло по двум направлениям. Главное направление выразилось в создании А. М. Бутлеровым теории химического строения (1861), сыгравшей огромную роль в развитии органической химии. Второе направление, носившее умозрительный, гипотетический характер, возникло в связи с попыткой теоретического объяснения природы валентности и ее закономерностей.
ГЛАВА ВОСЬМАЯ
$
ПЕРИОДИЧЕСКИЙ ЗАКОН
Открытие периодического закона было подготовлено всем предшествующим развитием химии, главным образом развитием атомно-молекулярной теории и учения о химических элементах.
К концу 60-х годов XIX в. выявились следующие научные предпосылки открытия периодического закона: установлены близкие к современным атомные массы многих химических элементов; развито учение об атомности (валентности) химических элементов; открыто сходство кристаллических форм (изоморфизм) различных химических элементов; разработано учение о химическом соединении, основанное на унитарных и молекулярных представлениях.
Многие из тех, кто описывал историю открытия периодического закона, отмечали факт, подчеркиваемый не раз и самим Д. И. Менделеевым * *, что о некоторых работах своих предшест-
Примечание. Литературу см. стр. 413—415.
* Дмитрий Иванович Менделеев родился 27 января (8 февраля) 1834 г. в Тобольске в семье директора гимназий и училищ Тобольской губернии. В 1841—1849 гг. учился в Тобольской гимназии. После ее окончания был принят студентом Главного педагогического института в Петербурге на физико математический факультет. Под влиянием таких профессоров института, как химик А. А. Воскресенский, физик Э. X. Ленц, геолог С. С. Куторга, Менделеев глубоко заинтересовался химией. Будучи студентом, он напечатал работу «Об анализе ортита и пироксена из Финляндии» (1854), а по окончании курса представил дипломную работу «Изоморфизм в связи с другими отношениями кристаллической формы к составу» (1855). 9 сентября 1856 г. Менделеев защитил диссертацию «Удельные объемы» на степень магистра химии и в 1857 г. стал приват-доцентом химии Петербургского университета. Весной 1859 г. он отправился в двухгодичную научную командировку за границу, в Гейдельберг, для дальнейшей самостоятельной экспериментальной работы по химии. В 18G1 г. вернулся в Петербург и приступил к чтению курса органической химии в Университете. В 1865 г. защитил докторскую диссертацию «Рассуждение о соединении спирта с водою» и с 1867 г. стал профессором общей химии Петербургского университета, который вынужден был покинуть в 1890 г. вследствие конфликта с министром народного просвещения, отказавшимся принять от Менделеева петицию студентов. 10 ноября 1892 г. Менделеев был назначен ученым хранителем мер и весов в Депо образцовых мер и весов, которое он в 1893 г. реорганизовал в Главную палату мер и весов, начав в пси важные метрологические исследования»
334
ЙЁЁЙОДЙЧЁСЙИЙ ЗАЙОЙ
А
всшшков Менделеев вообще не знал (о них он узнал после 1869 г.) 4. Сам Менделеев в своих обобщающих работах и вылуплениях называет, однако, Л. Гмелина, Дж. Гладстона, Э. Лен-сена, Ж. Б. Дюма, Дж. Кука, П. Кремерса, М. Петтенкофера и А. Штреккера, чьи заслуги в разработке систематизации элементов он считал значительными. Заметим, что ни А. де Шан-куртуа, ни Дж. Ньюленде, ни Л. Мейер не стоят в этом ряду! В необъективности или «сокрытии истины» Менделеева вряд ли можно обвинить. Дело тут в том, что в «поисках» системы элементов Менделеев шел последовательно, через изучение ряда проблем (исследование взаимосвязи свойств, изучение фундаментальных понятий, возникших на базе атомно-молекулярной теории). Недаром и на Фарадеевском чтении, и в других выступлениях Менделеев отмечает заслуги тех ученых, которые способствовали развитию представлений о системах атомных и молекулярных весов (Ш. Жерар, С. Канниццаро, А. Авогадро, А. Реньо), о свойствах редких элементов (Ш. Мариньяк, К. Раммельсберг и др.), об изоморфизме, атомных объемах и других свойствах, которые он называл «коренными» [1, с. 210—214].
В предисловии к книге «Два лондонских чтения» (1889) Д. И. Менделеев дает краткий анализ основных течений в химической науке в Европе и указывает, что в Англии стали традиционными исследования в области структуры и свойств материи (И. Ньютон, Дж. Дальтон и их последователи). «Английские химики с Вильямсоном, Франкландом и Роско во главе, не придерживаясь односторонних убеждений времени... в сущности ждут, ищут таких дальнейших новых, верных и общих начал, которые были бы пригодны для реального постижения и обладания природой химических изменений вещества...» [2, с. 427]. Хотя в субъективности этой оценки не приходится сомневаться, нужно признать и справедливость менделеевского прогноза. Достаточно назвать имена У. Рамзая и Э. Резерфорда, чтобы убедиться в сохранении отмеченной Менделеевым традиции. Будто имея в виду это будущее направление развития, Д. И. Менделеев сказал на Фарадеевском чтении: «Связав понятие о химических элементах новыми узами с Дальтоновым учением о кратном или атомном составе тел, периодический закон открыл в естественной философии новую область для мышления. Канту казалось, что в мире есть «два предмета, постоянно вызывающих людское удивление и благоговение: нравственный закон внутри нас и звездное
Здесь он работал до конца жизни. Умер Д. И. Менделеев 20 января (2 февраля) 1907 г. в Петербурге.
О жизни и деятельности Д. И. Менделеева см.: Младенцев М. Н., Тищенко В. Е. Дмитрий Менделеев. Т. 1. М.; Л.: Изд-во АН СССР, 1938; Фи-гуровский Н. А. Дмитрий Иванович Менделеев. М.: Изд-во АН СССР, 1961.— Прим. отв. ред.
1 См. главу пятую.
335
ГЛАВА ВОСЬМАЯ
небо над нами». Вдумываясь в природу элементов и в период^ ческий закон, следует сюда присовокупить третий предмет: «при' роду элементарных индивидуумов — рядом фактов всюду выра' женную», так как без них немыслимо само звездное небо и так как в атомах единовременно открывается и своеобразность индц~ видуальностей, и беспредельная повторяемость особей, и подчиненность кажущегося произвола индивидуумов общему гармони-ческому порядку природы» (подчеркнуто нами.— А. М.) [3, с. 218]
Таким образом, открытие периодического закона осуществи! лось как на той цепи научных открытий, которая некоторым историкам кажется единственно логичной,— Дальтон—Одлинг— Ньюленде—Менделеев, так и на параллельной линии: Дальтон-Канниццаро—Вильямсон (+ Франкланд + Роско) —Дюма—Менделеев.
Центральным понятием второй логической цепи представлений является понятие об элементах-аналогах и учение о формах соединений.
Понятие об аналогии в химии и понятие об индивидуальности химических элементов исторически оказались связанными с понятием об элементах-аналогах, которое сформировалось еще в первой половине XIX в. При этом нередко химическая аналогия (или аналогия физико-химических свойств) рассматривалась как результат внутренней аналогии между атомами, которая, в свою очередь, постулировалась в результате изучения атомных весов элементов и построения на их основе различных гипотез о сложности атома. Многие из этих гипотез были механистическими [4].
Аналогия как метод познания использовалась и раньше. Достаточно упомянуть отмеченную X. Гюйгенсом еще в XVII в. аналогию между звуковыми и световыми явлениями (волновой характер явлений). Но, пожалуй, XIX век оказался наиболее богатым примерами подобного рода в различных науках, что позволило вскрыть содержание понятия «аналогия». Особенно плодотворным оказалось применение этого метода в химии, о чем свидетельствует такое фундаментальное обобщение, как периодический закон и периодическая система химических элементов.
Многие понятия химии оказались тесно связанными с понятием «аналогия» (от греческого — «сходство», «соответствие»). Так, в органической химии сформировалось понятие о гомологических рядах, сходство отдельных представителей которых заключается в общности принципа химического строения, несмотря на наличие гомологической разности (—СН2—). Одинаковыми (сходными) *по составу, но обладающими различными физико-химиче-• скими свойствами являются изомеры. В неорганической химии широко исследовались явление изоморфизма и, как уже отмечалось, элемент-аналогия по химическим свойствам.
Все это были различные случаи аналогии, хотя как признаки сходства и отличия, так и причины их появления были разными
336
ЙЁРЙОДИЧЁСЙЙЙ ЗАЙОЙ
в отмеченных случаях, что отражалось на классификации объектов. Разбирая этот вопрос, Д. И. Менделеев писал в учебнике «Органическая химия» (1861): «...все разнообразие органических соединений подчиняется некоторым общим законам аналогии и происхождения (подчеркнуто нами.—Л. 71/.), законам, которые затруднительно формулировать, по можно ясно уже видеть при обзоре нескольких рядов...
...Группировка соединений может быть весьма разнообразна. Цо ни одна система не может выразить в последовательном изложении всей последовательности в происхождении и всех аналогий, потому что данное соединение имеет связь не с двумя только соединениями, а с множеством соединений (подчеркнуто нами.— A. MJ): с одними по гомологии, с другими по аналогии, с третьими по замещению, с четвертыми по типическим реакциям, с пятыми по происхождению... и т. д. Геометрически можно выразить сказанное таким образом: всякое изложение представляет линию, а система требует телесной формы...» [5, с. 222].
Приведенное суждение Менделеева очень показательно. Во-первых, оно свидетельствует, на наш взгляд, о понимании русским ученым многозначности понятия «сходство», а во-вторых, сама возможность выделения различных признаков для осуществления суждений по аналогии свидетельствовала об определенном уровне состояния химической науки. Анализируя исторический опыт развития теоретических представлений в химии, Дж. Бернал назвал этот метод мышления по существу своему биологическим или социологическим [6]. В этом названии отразился тот факт, что в химии невозможно было применить методы математического анализа. «История открытий и особенно изобретений,— писал Д. И. Менделеев,— явно показывает, что новые механизмы изобретаются легче и ранее всего, а новые химические процессы являются на свет божий труднее и позднее всего... Причину такого различия следует искать в том, что механические явления наиболее явны и ощутимы, физические уже труднее уловимы, а химические, по существу, скрыты и нередко охватываются только по их истечении» [7].
Хорошо известно, что аналогии оправдываются и метод суждения по аналогии оказывается плодотворным тогда, когда существенные признаки предметов пли явлений, поставленных в ряд сравнения, совпадают. По, как явствует из слов Менделеева, именно эти внутренние признаки нередко скрыты от исследователя. А по внешним признакам не всегда можно сделать правильные выводы2.
Пример тому — предсказание И. Ньютоном того факта, что алмаз является горючим веществом, сделанное на основании наблюдаемого положения: горючие тела отличаются большой светопреломляющей способностью. На самом деле среди веществ подобного рода есть и негорючие.
337
ГЛАВА ВОСЬМАЯ
РАННИЕ РАБОТЫ МЕНДЕЛЕЕВА КАК ЭТАП НА ПУТИ К ОТКРЫТИЮ ПЕРИОДИЧЕСКОГО ЗАКОНА (1856—1868)
Первые теории органической химии (теории радикалов и ти-пов) были основаны на аналогии между химическими элементами и радикалами либо между основными типами неорганических соединений (Н2, Н2О, NH3, СН4) и простейшими органическими молекулами. Однако в дальнейшем выяснилось, что это были только внешние аналогии. Теоретическая химия первой половины XIX в. много внимания уделяла изучению атомных и молекулярных объемов. По аналогии с законом Авогадро, справедливым для газообразных веществ, для тел жидких и твердых устанавливали такие же правила («закон равноостаточности»). Менделеев был одним из тех ученых, кто исключительно вдумчиво подходил к такому выводу [8, с. 240]. Разбирая закон равноостаточности для окислов, он отмечает, что общее правило
”0” — ”МеО” — ”Ме” = 5
(т. е. объем кислорода, равный объему окисла за вычетом объема металла) верно только для ограниченного числа соединений, а именно таких, которые характеризуются «малой химической энергией». В его рабочем дневнике в связи с этим появляется такая запись: «Надо отличать два крайних случая химического соединения: 1) Проникновение. Объем соединения равен объему одной из составных, [частей] — это есть минерализующий элемент — С1 таков и есть. Качества элемента скрылись. По О, Н и S не имеют этого значения. 2) Сопоставление, когда объем соединения = сумме объемов составных частей. Таковы сплавы, изоморфы, молекулярные, органические соединения...» [8, с. 549].
Изоморфизм, удельные объемы, формы соединений, принадлежность к классу определенных или неопределенных соединений — все эти данные Менделеев использовал для своих первых обобщений.
Изучая работы и лекции Менделеева, мы можем видеть, как постепенно идея классификации из вспомогательной (при систематизации материала или в процессе обучения студентов) становится одним из важнейших инструментов познания еще не открытого, в том числе и внутреннего, мира химических превращений.
По-видимому, первой таблицей, которая объединила сходные элементы [9, с. 37], была таблица Т. Грэма (1847), взятая из учебника Т. Грэма и Ф. Отто [10], где изложен изоморфизм соединений:
1) S, Se, Те и О; 2) Mg, Са, Мп, Fe, U, Со, Ni, Zn, Cd, Си, Н, Сг» V, Al, Be, Zr, Yt, Er; 3) Ba, Sr, Pb; 4) K, Na, Am; 5) Cl, Br, F,
338
ПЕРИОДИЧЕСКИЙ ЗАКОН
Су; 6) N, Р, As, Sb, Bi; 7) Sn, Ti; 8) Ag, Au; 9) Pt, Pd, Ir, Os; 10) W, Mo; 11) С, B, Si.
i
(К символу мышьяка была сделана сноска, где говорилось о том, что As и S считали изоморфными, но что Густав Розе доказал противное.)
Для характеристики этой систематики, основанной — повторяем — на изоморфизме самых разнообразных (простых и сложных) соединений, нелишне привести следующие за таблицей рассуждения: «Между каждыми двумя группами есть посредствующие, переходные тела; так, между 2 и 10 — Сг, 2 и 3 — Са, 2 и 4 — Mg, 6 и 7 - Bi, 4 и 8 - Ag и т. д. Хлор, фтор и сходные с ними иногда заступают место кислорода; по крайней мере, так признают это многие химики и минералоги, например: Герман, Шерер и др.»,— отмечает Менделеев [9, с. 38].
Он приводит мнение Э. Митчерлиха, согласно которому изоморфизм простых тел должен служить объяснением изоморфизма соединений и наоборот [там же].
Приведя далее ряд примеров, подтверждающих сказанное, в том числе и таких, которые в будущем окажутся очень важными для Менделеева (изоморфизм кислот серы и селена, с одной стороны, и хрома, молибдена, вольфрама —с другой), Менделеев останавливается на более сложных случаях; так, углекислота и кремнекислота многими не признаются изоморфными («не выяснена еще формула кремнекислоты»,—пишет Менделеев), а борную кислоту многие считают изоморфною кремнекислоте. «Вообще там, где трудно решить (и не решено), какую формулу должно дать окислу, трудно определить и изоморфизм его с другими окислами. Очень вероятно, что в различных соединениях одному и тому же окислу должно придать различные формулы и потому различные круги изоморфности» [9, с. 38—39].
Таким образом, в 1854—1856 гг. Менделеев был далек от того вывода, который он сделал несколько позже, на основании других количественных данных: изоморфизм служит критерием сходства только для определенных ступеней окисления элементов.
Развивая идеи об изоморфизме в диссертации «Удельные объемы», Менделеев вводит понятие о химическом попарном замещении (например, А1+Са вместо K+Si), основываясь на таком ряде окислов, где они стоят по степени усиления кислотных свойств:
К2О, Na2O, MgO, CaO, FeO, Fe2O3, A12O3, SiO2.
Комментарий Менделеева таков: «При замещении группами сумма тел, стоящих по краям, замещается суммою тел, заключающихся между ними» [11, с. 218]. При этом «сходственными соединениями» Менделеев называет те, «которые представляют Целый ряд одинаковых реакций» [И, с. 112],
339
ГЛАВА ВОСЬМАЯ
Следующий этап в поисках систематики элементов связан с использованием величин удельных объемов. В «Положениях избранных для защищения на степень магистра» (1856) об этом сказано совершенно ясно: «...19) сходственные тела очень часто имеют близкие удельные объемы. Например, a) Ni2 — 42 4-Со2 —42,7; Си2 —44,7; Fe2 —44,6... 20) Многие сходственные тела имеют удельные объемы (У), постепенно увеличивающиеся с уве-личением пая (П). Например: a) Li2 — У=136, 11=81; Na2—7=^ =404, П=289; К2—F==561, П=488... 21) Эти два закона (подчеркнуто нами.—Л. М.) дают твердую опору естественной классификации. Их указал Дюма в 1821 и 1854 годах» [9, с. 317].
Наконец, Менделеев составляет общую классификацию соединений п радикалов (органических и неорганических), которая основана на удельных объемах и «согласовании свойств». Вывод из таблицы, сделанный Менделеевым, таков: «...величина удельного объема постепенно изменяется с изменением состава или с замещением и с изменением свойств».
Некоторые историки химии справедливо усматривают в этих работах Менделеева «Изоморфизм в связи с другими отношениями кристаллической формы к составу» и «Удельные объемы» ясные шаги на пути к открытию периодического закона [12]. И хотя Менделеев, излагая план работы «Удельные объемы», говорит о необходимости рассмотреть естественную классификацию минеральных и органических тел, т. е. соединений [9,с.256], тем не менее можно сказать, что это действительно шаг к систематизации элементов.
Этот вывод подтверждается мнением Менделеева, высказанным в 1869 г. в статье «Соотношение свойств с атомным весом элементов», которая начинается словами: «Систематическое распределение элементов подвергалось в истории нашей науки многим разнообразным превратностям. Наиболее распространенное разделение их на металлы и металлоиды опирается как на физические различия, замечаемые между многими простыми телами, так и на различия в характере окислов и соответственных им соединений» [3, с. 10].
Какие же классификации простых веществ и соединений были наиболее распространены? Менделеев перечисляет их: системы, основанные на отношении к водороду и кислороду (или по относительному сродству), по электрохимическому порядку, а также основанные на атомности. К ним надо прибавить многочисленные попытки рассматривать группы сходных элементов в соответствии с изменением атомных весов. Разбирая каждый из этих способов, являющихся по существу классификацией или простых тел, или соединений, Менделеев и пришел к выводу о необходимости разработать классификацию элементов.
В подтверждение сказанному приведем те слова, которыми Менделеев характеризует свое отношение к делению элементов
340
ПЕРИОДИЧЕСКИЙ ЗАКОН
(простых веществ) па металлы и неметаллы: «...то, что казалось при первом знакомстве с предметом ясным и абсолютным, то при ближайшем знакомстве с ним совершенно потеряло свое значение, f тех пор как стало известным, что один элемент, как, например, фосфор, может являться и в состоянии металлоида, и в метал-пическом виде, стало невозможным опираться на различия в физических признаках. Образование основных и кислотных окислов це представляет также ручательства сколько-либо точного, по той причине, что между резко основными и кислотными окислами существует ряд окислов переходных, куда, например, должно отнести окислы: висмута, сурьмы, мышьяка, золота, платины, титана, бора, олова и многих других. Притом аналогия соединений таких металлов, как висмут, ванадий, сурьма и мышьяк, с соединениями фосфора и азота; теллура с селеном и серой; гак же как кремния, титана и циркона с оловом, не позволяет уже ныне строго держаться, в разделении простых тел, различия между металлами и металлоидами. Исследования металлоорганических соединений, показавшие, что сера, фосфор и мышьяк образуют соединения совершенно того же разряда, как и ртуть, цинк, свинец и висмут, служат еще более ясным подтверждением справедливости предыдущего заключения» [3, с. 10—11].
Таким образом, Менделеев столкнулся с относительностью любой классификации, которая распространялась бы на простые пли сложные вещества. Для того чтобы найти взаимосвязь между ними, требовалось ввести такое общее понятие или установить такой общий закон, которые позволили бы не искусственно, а естественно соотнести их друг с другом, подчинив этому общему.
В цитированной статье Менделеев объясняет, почему такое понятие, как валентность, казавшееся одним из самых общих и получившее широкое распространение, или такой закон, как закон четных паев, составивший «столь важную опору при изучении всех органических соединений» [3, с. 14], не могут быть признаны достаточно общими.
Только понятие атомного веса как постоянной количественной характеристики элемента и понятие элемента как «коренного» понятия химии в целом, по мнению Менделеева, могут быть такими общими понятиями. Он писал в той же статье: «...всякий из нас понимает, что, при всей перемене в свойствах простых тел, в свободном их состоянии, нечто остается постоянным, и при переходе элемента в соединение это нечто — материальное и составляет характеристику соединений, заключающих данный элемент. В этом отношении поныне известно только одно числовое Кнгное, это именно атомный вес, свойственный элементу. Величина атомного веса, по самому существу предмета, есть данное, °тносящееся не к самому состоянию отдельного простого тела, в к той материальной части, которая обща и свободному просто
341
ГЛАВА ВОСЬМАЯ
7
му телу, и всем его соединениям. Атомный вес принадлежит углю и алмазу, а углероду» [3, с. 17].
Сделав понятие «элемент» центром новой системы представ лений, Менделеев осуществил и следующий шаг — открыл закон периодичности свойств элементов.-
Анализируя взгляды Менделеева на основные положения атомно-молекулярного учения и его выступления по вопросам химического соединения, мы отмечали, что Менделеев систематизировал данные о сходстве элементов по изоморфизму и удельным объемам для решения проблемы классификации веществ. Так, если сравнить таблицу Грэма и таблицу Менделеева, основанную на данных по удельным объемам, то можно увидеть, что за два года сделан значительный шаг вперед. В таблице Грэма вторую строку составил длинный перечень элементов:
Mg, Ca, Мп, Fe, U, Со, Ni, Zn, Cd, Си, Н, Cr, V, Al, Be, Zr, Yt, Er, свидетельствующий о недостаточном выборе критериев сходства и различия.
В «Удельных объемах» и «Положениях...» к защите магистерской диссертации, где говорится уже о наличии двух законов (Ж. Б. Дюма) изменения величин удельных объемов и атомных весов сходных элементов — сходных по совокупности физических и химических свойств простых веществ и соединений,— эти элементы распределены в две разные совокупности.
Идея о двух «разрядах элементов», или двух разных совокупностях, каждая из которых характеризуется своим законом изменения свойств,— центральная идея на том этапе, который предшествовал открытию периодического закона (1856—1869). Начиная с 1856 г. Менделеев ищет общее свойство и общий закон, который позволил бы объединить эти две совокупности элементов (хотя сами границы этих двух совокупностей еще не очерчены ясно).
В цитированных выше «Положениях...» к защите диссертации упоминается о двух статьях Ж. Б. Дюма (1800—1884), одного из крупнейших французских ученых. После смерти Дюма в 1884 г. в своем выступлении на заседании Русского химического общества Менделеев сказал, что Дюма «оставил такую массу важных научных работ и оказал такое влияние в химии, что его имя останется бессмертным, пока существует паука» [13, с. 337]. Перечислив заслуги Дюма в области органической, физиологической и прикладной химии, а также в разработке философии пауки, Менделеев продолжал: «...имя Дюма соединяется с понятиями, столь основными в химии, как металепсия (1838 г., хло-роуксусная кислота), как химические типы замещения (1840) я как правильности в атомных весах элементов... я признаю, что современное положение вопроса об отношениях атомных весов химических элементов определено трудами и идеями Дюма боле®»
342
ПЕРИОДИЧЕСКИЙ ЗАКОЙ
чем кого-либо другого (подчеркнуто нами.—Л. Л/.)» [13,
с. 337-338].
Итак, мы обратили внимание, что работы Дюма, по свидетельству Менделеева, «побудили искать действительный закон» [14, с. 288]. Будущий автор периодического закона в процессе сопоставления свойств химических соединений заинтересовался вопросом о составе частиц, т. е. молекул. Это сопоставление началось с выяснения понятия об удельных объемах негазообраз-вых тел.
В 1850—1860-х годах проблема определения состава и выражения его с помощью химических формул широко обсуждалась химиками и физиками.
Подводя итоги «историко-химического исследования», Менделеев, как уже говорилось, обратился к различным физико-химическим свойствам и с этой целью анализировал работы Дюма: «Дюма показал, что растворимость зависит от величины сжатия элементов при их соединении. Это он нашел над сернокислыми солями Mg, Ca, Ba, Sr. Зависимость растворимости от удельных объемов аналогических (подчеркнуто нами.—Л. М.) тел видна уже из того, что Горсфорд, с одной стороны, показал зависимость ее от величины пая, а Кремере, с другой стороны,— от удельного веса» [9, с. 255].
Обращает внимание Менделеев и на работу Дюма, в которой рассматриваются «удельные объемы аналогических тел» [15].
Не случайно уже во второй части диссертации «Удельные объемы и состав кремнеземных соединений» Менделеев рассматривает понятия «аналогия», «гомология» и «сходство» «в тесном смысле этого слова» [И, с. 115] и обращается к выводу Дюма: «Дюма, вслед за многими другими, старался найти между величинами паев таких соединений такое же отношение, какое существует для гомологов и аналогов, и успел показать несколько замечательнейших примеров» [там же].
Под аналогами — в соответствии с классическим определением этого понятия — Менделеев понимает сходство в определенном отношении таких предметов, которые в целом различны (пример Менделеева — акриловая и коричная кислоты), а под гомологами — соединения сходного «происхождения» или класса, отличающиеся друг от друга постоянной «гомологической разностью» (пример Менделеева — два непредельных спирта).
И далее, говоря о законах изменения объемов, относящихся «до сходных соединений», Менделеев пишет: «Многие сходные соединения (но никогда аналоги и гомологи) имеют близкие удельные объемы» (со ссылками на работы Персо, Коппа, Шродера и многих других). «Это показывает, что некоторые сходные соединения стоят в ином отношении друг к другу, чем аналоги й гомологи.
343
fttfAfiA ЙОСЬМАЙ
Никель 42,4; кобальт 42,7; железо 44,6; медь 44,7
Серебро 128,3; золото 127,1
Платина 58,4; палладий 57,3; иридий 56,7
Хлор 334; бром 310 и иод 321
...Сличение удельных объемов многих других сходных соеди, нений показало, что для них увеличение и уменьшение пая влечет увеличение и в объемах. Этому закону подчинены гомологи аналоги и многие сходные соединения. Этот закон... в первый раз высказан был в 1854 г. Дюма по поводу предложения графического метода для открытия соотношения между весом пая и физическими свойствами» [11, с. 117—118].
По-видимому, не будет преувеличением сказать, что вслед за Дюма Менделеев стал искать законы связи между различными соединениями одного элемента и сходными (по форме) соединениями разных элементов.
Внимание Менделеева привлекла также работа американского ученого Дж. П. Кука, поскольку в ней получили развитие идеи Дюма (на них Кук прямо ссылается в своей статье, причем подобие элементов с органическими радикалами было едва ли не исходной идеей [16]. По-видимому, поиск закона, объединяющего элементы с разными свойствами, особенно занимал Менделеева (как следующий шаг после обобщения в виде двух законов Дюма). Не случайно поэтому рассуждения Кука он использовал в лекциях по теоретической химии, когда читал о законе четных паев [17].
Находясь в заграничной командировке в Германии (1859— 1861), посещая города Франции, Италии и Швейцарии, Менделеев, бесспорно, познакомился со многими учебниками, монографиями и статьями, в которых рассматривались способы определения атомных весов элементов и делались попытки осуществить различные систематизации. В учебнике «Органическая химия», написанном после этой командировки (1861), Менделеев привел таблицы атомных весов элементов, дал сведения о способах определения атомных весов.
Особую роль в определении атомных весов он отводил идеям С. Канниццаро и работам А. Реньо. Привлекла внимание Менделеева работа А. Штреккера [18] (эту брошюру Менделеев привез из заграничной командировки), в которой был дан обзор работ Дюма и других ученых по изучению разностей атомных весов элементов естественных групп и были сделаны попытки связать их с аналогиями в свойствах всех известных тогда элементов. Штреккер обращает внимание на изоморфизм солей многих металлов: «Мы можем, наконец, в группе следующих изоморфных металлов заметить почти равную разность:
Сг = 26,2 Мп = 27,6 Fe = 28 Ni = 29
Со = 30 Си = 31,7 Zn = 32,5» [18]. 1
344
ПЕРИОДИЧЕСКИЙ ЗАКОН
jfjTpeKKcp явно догадывался о наличии какой-то, еще неясной, закономерности. Он отмечал, что «отношения между атомными расами тогда только имеют постижимый смысл, если вещества находятся сами среди таких же (им подобных), а ряд, например
0 = 8, N=’14, Са = 20, Сг = 26,
где разность всегда 6, является без всякой внутренней связи» [18]. Эта фраза была подчеркнута Менделеевым в экземпляре, хранящемся в его личной библиотеке [19]. Но еще большее внимание привлекла такая мысль Штреккера: «Вышевыставлен-ные отношения между атомными весами (или эквивалентами) химически сходственных элементов, конечно, едва ли могут быть приписаны случайности. Но ныне мы должны предоставить будущему отыскание законности, проглядывающей между указанными числами». (Эту фразу мы даем в переводе Менделеева, приведшего ее в своей Фарадеевской лекции [3, с. 212]).
После возвращения в Россию в 1861 г. Мендеелев делает пространные выписки из сочинений Ж. Б. Дюма и Э. Ленсена. (По-видимому, он использует их в лекциях 1861 г.)
В те же годы Менделеев разрабатывает представление о формах химических соединений. Возможно, определенное влияние здесь оказала работа ученика Я. Берцелиуса шведского химика и минералога II. Норденшельда «Об атомно-химической системе минералов» [20]. В основу системы минералов шведский ученый положил соответствующие формы окислов3:
Н2О МпО А12О3 СО2 Sb2O5 SiO3 Mn2O7 OsO4.
Отношение Менделеева к ряду важных понятий химии, обсуждавшихся в то время, определяется тем обстоятельством, что он занимался изучением веществ в твердом состоянии. Как известно, в этом случае особенно остро вставала проблема определения истинного молекулярного веса. В учебнике «Органическая химия» Менделеев отмечал, что «атомность есть краткое выражение состава множества тел и что формулы, выражающие один элементарный состав (простейшие формулы.—Л. Л/.), недостаточны для сравнения и систематического описания тел». При этом Менделеев имел в виду явление полимеризации. В дальнейшем, в «Основах химии», он отмечал, что в газообразных водородистых соединениях, по которым определяли валентность элемента, находится только одни его атом: СН4, NH3, ОН2, С1Н, но в окислах металлов, например, необходимо учитывать полимерию: (МпОп)^.
В годы, предшествовавшие открытию периодического закона, Менделеев сумел найти некоторые принципы классификации соединений, которые он затем использовал для сопоставления от
3 Формула кремневого ангидрида определена неверно; соединение OsO4 открыто английским химиком С. Теннантом в начале XIX в.
345
ГЛАВА ВОСЬМАЯ
дельных элементов и их совокупностей. Он правильно оценил глубокие причины, определяющие их сходство и различие, отражением которых явилась специфика их соединений и учет агрегатного состояния.
Так, например, Менделеев сравнивает соединения кремния и углерода: «...большая часть их соединений столь резко отличает-ся, что составляет два противоположных предела известных нам сложных тел» [И, с. 175]. И продолжает далее: «...в телах органических несомненно существование сложных радикалов, в кремнеземистых соединениях нет нужды признавать их... Разнообразие органических тел, при небольшом числе элементов, изъясняется сложными радикалами, т. е. разнообразием расположения частей, что доказывается изомерностью. Разнообразие кремнеземистых соединений должно объяснять только количество и качество входящих элементов» [И, с. 215].
Разнообразие классификаций соединений и простых веществ затруднило выбор критерия для выявления классификации элементов в целом. Законы изменения свойств в группах и рядах, как теперь хорошо известно, был далеко не одинаковым.
ОТКРЫТИЕ ПЕРИОДИЧЕСКОГО ЗАКОНА
Задачу классификации Менделеев решил в 1869 г., открыв периодический закон. Это было подлинно революционное событие в науке, потребовавшее от ученого большой смелости, ибо нужно было выйти за рамки привычных представлений, совершить известную абстракцию, подняться от понимания «единичного» (специфика свойств одного элемента) через «особенное» (специфика изменения свойств определенной совокупности элементов) ко «всеобщему». Менделеев в решении проблемы систематики элементов исходил из возможности установления взаимосвязи того или иного элемента с другими, из представлений о том, что каждый элемент занимает определенное «место» в совокупности других и что существует определенный закон в изменении этих взаимосвязей. Расположив элементы в порядке нарастания атомных весов и приняв во внимание изменение свойств простых веществ и соединений (прежде всего, величины атомных объемов, изоморфизм и формы соединении), он убедился, что этот закон является периодическим.
Чтобы оценить значение этого открытия, нужно вспомнить, что на‘пути к его установлению лежало немало трудностей: и разные значения величин атомных весов у почти одной трети известных в ту пору 60 элементов, и прокрустово ложе учения о постоянной валентности, и пеизученпость отдельных элементов, особенно редких, и неудачи предшественников в решении проблемы систематики элементов.
346
ПЕРИОДИЧЕСКИЙ ЗАКОН
ОПЫТЪ СИСТЕМЫ ЭЛЕМЕНТОВ!),
ОСНОВАННОЙ НА ИХЪ АТОМНОМЪ ВЪСЬ И ХИМИЧЕСКОМЪ сходства.
Т1 = 50 Zr = 90 ? = 180
V = 51 Nb= 94 Ta=182
Сг = 52 Mo = 96 W = 186.
Mn = 55 Rh = 104,4 Pt = 197,4
Fe = 56 Ru = 104,4 k = 198
Ni=Co = 59 PI = 106,6 Os=199
Н= 1 Cu = 63,i Ag=108 Hg = 200
Ве= 9,«Mg = 24 Zn = G5,2 Cd = 11 2
В= 11 Al = 27,4 ?=68 Ur= 116 Au= 197?
С= 12 Si = 28 ?=70 Sn= 118
N = 14 P = 31 As = 75 Sb=122 B1 = 21O?
0=16 S = 32 Se = 79,< Те =128?
F = 19 С1 = 35,зВг = 8О 1=127
Li = 7 Na = 23 K = 39 Rb = 85,4 Cs= 133 TI = 204
Ca = 40 Sr = 87,6 ?=45 Ce = 92 ?Er = 56 La = 94 ?Yt = 60 Di=95 ’In = 75,6 Th = 118? Ba=137 Pb=2O7
Д. Мендел'Ьевъ
«Опыт системы элементов...», разосланный Д. И. Менделеевым русским и иностранным ученым
Но, конечно, самой большой трудностью, которая сказывалась не только в процессе открытия, но и развития периодического закона, являлось незнание истинных причин периодического изменения свойств. По этому поводу Менделеев вынужден был заметить: «...периодическая изменяемость простых и сложных тел подчиняется некоторому высшему закону, природу которого, а тем более причину ныне нет еще средства охватить. По всей вероятности, она кроется в основных началах внутренней механики атомов и частиц» [2, с. 384].
Умение учесть отражение внутренних причин периодичности, Которые неизбежно должны были проявиться в физико-химиче-
347
ГЛАВА ВОСЬМАЯ
III Jy . и названному
ских свойствах элементов, Менделеев показал при составлен таблиц, выражающих открытый им закон. Обратимся к первом варианту, опубликованному 1(13) марта 1869 г. и названном^ «Опыт системы элементов, основанной на их атомном весе химическом сходстве» [3, с. 9]. Если мы посмотрим па эту табли цу, то она поразит нас своей необычностью: все последующ^ варианты длинной формы построены по-другому. Возникает вон, рос: почему эта таблица имеет такую «причудливую» форму?
Есть основания утверждать, что «Опыт системы элементов.. >> выражает, с одной стороны, идею противопоставления двух наибе
лее полярных по своим свойствам групп — щелочных металлов и галогенов, а с другой стороны — идею постепенного перехода от совокупности элементов так называемого I разряда к сово-купности элементов II разряда. Эти два разряда были намечены Менделеевым в результате изучения зависимости свойств от атомного веса: «Все группы сходных элементов могут быть разделены на 2 главных разряда: в одних из групп сходные (подчеркнуто нами.— А. М.) элементы представляют значительное
различие в величине атомного веса... Другой разряд групп сходных элементов составляют такие, которые имеют близкие атомные веса» [3, с. 32, 34]. Нетрудно убедиться, что в первом случае речь идет об элементах-аналогах, которые составляют ныне
главные подгруппы системы, а во втором — об элементах «встав-
ных декад», т. е. элементах-аналогах, стоящих в одном ряду. Понимая возможность представить обе эти совокупности иным образом (другой таблицей), Менделеев писал: «Может быть система распределения элементов по группам, вследствие ближайшего изучения этих групп, изменится таким образом, что в определенных местах системы сходство будет наблюдаться между членами горизонтальных рядов, а в других частях системы между членами вертикальных столбцов» [3, с. 28].
Проявление различий в свойствах элементов указанных двух совокупностей есть результат отражения непознанных тогда еще внутренних особенностей атомов (с одной стороны s- и /?-элемен-ты, а с другой стороны — d-элементы). Так, в «первом разряде» оказались такие элементы, которые обладали близкими химическими свойствами, а разности атомных весов у элементов-аналогов были при этом значительными:
Li Na К Rb Cs
Al 23 39 89 133
АЛ 16 16 50 44
В пределах всей совокупности в целом наблюдались специфические особенности изменения свойств простых веществ и
348
ПЕРИОДИЧЕСКИЙ ЗАКОН
У;^г& V^$7 Ci^£
ji'^ty
У*" D—~ 5Я, /2^/^/
#-// .?-Л
C^/t J'./з/ jx w л* н ра ?/ ^4=^ f=/g /li~&
JQ^»i ХъЗу. Ai=tyf
?£,^f ?y&40?
?=» /Й. ж=/л £_-//< &)- //o' ^=/Z$V do-S/tf Sti^jf,
^~=l//^
G*sm
Черновик «Опыта системы элементов...»
ГЛАВА ВОСЬМАЯ
Фрагмент рукописи таблицы из седьмого издания «Основ химии»
соединений: переход от металлов к неметаллам — для простых веществ, зависимость кислотно-основных свойств гидроокисей от формы соединения и главным образом от природы элементов.
Во «втором разряде» оказались элементы, которые обладали близкими физическими свойствами (простые вещества) и близкими химическими свойствами (при степени окисления, равной 2) 4. В пределах этой совокупности также наблюдались особенности изменения свойств, в частности большое разнообразие форм соединений и зависимость свойств не столько от положения элемента в «разряде», сколько от формы соединений.
Эту же мысль Менделеев развивает в первом издании «Основ химии»: «...в ряду элементов есть два класса, сходственных между собой, в одном классе элементов сходственные вещества представляют постепенное увеличение в атомном весе, сообразно с постепенным изменением в характере и в свойствах соединений. Пример этому мы знаем уже в галоидах, щелочных металлах, в металлах щелочных земель и будем видеть еще над многими другими простыми телами. Другой разряд сходственных элемен
4 Валентность 2 нередко признавалась для них основной.
350
ПЕРИОДИЧЕСКИЙ ЗАКОН
тОв характеризуется тем, что при том большом сходстве, какое здесь существует, нет различия, или, правильнее сказать, нет значительного различия в величине атомного веса сходственных элементов. Причина различия в первом разряде сходственных элементов весьма понятна из значительной разпости в весе атомов сходных элементов, но для металлов второго разряда причина замечаемого различия не лежит уже в величине и в весе атома, а, конечно, в других внутренних различиях материи, входящей в состав атомов таких сходственных элементов, подобно тому различию, какое замечается между изомерными сложными телами. Между последними известна изомерия нескольких родов; один вид такой изомерии, называемый полимерностью, весьма легко понимается, потому что вес частицы полимерных тел пе одинаков... но есть другой род изомерии, называемый метамерностью. Метамерные тела имеют один и тот же вес частицы, по, между том, в пих распределение частей или атомов внутри частицы, несомненно, пе одинаково... Число подобных примеров в органической химии весьма велико... Так и между элементами, имеющими близкие атомные веса. Натрий (23), магний (24), алюминий (27), кремний (28), фосфор (31), сера (32) и др. представляют близкие атомные веса и при этом ясное различие в химическом характере, тогда как церий, лаптан и дидимий, так же как железо, марганец, пикель и кобальт, имеют столь же близкие атомные веса и, между тем, представляют много сходного в своем характере. В особых последующих главах мы увидим более разработанные примеры таких групп, чем та группа, о которой мы говорили выше» [21, с. 191—192].
Уже па этой стадии Менделеев делает вывод: «Элементы, расположенные по величине их атомного веса, представляют явственную периодичность свойств» [3, с. 30]. Об этом же он пишет впоследствии: «Первые мысли о периодичности вложены мною в листок, который 1 марта 1869 г. был послан многим ученым» 5 [22, с. 53]. Более пространная формулировка открытого им закона содержалась в «Основах химии»: «Физические и химические свойства элементов, проявляющиеся в свойствах простых и сложных тел, ими образуемых, стоят в периодической зависимости... от их атомного веса» [21, с. 907].
Результаты первого этапа работы Менделеев сообщил в статье «Соотношение свойств с атомным весом элементов», которая была Доложена от имени автора6 Н. А. Мепшуткипым 6(18) марта 1869 г. па заседании Русского химического общества. Примечательно, что в этой статье содержится уже фрагмент будущей
5 «Опыт системы элементов...» был напечатай на русском и французском языках.
Менделеев находился в это время в отъезде: по поручению Вольного экономического общества он занимался обследованием артельных сыроварен,
351
ГЛАВА ВОСЬМАЯ
«Естественной системы элементов»:
Si = 28 Ti = 50 ? = 70
Р =31 V = 51 As = 75
S = 32 Сг = 52 Se = 79
Cl = 35,5 Mn = 55 Br = 80 [3, c. 26].
Это позволило Менделееву в конце статьи написать: «Цель моей статьи была бы совершенно достигнута, если бы мне удалось обратить внимание исследователей на те отношения в величине атомного веса несходных элементов, на которые, сколько то мне известно... не обращалось почти никакого внимания» [3, с. 31],
Таким образом, Менделеев наметил переход от стадии «особенного» к стадии «всеобщего» в отыскании наиболее полной взаимосвязи между всеми элементами. Эта взаимосвязь между двумя совокупностями элементов проявляется, как он показал позднее, в высших формах соединений. Что же помешало распространить сразу этот принцип па большее число элементов, а не только на те, которые он использовал для приведенного выше примера? Менделеев дает ответ и на этот вопрос. Он пишет, что не решился предложить составление подобных же столбцов, так как убедился, что «останутся аналоги, несомненно принадлежащие к разным рядам. Достаточно указать на то, что Mg, Zn и Cd представляют много аналогий с Ca, Sr и Ва, а между тем перемешать эти тела в одну группу Mg=24, Са=40, Zn=65, Sr = 87,6, Cd = 112, Ba = 137 — значит нарушить естественное сходство элементов, как мне кажется» [3, с. 26—27]. В самом деле: между хлором и марганцем существуют принципиальные отличия, когда мы сопоставляем простые вещества и какие-либо сложные вещества, которые образуют указанные элементы. Но в одном случае, при образовании высших форм соединений, это различие максимально устраняется: налицо сходство свойств окислов, кислот, изоморфизм солей. В приведенном же примере с элементами будущей второй группы (а также и первой) такой близости свойств высших форм соединений не наблюдается, скорее наоборот: известная близость наблюдается у простых веществ (металлы). Не потому ли, желая проследить типы аналогий. Менделеев писал в уже упомянутой статье: «...я сам, лишь только дозволит мне время, обращусь к сравнительному изучению лития, бериллия и бора» [3, с. 31] (см. также [17, с. 18]). В той же статье он указывает на возможное обнаружение неизвестных еще элементов, следующих за алюминием и кремнием (кроме того, в «Опыте системы элементов...» значится еще аналог бора: ?=45), обращает внимание па возможность изменения атомных весов некоторых элементов (теллур) и выявление новых аналогов (уран как аналог бора и алюминия). Кроме того, в примечании, которое было внесено уже в корректуру статьи, он подчеркивает важное теоретическое значение своей таблицы
352
OFb АТОМНОМ! ОВЬЕМ! ПРОСТЫХ!» ПЛЪ.
Д Шядда^вйд,
М<=М <Ж«4» Жж«еву| 8? зх 8?,«
Ota- W>4 HbxsSM
14 Всеобщая история химии
ПЕРИОДИЧЕСКИЙ ЗАКОН
Ten» кжкъ рждЬдевнг 4дгмедтоеъ вл оеодвшй «нектрмчегких* м метллличгскт» смйетг», также к*г» и в* исжтши сшм-абвн-етв {«хшгагв воду совершенна вс удометждржетд. ecreemwojn ехидству, существ ввел у вежду жкипгжя ш мнх». ж такт, кшсъ лКшве ллемейтиг* в* «саовавш мхъ. такт, называемой. жтомит тк иоювамо м* сиверввпв» условных» донпцешахч., а «итожу оно я соедвялеп, eeylju» в» одму груват twt. ражшчиые лдежемты. «мт» К, Хж, Cl. F, г» в доохво считать иредшжашым до »нхъ верь системы аяемеэтом» кап» жжусствежяыж. т. е, оевгнпишыв ня одиожт. нля же мяишх» нрминакас». Рнснре.дкнчие алемгвтвп 1И> Величии! ИХ» ЯТОМИЖГ» BtCA МЪ ТОК* BH1L ьлгь (W били иреддожено ямов вь йровмежт» году. удожлетмрлегь требокандяжъ •WW стрнгей системы. win прединчтвоналмшж. но идмаки оно надставляло два важных* недостатка; войерних», — т.дтъ, что часть .иемегпнгъ,иямевж» «ернтияме члемепы. уран »> a наддй не находили вадлржашяго жЬста ю. зпЛ гнетем h. » «пому можно било девать, что нрняцпл* нертич»тк«Л завнеммистн смнВгтм* оп» величаем йтожкаго trfn* лежаний м, осш>«1, преддоженной ин»» системы. Не «тл«чается тою «бидноетъ». кака» д<ыжн& «читакт* i гайспи» нрввиввовъ естественной ок гемы; я жн»то|>им>. вт> т*»й 4**рм1. которая какались мн!> тогда киШк у.тобнн» для выражены» всЬх» (чмегпошснШ ирехгожениое «ноя» рвгяре.гКычпе <ut-менгнн* вред» тая.»жю блншин* пнюстив.н'ий' тдьнхт, »ндгь греки* зАгментоп»- каяь «(елочные металлы и галон w. которые »♦> химическому характеру напоил!» отличны между гобо», Оупн. гул. «|»слгтавяте1Н нгтиииыхъ ммадлою,, а други* самым* р1зк«\ъ чегаллондовъ; одни e«c,tiffiatwx ГН неЛсылиамт. ыднемчттмг. к»-«’Дорада» a друпе «т* «данвтсльныжъ; одни «^вмуют* гое адвент г* ВеДорОДоП», Я Ц»уна WC ДА»»П* ТйКНХ’Ь CW (й«ГН1Й -Ив два НС' достатка той Системы а»сМ«нтонъ. котором нцла нредлнжева мвнв «врвдншяяльже. жь «вегимадев время уже могут» быть устрамсам
Первые страницы раиит -посвященных периодическому закону
Пэ. гттД, дямЛжемжА л жуувкаХ Fyemrc Хюнме^аев >бшмт«а нкх» ид. стр. <i»i, « cnpatai «кж*»п wpfcjmeatjF* аамюшосп между сжЛстм» шежчтад « мемчадлп sen »wb*to Лж Вт иредлгвмю» cani » вжмДрвяд аоевлажт». ажэдяш*?
BHt груит» стеднагь «асмемтагь могут* бит* раахкммы жа 3 гялю» рвения ш ндвАхт яп n*F^* етвхвме ямямпи ^txmuuuiim амчжпэджа» рнлиж я* м-дж-wirt »тздпя» Ле»; сжим яранцмшкт Лммтястяо прайм»* Лхь « «*я могут* «ян* ]»е1фвд»4«ам во яигоогк «««мне Лс* г* труави еож.умяжме ежмнрвчеек». жяо пеайыамчдй а^одачеогу» аямежавет* ежФ-m от* явлжшжм j»uB—ry Лсж «да m кдяо м* «фшагммп» дфоИф»
Эя грунвы МММ 4м» 4» мпм* ЯМЙКМВ гамавпммяа**. «ямбы ме
ли» п mat* тмм тагам, морга умфем^вт* «миДжжтикмет* в* ямНймяав сжЛстя* Тщ* Я* <! »|»а--»Wim»l* «» и. ««К»Hw»* Ww>« J»*»». Ж »ИП|ЖяП
жкаСвя гаяиз мп дт » едаие» » X40J
22. Естественная система элемеятовъ и прнм$нен!е ея кг указажю своЙст неоткрытмхг элементт.
Mj»» м$о »...,
3, /А ГхиалЬу»#?» е«»бяил* о «еяла^аяж м . Сидолт. слумгаий для ткдучети ксиодн» бил* ; Ш-пр<> «докгядлда. Кгелидда* кявип. ври 2IC , уд. jy?,5. Ап»'твс< илвдх ялаватса иу»и 123 \ «внидг нр» Гамает* дфезрячдмй* жристалли, Ияедйитвы к« ♦йМдврохиая. шавелеваа и изотйая сели Вря в»м i (»лгтвмои,н л»е было врйяждеяо в<ч |.'л«л «стам*», «адае количество кряеталлт
/НСХЯ6^
ягрспДгв в врезвлеит» »г._, Оедоро»к/5>Й^«йй%р^>е-, атов* зшЛджшя сделкам елДдуюнйя сооСаммйа.
X Mweyteitee* от* жжеяя IT. II 3wwta доп<инветь,с»и>б; я* а»с1дшм» 11-го гсвткбра ’X смякши жъ«»илл <йг» ирв 160—1 во'' м ивмяляЭ». ЙроаеходмаиЙ, по pff ,НЛА «н.о «• с,й.о •+• с,,а,лл.«««дь г„н,Л<>., милигт* аадай ямяд* форяоведаяжжоД «моты:
ад.»Д ~ С..Н а гм!г 2Н»0.
1 Д. МеиЛглвга* соибйоипь, что жолжчсство кясдорода г* солевых* «кастах* определяете» воридв
-* «х* атома, что ввдио »* врммГрк ««PS fl
*= п. Cfto,
НЬШЪ ВЪСОМЪ Зл^.,
Д. Мендслгсйл, «о, ЗЁтьы pvetwo Х«пмн»»ж*г>» О**» че»* a * f
- «ал
1Чж»д«маТ«*Ъ'мое расер/дДлеяй» *•»«»..
рЖмте* «аука «мням* ршаопбрамшк* ар»вр»...
дЛ рмжрострвмеямое ршблемй» ах* Ш лимилм и *.»»««.. «ЛфМЗД ж»к* в» фя'жмежм р*а»*1», жыгДчввмыа жжц г йпм вроетыш гЬгам, таг* • ла раиячм в* хараатер» «Зия* я «иитктстижых* мм-u соедййеаД Ното, w кзлм<»с бермж* амяоястя» с* иредмет-'М*, *е«шм* жябоолотяы**, 1 &хяжа*м««* «каквмагЛ с* вим* ссяврвеяяо вотеряло ем»%'
Л»* »°P> *»»* стало* -* '»»* а»
мяр. фойфор*, мпжет* мала
-~*оска«* вмд Ь, стаж»
I
ГЛАВА ВОСЬМАЯ
ОСНОВЫ
2С 2££С JVC 1JE22 !
СО. чАСен^елгье^а,
ПГВ*ВССОРЛ И. fns УМЯЯГРСИГВ1А.
ЧАСТЬ ПЕРВАЯ, съ 151-мъ политипажемъ
С> Л КТКРВУГГЪ.
1S69.
Титульный лист первого издания книги Д. И. Менделеева «Основы химии» (1869)
ных свойств в группах и рядах системы. Это делалось как при подготовке открытия, так и после его осуществления. Так, например, знание закономерности изменения атомных объемов в рядах системы позволило изменить положение урана и поста-
и работы, затрагивающих «основные вопросы химии» [з с. 30], и отмечает, что ранее предлагавшиеся таблицы (па-пример, Одлинга) пе имели таких последствий.
В дальнейшем Менделеев публикует целую серию статей в которых развивает идеи, высказанные в марте 1869 г. В августе 1869 г. на II Съезде естествоиспытателей в Москве он делает доклад «Об атомном объеме простых тел», осенью того же года пишет статью «О количестве кислорода в соляных окислах и об атомности элемента» (публикуется в первом выпуске Журнала Русского химического общества за 1870 г.). Второй том «Основ химии», посвященный изложению свойств элементов, составляется на базе периодического закона.
Большое внимание Менделеев уделил изучению закономерностей изменения различ-
7 Понятие «атомный объем» было введено в 1821 г. Ройе и Ж. Б. Дюма. Величина атомного объема определялась путем деления значений атомного веса химического элемента и удельного веса образуемого им простого вещества. С введением понятия «молекула» (или «частица») было внедрено и понятие «молекулярный объем», и возник вопрос об аддитивности этого свойства. После того как С. Канниццаро (1858, 1860) предложил удельный вес считать свойством молекул, из которых состоят все вещества, стала ясна зависимость атомного объема от расстояния между атомами в молекулах. Эти расстояния, как очевидно, могут меняться в зависимости от агрегатных состояний и от аллотропии простого вещества.
Когда в 20-х годах нашего века путем рентгеноскопических исследований были получены новые значения атомных (ковалентных) и ионных радиусов, то выяснилось, что «так называемые атомные объемы не имеют реального значения, будучи частными от деления двух несоизмеримых величин: атомного веса химического элемента и удельного веса простого тела» [23, с. 309].
354
ПЕРИОДИЧЕСКИЙ ЗАКОН
в1ць на это место индий, а учет различий в ходе изменения точки ^давления простых веществ по группам системы позволил правильно предвидеть аномалии в точках плавления будущих гал-дия и германия.
Результаты проделанной за эти два года работы Менделеев сообщил в статьях «Естественная система элементов и применение ее к указанию свойств неоткрытых элементов» (ноябрь ^870 г.) и «Периодическая законность химических элементов» (1871). Вторая статья была опубликована в немецком журнале «Liebig’s Annalen». Получив оттиски этой статьи, 15 ноября 1871 г. Менделеев послал ее Ж. Дюма, А. Вюрцу, С. Канниццаро, Ж. Мариньяку, У. Одлингу, Г. Роско, К. Раммельсбергу, А. Байеру, К. Бломстранду и другим химикам 8. В письме к Дюма он писал 17 ноября 1871 г.: «То лестное для меня внимание, с каким Вы в Женеве встретили мои мысли о соотношении свойств элементов с их атомным весом, дает мне смелость препроводить при этом письме немецкий перевод всей статьи, только что присланной мне редакцией). Мне было бы весьма драгоценно узнать Ваше компетентное суждение теперь, когда Вы имеете возможность обсудить мои мысли в том направлении, какое видно из моей статьи». Письменных ответов ни от Дюма, пи от других химиков по существу не последовало.
Отношение к менделеевскому открытию было двойственным. Одним оно дало толчок к развитию их собственных работ и потому было встречено с интересом. Другим это открытие показалось чересчур смелым, и они старались находить в нем изъяны. Со временем число сторонников Менделеева во многих странах резко возросло и, как это нередко бывает в подобных случаях, возникли даже споры... о приоритете в открытии этого закона.
ФОРМИРОВАНИЕ И РАЗВИТИЕ ПОНЯТИЯ
О ПЕРИОДИЧЕСКОЙ СИСТЕМЕ КАК ФОРМЕ ВЫРАЖЕНИЯ ПЕРИОДИЧЕСКОГО ЗАКОНА
Закон, система и таблица — вот три основных понятия, связанных воедино и относящихся к проблеме взаимосвязи между элементами. Последовательность обнаружения этих категорий Менделеевым можно выразить так: разряды элементов, идея периодичности, опыт системы элементов, периодический закон, периодическая система.
Изучая работы Менделеева, можно проследить, как короткий вариант системы — в согласии с периодическим законом элемен
8 Летом 1871 г. Менделеев ездил в Германию и Швейцарию, где встречался со многими химиками, а также покупал нужные ему для исследования минералы.
355
14*
ГЛАВА ВОСЬМАЯ
тов — постепенно занимает главенствующее положение среди дру^ гих таблиц, как строится вся система понятий по отношению этой таблице.
В статье «К вопросу о системе элементов» (1871) Менделеев показал значение периодической системы: «Было бы правильнее мою систему называть «периодической», потому что она вытекает из периодического закона... В сопоставлении несходных элементов заключается также, как мне кажется, важнейший признак которым моя система отличается от систем моих предшественников. Как и эти последние, я принял, за небольшим исключением, те же группы аналогичных элементов, но при этом я поставил себе цель исследовать закономерность во взаимном отношении групп» [3, с. 388].
В третьем издании «Основ химии» Менделеев писал: «...важнейшее качество элемента познается не по низшим, а по высшей степени окисления... По этим причинам важнейшее значение для характеристики элемента имеют высшие формы образуемых им соединений... Хлор и марганец, металлоид и металл, образующие в низших окислах кислотный и основный гидраты С1(ОН) и Мп (ОН)2, в высшем дают одинаковые кислоты: хлорную С1О2<3>ОН и марганцевую МпО: 3 ОН, соли которых сходны и даже изоморфны... Так точно ванадий в своих высших формах VX5 очень сходен с фосфором РХ5, хром с серою... Так, каждый элемент, образующий несколько форм, представляет в каждой из них самостоятельные свойства и сходство с соответственными формами некоторых других элементов» [2, с. 343—344]. Эти методологические указания Менделеева дают недвусмысленный ответ на вопрос о том, как автор закона смотрел на иллюстрацию взаимоотношений между элементами. Поиск такого рационального варианта, который наиболее полно выразил бы основную идею — идею периодичности,— отражает путь нахождения специфических закономерностей (два разряда элементов), а затем и общего закона, объединившего все элементы (восемь типов форм соединений). Вот слова самого Менделеева: «При столь разнообразных отношениях, какие существуют между простыми телами, нельзя и думать систему их представить в виде одного непрерывного ряда» [3, с. 11]. Это не что иное, как попытка перейти от подчеркивания индивидуальности каждого элемента (непрерывный ряд) к выделению двух совокупностей элементов: «Может быть система распределения элементов по группам... изменится таким образом, что в определенных местах системы сходство ж будет наблюдаться между членами горизонтальных рядов, а в других частях системы между членами вертикальных столбцов» [3, с. 28].
В статье «Соотношение свойств с атомным весом элементов» Менделеев предложил несколько таблиц, но ни одна из них не была завершенной. Высказано и такое замечание: «Мне кажется
356
ПЕРИОДИЧЕСКИЙ ЗАКОН
притом наиболее естественным составить кубическую систему (предлагаемая есть плоскостная), но и попытки для ее образования не повели к надлежащим результатам». Тут же сказано 0 разных вариантах: «Подобных распределений возможно боль-jiroe число. Они не изменяют существа системы. Все, что выражается в этих системах, проглядывает и в тон, которую я выставляю как опыт подобных систем» [3, с. 22, 24]. Последние слова Менделеева также надо учитывать при оценке попыток, предпринимавшихся в то время. Все они были еще несовершенны, так как основные контуры системы (число элементов), да и принципы ее построения, ее структуры находились в стадии решения, поскольку еще продолжалось проникновение в сущность периодического закона (на первом, химическом, этапе).
Решающий шаг в обобщении этих попыток и в осуществлении перехода от стадии специфического, присущего той или иной совокупности элементов, к стадии всеобщего, характерного для всех элементов, в изучении законов изменения свойств был сделан Менделеевым в 1870 г. Для понимания этого момента особенно важны слова Менделеева: «...признают чересчур многое индивидуальным... связать эти индивидуальности общею идеею — цель моей естественной системы» [8, с. 618]. Короткая форма системы явилась прямым выражением этой идеи, этого перехода.
Как явствует из главных работ Менделеева, черты, фрагменты таблицы были ему ясны уже в самом начале работы над проблемой систематики элементов (это подтверждает и факт существования «полудлинной» таблицы).
Одним из важнейших вопросов на этом этапе являлось размещение в системе «переходных элементов».
Как уже отмечалось, выдвигая принцип объединения несходных элементов — повое по сравнению с предшественниками,— Менделеев столкнулся с той трудностью, что в случае Mg, Zn, Cd—Са, Sr, Ва пришлось бы объединить довольно сходные по химическим свойствам элементы.
Если обратиться к рассмотрению рукописи «Опыта системы элементов...», то, имея в виду только что отмеченное обстоятельство, можно предположить, что при распределении групп элементов и отдельных элементов в соответствии с атомными весами Менделеев руководствовался следующими взаимно согласующимися положениями:
1. Хотя первый вариант («Опыт системы элементов...») является так называемым длинным вариантом, тем не менее размещение некоторых, уже предсказываемых, элементов заставляет предположить и о существовании короткого варианта, хотя и не окончательно отработанного. Таким образом, как это явствует из ранее цитированной статьи «Соотношение свойств...», Менделеев начал осуществлять разрыв естественных групп и помещать в °Дну группу с ними элементы нынешних дополнительных под-
557
ГЛАВА ВОСЬМАЯ
групп. Эта попытка была еще не закончена к моменту опублц кования «Опыта системы элементов...», поскольку выявились трудности в размещении элементов I и II групп, часть элементов оказывалась за «контурами» системы (триады «VIII группы») не был решен вопрос о положении редких земель. ’
Если в предполагавшемся уже коротком варианте решились бы две первые проблемы, то церитовые металлы при таком рцс, пределенип оказались бы в центре системы. Вот, собственно когда возникла проблема их распределения в системе. Это-то и придало первому варианту «Опыта системы элементов...» столь причудливый характер. Следовательно, первая попытка решить возникшую задачу основывалась на прямом сопоставлении всех четырех выявленных триад, но не по химическим функциям, конечно, а по типу сходства.
Отсюда вытекает второе положение, из которого, как нам представляется, Менделеев исходил, составляя «Опыт системы элементов...».
2. Составляя «Опыт системы элементов...», Менделеев стремился наглядно выделить элементы со сходством второго типа, группируя их по краям системы, точнее ее костяка из повторяющихся — если взять короткий вариант — семи групп. Это оказалось возможным только по отношению к семействам Fe, Ru и Os. Но распределить все четыре триады можно только так, как это Менделеев представил в «Опыте системы элементов...», т. е. в верхней и нижней частях таблицы. Наличие пока в верхней части подгрупп Ti, V, Сг и Мп не должно смущать, они в дальнейшем займут места в соответствующих группах. Тогда в верхней части остаются только элементы будущей VIII группы. Что касается нижней части системы
? =45 ?Ег = 56 ?Yt = 60 ?In = 75,6 Се = 92 La = 94 Di = 95 Th = 118 [3, с. 9],
то, по мнению некоторых историков науки, Менделеев здесь расположил элементы, которым не нашел пока ясного места в системе.
Однако есть и другая точка зрения, основывающаяся на третьем выводе, содержащемся в статье «Соотношение свойств...», где Менделеев совершенно ясно указал, что сопоставление элементов выявило необходимость выделения ряда
Li, Be, В, С, N, О, F
и что свойства элементов этого ряда так или иначе повторяются в других рядах. В той же статье он пишет, что «...существует как бы период свойств простых тел
358
ПЕРИОДИЧЕСКИЙ ЗАКОН
Li= 7; Be—9,4; B=ll; С=12; N=14;
Na=23; Mg=24; Al=27,4; Si=28; P=31;
К=39; Ca=40; — Ti=50; V=51;
0=16;
S=32;
F=19
Gl=35?5
Ag=108; Gd=112; Ur=116; Sn=118; Sb=122; Te=128; J=127>
[3, c. 18].
Из дальнейшего изложения станет яспо, с какой целью он нарушает это очевидное — согласно периодической повторяемости — распределение и изменяет периоды так, как это представлено в «Опыте системы элементов...», где в одном периоде, отражающем по-прежнему костяк системы, находятся уже не семь, а восемь и девять элементов. Только так можно отразить два типа сходства (два разряда элементов), не нарушая основных взаимосвязей между элементами, составляющими костяк, т. е. элементами первого типа сходства. Правда, ври таком расположении сильно проигрывает наглядность тех, пожалуй, наиболее полных связей, которая достигается в коротком варианте при длине периодов в семь элементов. В этом и заключена альтернативность решения всей проблемы в целом.
Более того, появлялось как бы новое звено среди элементов второго разряда. Вот что пишет сам Менделеев: «Должно заметить, сверх того, что верхние члены четвертого столбца (Мп, Fe, Со, Ni, Zn) представляют переход (подчеркнуто нами.—Л. М.) к нижним членам того столбца, в котором находятся Са, К, С1 п т. п.; так, кобальт и никель, хром, марганец и железо представляют по свойствам и по атомному весу переход (подчеркнуто нами.—Л. М.) от меди и цинка к кальцию и калию. Может быть их положение должно быть вследствие того изменено, и они, вместо расположения в верхних рядах, поместятся вниз; тогда здесь получилось бы три столбца элементов, представляющих сходство во многих отношениях, а именно столбец, заключающий кобальт, никель, хром, марганец и железо; другой столбец — церий, лантан и дидимий, палладий, родий, рутений; наконец третий столбец, заключающий платину, иридий и осмий» [3, с. 27]. И далее: «Множество вопросов рождается при сопоставлениях в одно целое всех элементов, но самый интересный, мне кажется, вопрос составляет: распределение таких элементов, которые сходственны с железом, церием, палладием и платиной,— потому что здесь близкие по природе элементы представляют и близкие атомные веса... Может быть система распределения элементов по группам, вследствие ближайшего изучения этих групп, изменится таким образом, что в определенных местах системы сходство будет наблюдаться между членами горизонтальных рядов, а в других частях системы между членами вертикальных столбцов» [3, С. 28].
Приведенные выше слова Менделеева подтверждают нашу мысль. Действительно, можно предположить, пто Менделеев не
359
ГЛАВА ВОСЬМАЯ
случайно подставил Се=92, La=94, Di=95 (без знаков вопроса!) к Sr=87,6 — он столкнулся с нарушением последовательности атомных весов. Кстати, при таком расположении Се выступает как аналог неизвестного еще элемента с атомным весом 45 (что несомненно, могло быть рассчитано только на основании короткого варианта). Таким образом, Менделеев мог предположить наличие за элементом «? = 45» еще несколько элементов, аналогичных La, Di, Th= 118?. Однако вопросы справа и слева около символов Ег=56, Yt=6O, In=75,6 указывали, по нашему мнению, на сомнения Менделеева и относительно атомных весов, и относительно положения перечисленных элементов именно в этой части системы. Вот эти элементы, пожалуй, и относятся к тем, относительно которых можно сказать, что «они не нашли ясного места в системе».
Существенным подтверждением сказанного выше является изложение Менделеевым материала во второй части первого издания «Основ химии». По свидетельству самого Менделеева, «первые главы этого тома написаны в начале 1869 г. ...». Изучение этих глав дает отчетливое представление о том, что они обдумывались и писались Менделеевым именно в момент составления «Опыта системы элементов...» и вскоре после опубликования. В них чувствуется «поисковый» характер выяснения взаимосвязей между элементами. Так, пятая глава носит название «Цинк и кадмии (индий, цериты и гадолиниты)». В ней развивается идея разной степени сходства некоторых элементов с магнием. В предшествующей главе, правда, магний рассмотрен совместно с щелочноземельными металлами. Теперь же, при рассмотрении взаимосвязи с другими металлами, Менделеев указывает на связь с магнием таких металлов:
(I) Ba Sr Са<—Mg<—Zn Cd
(II) Си11 J FenMnnConNinl
(III) : In
: Ce La Di
: Yt Er Th
(Стрелками отмечено указанное Менделеевым изменение активности в ряду I.)
Свойства Fe ... Ni (ряд II) Менделеев подробно разбирает в следующей, шестой, главе. Наряду с указанием об определенном сходстве между этими металлами (изоморфизм солей М11) существуют и различия. «Это разнообразие в степенях окисления названных металлов,— пишет Менделеев,— придает им совершенно самобытный характер и заставляет группировать их в отдельности от магния и тому подобных металлов, неспособных давать различных степеней окисления» [21, с. 254]. Наконец, в третий ряд (In и др.) Менделеевым включены металлы, сходные «по всей
ПЕРИОДИЧЕСКИЙ ЗАКОН
вероятности, с магнием». Недостаточная изученность к этому времени индия, TR и тория не позволила Менделееву сделать окончательные выводы (TR — редкоземельный элемент).
Характерно, что все эти металлы концентрировались около двухвалентного магния (промежуточного по своей активности в ряду двухвалентных элементов), и задача состояла в том, чтобы выделить те отличительные признаки, которые позволили бы наиболее четко охарактеризовать место этих элементов в системе. Любопытно также, что в шестой главе Менделеев совершенно определенно указывает на переходный характер металлов ряда:
ВО сходно с Mg О
Ti V Cr Mn Fe^Co Ni Си
дают RO3
Об этом ряде Менделеев пишет, что Со и Ni по всем физическим и химическим свойствам составляют переход от Fe к Си. С другой стороны, Мп и Сг составляют переход от Fe к V и Ti. Указанные элементы дают окислы разной устойчивости: Fe и в особенности Сг образуют неустойчивые RO не только в свободном виде, но и в соединениях. Mn, Со, Ni образуют устойчивые окислы RO. Cr, Мп, Fe образуют R2O3, для Со и Ni эта форма неустойчива. Си ее вообще не дает. Cr, Мп, Fe дают и более высокие формы окислов, устойчивые для Сг и Мп и неустойчивые (RO3) для Fe.
Так намечается представление о формах соединений и их роли в понимании взаимосвязи элементов, в понимании количественных характеристик места элемента в системе.
Учитывая все изложенное выше, можно прийти к следующему выводу: идея периодической повторяемости, лежащая в основе «Опыта системы элементов...» и достаточно ясно выраженная в статье «Соотношение свойств...», наиболее четко выражалась графически — в длинном или коротком варианте — при исключении из рассмотрения церитовых металлов. Менделеев выдвинул идею о металлах, осуществляющих переход от одного периода к другому. Особую роль в этом переходе играли звенья особенно сходных элементов (будущие триады VIII группы).
Возникали и другие вопросы, требующие своего разрешения. Главным среди них был вопрос о свойствах и месте церитовых металлов, что должно было привести, в свою очередь, к решению проблемы строения всей системы в целом. Возникал вопрос о изображении связи между элементами одной группы (в дальнейшем: четные и нечетные ряды — по Менделееву, главные в дополнительные — по Л. Мейеру).
Обратимся теперь к выяснению того, как представление о «переходных» элементах позволило отразить идеи периодичности в периодической системе элементов.
361
ГЛАВА ВОСЬМАЯ
Во второй части «Основ химии», написанной в самом начале 1869 г., Менделеев отмечал: «Галоиды и щелочные металлы составляют в некотором смысле самые крайние по характеру элементы» [21, с. 95]. Надо заметить, что при составлении плава «Основ химии» Менделеев исходил из возможности разделать большинство элементов в соответствии с их атомностью на 4 группы. Правда, при этом он замечал: «Однако нельзя резко различить элементы по их сходству с II, О, N и С, или, как говорят, во их атомности, потому что в природе элементов больше разнообразия, чем в четырех описанных нами» [23а, с. 651].
Убеждение в этом росло у Менделеева тем более, чем чаще оц обращался к другим свойствам (помимо атомности) и к сравнению изменения этих свойств в зависимости от атомных весов элементов.
Решающим шагом в сопоставлении исходных элементов явилось сравнение Менделеевым свойств К и С1, а затем и целых групп щелочных металлов и галогенов [24].
Еще в первых главах второй части «Основ химии» (подготовленных до создания «Опыта системы элементов...») и в ряде черновых набросков Менделеев пытался представить себе взаимосвязь между некоторыми известными естественными группами со стороны перехода от одной группы к другой через некоторые «переходные металлы». Как мы увидим далее, эта мысль связапа пока с делением элементов по атомности. Он, например, пишет в начале 4-й главы, что к щелочным металлам примыкает ряд металлов, переходных к двухатомным, точно так же к двухатомным щелочноземельным металлам примыкает «ряд переходных металлов». Несколькими строками дальше Менделеев делает знаменательное замечание об отличии в некоторых характеристиках, с одной стороны, щелочных и щелочноземельных металлов, а с другой — переходных металлов: «Было бы, конечно, последовательнее описать сперва эти переходные (от щелочных к щелочноземельным.— А, М.) металлы, но мы этого не делаем по той причине, что настоящие щелочноземельные металлы имеют и большее распространение в природе, и более явственную характеристику, для них пет тех сомнений, какие существуют иногда для переходных металлов. Точно так же, как между щелочными и щелочноземельными металлами существуют переходные члены, так точно между щелочноземельными металлами и металлами, образующими еще высшие степени окисления, еще более сложные атомные соединения, существует ряд переходных элементов, п только принимая во внимание эти переходные члены, есть возможность составить себе ясное, представление о соотношениях различных групп металлов между собой» [21, с. 120—121]. •
Это замечание Менделеева интересно в двух отношениях: с точки зрения сложностей, с которыми он столкнулся затем при «сортировке» элементов будущих дополнительных подгрупп по группам системы (па основе валентности), а также с точки зрения
362
ПЕРИОДИЧЕСКИ!! ЗАКОН
отыскания «короткого варианта», где костяком являлись бы семь (вначале четыре) основных групп [24, с. 345—349].
Таблицы, составленные Менделеевым 17 февраля 1869 г., ясно указывают на намерение ученого отойти от прежнего плана размещения элементов.
Как показывает «Опыт системы элементов...», датированный тем же числом, решение все же было найдено, однако, как мы уже отмечали выше, именно не выясненная до конца проблема размещения некоторых переходных металлов пе позволила представить окончательного варианта.
Наиболее устойчивым звеном, не претерпевшим каких-либо существенных изменений при составлении различных первоначальных вариантов, является звено из тех четырех групп, которые Менделеев рассматривал как исходные при составлении плана написания «Основ химии», а именно:
С Si — Sn
N Р As Sb Bi
О S Se Те
F Cl Вг J
Затем к группе галогенов (см. «Опыт системы элементов...») были подключены группы щелочных металлов, щелочноземельных металлов, а также ? — 45 и Се = 52 (последние два элемента Менделеев считал трехвалентными). Таким образом, основной костяк системы составляют группы с четко выраженными валентностями и расположенные по принципу нарастания атомных весов:
C=12 Si=28 ?=70 Sn =118
N=14 P=31 As=75 Sb=122
0=16 S=32 Se=79,4 Te=128
F=19 Cl=35,5 Br=80 J=127
Na=23 К=39 Rb=85,4 Cs=135
Ca=40 ?=45 • Sr=87,6 Ce =92 Ba=137
Сопоставление этой таблицы с приведенной на стр. 349 показывает, что наиболее трудным оказался переход от группы углерода к другим группам элементов с уменьшающейся валентностью. Затруднения вызваны пе только отсутствием некоторого числа элементов, прежде всего будущей третьей группы, по и неясностью валентности у бора и алюминия, которые нередко рассматривались и как трех-, и как четырехвалентные. Если учесть и тот факт, что следующие за ними элементы «подгруппы» цинка и меди рассматривались Менделеевым как «переходные элементы», то надо признать, что вся верхняя часть таблицы, стоящая вслед за звеньями триад будущей VIII группы, носит такой же переходный характер Жак элементы с переменной валентностью), хотя Менделеев в
363
ГЛАВА ВОСЬМАЯ
этот период и пытался уже, как мы видели, размещать некоторые из них по основным группам. О роли сходных свойств триад ре Ru и Os, а также «церитовых металлов» при определении их по’ ложения в системе мы уже говорили. ‘
Если принять во внимание сказанное выше, а также выявлен-ные Менделеевым, но нерешенные проблемы отражения взаимосвязей между элементами, то можно наметить в «Опыте системы элементов...» следующие три звена групп9 элементов, которые соответствуют замечаниям Менделеева, содержащимся в статье «Соотношение свойств...». Первое звено составляют основные группы, приведенные им на стр. 18 и 20 в [3], хотя они выписаны по-разному: на стр. 20 так, как даны в «Опыте системы элементов...»:
Са—40 Sr=87,6 Ba=137
Na=23 Ka=39 Rb=85,4 Cs=133
F=19 Cl=35,5 Br=80 J=127
0=16 S=32 Se=79,4 Te=128
N=14 Р=31 As=75 Sb=122
С=12 Si=28 Sn=118
а на стр. 18 — с учетом начинавшегося сдваивания рядов (см. стр. 359 данной главы). При этом явно бросается в глаза то, что Менделееву, по-видимому, оставалось недостаточно ясным выделение одной из основных групп — группы, состоящей из трехвалентных элементов. Поэтому в таблице на стр. 20 элементы этой группы не приведены, т. е. Менделеев ограничивается сопоставлением шести основных групп.
Второе звено составляют те переходные элементы, которые обнаруживают сходство второго типа: близость свойств при близких атомных весах, т. е. триады будущей VIII группы и церитовые металлы.
Третье звено — также переходные элементы, но те, которые Менделеев пытается за счет сдваивания рядов поместить среди основных, или естественных, групп.
Как же Менделеев осуществил сдваивание рядов и переход к короткой форме таблицы? Периоды (ряды), наметившиеся в «Опыте систем элементов...», как мы отмечали выше, сформировались именно в таком виде в результате попытки поместить на границах системы элементы со сходством второго типа (три триады Fe, Ru, Os и церитовые металлы). Однако при таком распределении основных* элементов в периодах Менделееву пришлось столкнуться . с «ненормальным ходом» изменения свойств. Мы имеем в виду его замечание в сноске [3, с. 21] к статье «Соотношение свойств...», где говорится о водородистых соединениях элементов. (Состав
9 Т. е. речь идет не о месте отдельных элементов, а о «групповом» месте, или месте звеньев в системе.
364
ПЕРИОДИЧЕСКИЙ ЗАКОН
NaH кажется Менделееву неестественным, он ставит вопрос, почему не образуется соединение состава NaH7.)
Все чаще Менделеев обращается к такому изображению системы, которое он намечает в таблице, помещенной на стр. 18 в [3]. Впрочем, там не случайно пропущены ряды, в которые по величине атомных весов должны попасть «церитовые» металлы 10.
Вопрос о сдваивании рядов и переходе к короткой форме таблицы нашел довольно полное отражение главным образом в работах Б. М. Кедрова. Опубликованные им в 1950 г. архивные материалы [25] позволили довольно точно представить основные этапы поисков системы, которые были предприняты Менделеевым в 1869—1871 гг. Мы хотели бы здесь остановиться на некоторых сторонах решения этого вопроса, тесно связанного с проблемой «переходных» элементов.
Обратимся прежде всего к таблице так называемых четно- и нечетноатомных элементов, составленной Менделеевым на одном из первых этапов работы над созданием короткой формы системы [3, с. 26] (см. стр. 359). На наш взгляд, составление такой таблицы и ее анализ потребовались Менделееву для решения вопроса о числе элементов в рядах (короткая форма), т. е. необходимо было определить границы между вторым и третьим типами эле
ментов.
В первом издании «Основ химии» и в первых работах о периодическом законе Менделеев отмечает, что такие элементы, как Ti и V, должны быть поставлены в число аналогов элементов естественных групп. Относительно Сг и Мп полной уверенности в правильности такого же решения (как единственного) не было. Многие предшественники Менделеева подчеркивали близость Сг и Мп, в особенности последнего, к Fe.
На первых порах Менделеев хотел решить вопрос, подсчитав число элементов в рядах. Анализируя разности в изменении атомных весов, он сопоставил число нечетно- и четноатомных элементов:
Нечетноатомные Четно атомные ДА
ДА
Li=7 В=11 4 о Ве=9 С=12 3
N-14 3 Г" 0=16 4 о
F=19 5 Mg=24 о
Na=23 4 Si=28 4 4
Al=27 4 S=32
Р-31 4 г Са=40 8
С1=35 4 Л Ti=50 (см.11) 10 а
К-39 4 Fe=56 о
10 Это, конечно, с учетом изменения атомных весов.
11 Атомный вес Ti тогда считался равным 50.
365
ГЛАВА ВОСЬМАЯ
Прежде всего обращало на себя внимание постоянство разности атомных весов у нечетноатомных элементов (АЛ=4). Разности между атомными весами четпоатомных элементов, стоящих в одном ряду, также равны 4, по при переходе к новому ряду они возрастали до 8. В каждом ряду было четыре печетноатомных и три четноатомных элемента, что можно схематически представить так:
Отмеченные выше скачки в разности атомных весов при переходе к новому ряду навели Менделеева на мысль о возможности существования еще одного четного элемента в первых двух рядах (Х=20 и Х=36). Не вступая в полемику по поводу интерпретации этих данных, мы хотели бы подчеркнуть предположение Менделеева о возможности нахождения в одном ряду равного числа четноатомных и нечетноатомных элементов. Однако, как видно из приведенной выше таблицы, изучение разностей атомных весов у элементов следующего ряда уже привело к определенным трудностям. Прежде всего, составляя таблицу, Менделеев включил три четноатомных элемента второго ряда (по аналогии с предыдущими) : Са=40, Ti=50, Fe=56. Первый четноатомный элемент ряда — кальций — имеет атомный вес, на единицу превышающий атомный вес предшествующего нечетноатомного элемента (К=39). То же наблюдается и в других рядах (Li=7, Ве=9; Na=23, Mg=24). По атомный вес Ti оказался непомерно высоким (Ti=50), что, даже с учетом в дальнейшем исправленного 12 на 48, представляет аномалию при сравнении с уже известными значениями АЛ, равными 4. Не потому ли в первое время Менделеев думал, что здесь не хватает нескольких элементов13, и отразил это в некоторых заметках? То, что Менделеев не случайно ставил железо третьим в ряду, т. е. рассматривал его как аналог О и S, находящихся в предшествующих рядах, подтверждается мыслями о свойствах железа, которые изложены Менделеевым в других работах («Основы химии», планы исследований).
Неясным также оставалось и положение хрома. Складывается впечатление, что Менделеев колебался в определении его характерной валентности: 6(СгО3) или 3(Сг2О3). По-видимому, не слу
12 Кстати, это подозревал уже Менделеев (см. [21, с. 765]).
13 Мысль, до некоторой степени оправданная тем, что при переходе от щелочноземельных металлов к металлам с большой валентностью Менделеев столкнулся с церитами, имеющими близкие атомные веса.
366
периодический закон
чайно в одном из черновых вариантов системы Менделеев пытается поместить хром в третью группу на место будущего скандия.
Анализ, проведенный Менделеевым, был поучителен. Он указывал на большую сложность изменения атомных весов при переходе к рядам с переходными элементами и как следствие — на недостаточность изучения одних разностей в атомных весах при определении места некоторых звеньев в системе.
При чтении первых глав второй части «Основ химии» складывается впечатление, что первоначально Менделеев принимал как бы два способа выражения сходства элементов в виде таблицы: не только четные и нечетные ряды (что намечается уже в «Опыте системы элементов...»: Na и Mg подключены к аналогам Си и Zn соответственно), но и деление на две подгруппы (как это впоследствии принял Л. Мейер). Так, в шестой главе «Медь и серебро» Менделеев пишет: «При сходстве атомного состава соединений закиси меди и окиси серебра с соединениями щелочных металлов представляется, однако, и значительная мера разницы между этими двумя разрядами элементов. Эта разность видна ясно в том, что щелочные металлы принадлежат к числу элементов, чрезвычайно легко соединяющихся с кислородом, разлагающих воду и образующих самые крайние электроположительные14 элементы, тогда как серебро и медь трудно окисляются, образуют в ряду металлов электроотрицательные члены и воды не разлагают... Различие тех и других видно далее в неодинаковости свойств многих соответствующих соединений» [21, с. 195]. Для сравнения с щелочными металлами Менделеев указывает на нерастворимость в воде окислов, хлоридов и ряда других солей серебра и меди.
Тем не менее можно считать, что в это время Менделеев не видел резких различий в природе элементов главных или дополнительных подгрупп (или четных, или нечетных рядов), хотя и высказал ряд соображений, с одной стороны, о сложности элементов и необходимости познать «внутреннюю механику атомов», а с другой — о различии некоторых закономерностей изменения свойств обоих разрядов элементов.
Сознавая наличие целого ряда индивидуальных отличий в свойствах групп, звеньев и отдельных элементов, Менделеев на основе такого общего свойства их, как формы соединений (высших кислородных, а также водородных), все же считал возможным дать графическое выражение общей взаимосвязи элементов в виде короткой формы таблицы. В 18-й главе «Основ химии», где он рассмотрел свойства Sn, Ti, Zr и Th, приводится часть такой таблицы. Она выделена, по-видимому, как не вызывающая сомнений:
14 Подчеркнуто нами. Это суждение Менделеева мы считаем крайне важным для решения вопроса о строении системы (длинный вариант).— А. М.
367
ГЛАВА ВОСЬМАЯ
R2O5 "1 Р=31 As=75 Sb=122 fa
V=51 Nb=94
ro2 ] ' Si=28 Ti=50 ?=72 • Zr=90 Sn=118 1 IV
R2O3 - f Al=27 ?=44 ?=69 . ?Yt=88 In-113 I HII
RO f Mg=2i Ca--40 Zn=-(»5 Sr=87 Cd=112' г11
Комментируя эту таблицу, Менделеев пишет: «При этом очевидно, что такие элементы, которые написаны в нижних строках групп, образуют при одинаковом содержании кислорода окислы более основного характера, кислоты — более слабые, чем окислы тех элементов, которые написаны в верхних строках каждой группы. Никакого нет сомнения, что такие отношения не случайны...» [21, с. 766].
И в следующей, 19-й, главе, где описаны платиновые металлы, он заключает: «...в каждой группе элементов, не считая вышеупомянутые типические элементы, встречаются элементы двух родов.
Таким образом, два ряда элементов образуют период: например, соответственно типическому ряду Li, Be, В, С, N, О, F, встречаем следующие два ряда: Rb, Sr, —, Zr, Nb, Mo как ряд более основных элементов и следующий за ним ряд Ag, Cd, Zn, Sn, Sb, Те, J — элементов более кислотного характера, что видно при сравнении соответствующих по форме окисления соединений Rb и Ag, Sr и Cd, Zr и Sn, Nb и Sb, Mo и Те... Таким образом, хотя в Ag, Cd и прочих повторяются многие (особенно количественные) свойства Rb, Sr..., так что от Rb до Ag замечается малый период элементов, ио оба ряда от Rb до J включительно образуют один большой период» [21, с. 781—782]. К этому Менделеев добавляет, что в первом ряду большого периода («малый период элементов») повторяются только формы окисления, наблюдаемые в типическом ряду, во втором же ряду повторяются и формы соединений с водородом. Триады VIII группы находятся в каждом большом периоде, т. е. повторяются через ряд.
Здесь же Менделеев приводит таблицу больших периодов, в одном из периодов он оставляет места для редкоземельных элементов, которые он предполагает рассматривать в плане уже выявленных закономерностей (типические элементы — элементы больших периодов). Правда, несколько ранее в статье «Естественная система элементов...», датированной 20 ноября 1870 г.15, говоря о больших периодах, Менделеев замечает, что если ранее (1869 г., «Опыт системы...») проблема больших периодов вызвала известные затруднения, то теперь она «служит главною причиною общности системы», и далее заключает: «Вероятно, что существует еще и больший
15 Последние главы «Основ химии» были написаны в начале 1871 г.
368
ПЕРИОДИЧЕСКИЙ ЗАКОН
период, состоящий из 4-х рядов... (подчеркнуто нами.— А. М.) [3, с. 78].
Таким образом, анализ основных свойств элементов (атомные веса, формы соединений), дополненный указаниями на характер дх изменения (в результате изучения изменений атомных объемов) и другими сведениями о сходстве и различии (изоморфизм, химические свойства простых тел и соединений), позволил Менделееву от «Опыта системы элементов...» перейти к «Естественной системе элементов». Нельзя забывать, что, публикуя таблицу под таким заголовком, Менделеев дал в «Основах химии» не только короткий, но и длинный вариант системы, так как,по-видимому, на основании изученных свойств он не имел возможности отдать предпочтение одному из них. Возможно, к этому он и не стремился. Короткий вариант подчеркивал близость элементов при сравнении таких общих свойств, как формы соединений, иллюстрировал эволюцию свойств с точки зрения форм соединений. Длинный вариант представлял большой простор для раскрытия особенных черт, присущих прежде всего звеньям переходных элементов, причем в варианте, представленном Менделеевым, это касалось, по существу, только триад восьмой группы.
СОЗДАНИЕ УЧЕНИЯ О ПЕРИОДИЧНОСТИ
В ноябре 1877 г. вышло в свет третье издание «Основ химии» Д. И. Менделеева. Это издание примечательно тем, что в нем впервые содержалась глава, специально посвященная изложению периодического закона. Опубликование этой книги является крупной вехой в формировании учения о периодичности. Напомним, что первое издание «Основ химии» появилось в 1868—1871 гг. и быстро разошлось. Второе издание, выпущенное в двух частях в 1872 и 1873 гг., мало чем отличалось от первого. В этих двух изданиях была дана формулировка открытого в феврале 1869 г. закона, прилагались «Опыт системы элементов...», а затем и «Естественная система элементов...»; в различных главах, посвященных описанию свойств элементов, приводились «некоторые мысли о периодичности» (использованы слова Менделеева, сказанные об «Опыте системы элементов...»).
По существу оба издания выражали ту информацию о периодическом законе, которая была получена в условиях «цейтнота», если иметь в виду обстоятельства открытия закона. При подготовке первого издания Менделеев использовал стенографическую запись курса, прочитанного им в 1868/69 учебном году студентам Университета. Более того, первоначальное содержание курса, который Менделеев впервые стал читать за год до этого, повторяло содержание курса, прочитанного А. А. Воскресенским в 1850/51 учебном году в Главном педагогическом институте. Об этом можно судить по конспекту лекций, составленному тогда Менделеевым, и допол
369
ГЛАВА ВОСЬМАЯ
нениям, внесенным в этот конспект позже, при подготовке собственных лекций.
Третье издание готовилось по-иному. За четыре года (1872— 1876), в течение которых велась подготовка нового издания «Основ химии», Менделеев проделал большую работу по углублению содержания сделанного им открытия, что и привело к формированию основ учения о периодичности.
В Предисловии к третьему изданию «Основ химии» Менделеев писал: «Знания, относящиеся к количественной стороне химических превращений, далеко опередили изучение качественных отношений. Связь этих двух сторон, по моему мнению, составит нить долженствующую вывести химиков из лабиринта современного уже значительного, но отчасти одностороннего запаса данных. Такая связь лежит в основе той системы элементов, которой подчинено все мое изложение. Когда (в 1869 г.) я предлагал ее в первом издании этого сочинения, во мне еще не было полной уверенности в общей применимости основного начала, выражаемого словами: свойства атомов и частиц зависят прежде всего от их массы. Теперь эта уверенность родилась» [2, с. 318].
Указав далее на осуществленные им в 1871 г. предсказания, на их подтверждение, выразившееся в исправлении атомных весов урана, индия и «церитовых» металлов, в открытии галлия, Менделеев продолжал: «Убедившись в правдивости основного начала, я провожу его в этом издании строже, чем было в двух предшествующих (подчеркнуто нами.—Л. М.). Но все же я понимаю, что истинный путь дальнейшего развития нашей пауки еще не найден, что скоро в ней должно ждать больших изменений» [2, с. 319].
Впервые сформулированная глава двадцать седьмая третьего издания «Основ химии» носила название «Сходство элементов и их система (Изоморфизм. Формы соединений. Периодический закон. Удельные объемы)» и была помещена между главами, посвященными элементам II группы (Двадцать шестая глава. Щелочноземельные металлы. Двадцать восьмая глава. Цинк, кадмий, ртуть). Для сравнения скажем, что в первом издании «Основ химии» эти вопросы рассматривались в разных главах, а также в «Заключении». Это одна из центральных глав, объемом 32 страницы [26, с. 829—861], которая в дальнейшем, при переизданиях, претерпела незначительные изменения (начиная с пятого издания изменилось название главы: «Сходство элементов и периодический закон»). То, что эта глава помещена в связи с изложением материала об элементах подгруппы цинка и подгруппы магния, не является случайным. Структура системы и обстоятельства открытия закона —вот, что отразилось в таком построении книги.
Названная глава начинается с рассмотрения вопроса о мере сходства и выборе измеримых количественных параметров. Таковыми признаны: изоморфизм, отношение объемов соединений, состав солеобразных соединений и отношение в весе атомов. Ока
570
ПЕРИОДИЧЕСКИЙ ЗАКОН
зывается, что эти четыре предмета и будут кратко обсуждены в рассматриваемой главе. При этом с самого начала Менделеев четко определяет свою методологическую позицию относительно соотношения между различными объектами: «...в химии... можно уже сравнивать не одни отдельные тела и их реакции: есть признаки, которые относятся к самим элементам. А понятие об элементе есть такое отвлеченное, как и понятие об атоме. Можно переносить сходство соединений данного элемента па самый элемент и проверять это сходство по такому признаку, как атомный вес (лучше говорить — элементарный вес), который принадлежит самому элементу, а не тем формам, в которых он является» [26, с. 829].
Этими словами Менделеев вновь, как и в первых работах по периодическому закону, ставит вопрос о коренных свойствах атома и взаимосвязи конкретных свойств простых и сложных веществ со свойствами атомов, из которых на первое место он ставит атомный вес.
При рассмотрении первого из измеримых параметров — изоморфизма — Менделеев особое внимание уделил сходству многих металлов с магнием. Этот вопрос теспо связан с самой историей открытия системы элементов, ибо, как нетрудно видеть из таблицы «Опыт системы элементов...», ко II группе примыкали как элементы «железного ряда», так и редкоземельные элементы, тогда признававшиеся двухвалентными.
Взяв фрагмент об изоморфизме из пятой главы второй части прежних изданий «Основ химии» («Цинк и кадмий»), Менделеев добавил теперь примеры, которые свидетельствовали о том, что изоморфизм служит признаком сходства только в определенной степени окисления (сравниваются Mg и Мп).
Рассмотрев различные случаи взаимосвязи кристаллической формы и состава соединений, а также приняв во внимание другие свойства и химические аналогии, Менделеев писал: «...ныне есть уже полное право сказать, что сходство форм, то есть изоморфизм и гомеоморфизм, наступает не только при полном сходстве атомного состава и свойств, но и при подобии свойств, отвечающем аналогии атомного состава, эквивалентности и преобладающему влиянию одних составных начал над другими. Этим дело усложняется, теряется та простота, которая господствовала при учении Митчерлиха о чистом изоморфизме. Но понятия Гаю были еще проще, а, однако, должны были уступить место тем усложнениям, которые введены знакомством с диморфизмом и изоморфизмом. Очевидно, значит, что все понятие об отношении между составом и кристаллическими формами подлежит еще дальнейшей разработке и усовершенствованию. Один изоморфизм не может выражать сущности этих сложных отношений, потому что не указывает перехода от одних форм к другим; понимание сущности дела только тогда и может наступить, когда будем знать ие только причины и
371
ГЛАВА ВОСЬМАЯ
меру сходства, но и явления, сопровождающие различие форм цри близости или известном отношении состава» [26, с. 839].
Завершая этот раздел главы 27-й, Менделеев выразил уверен-ность, что наступит время, когда эта область физико-химических исследований будет занимать важнейшее место в ряду текущцх вопросов науки.
Таким образом, можно констатировать, что на примере изоморфизма стала очевидной необходимость рассмотрения взаимосвязи свойств макроскопических тел со свойствами атомов и природой связи между ними. Только созданная на базе электронной теории строения веществ кристаллохимия смогла решить указанные Менделеевым задачи.
Аналогичные проблемы возникли при изучении форм соединений, в том числе молекул или кристаллов. «Как там, так и здесь оказывается немного форм, различных по существу... Там частицы слагаются в кристаллические формы, здесь атомы в частичные формы или в формы соединений. Там и здесьиз основной кристаллической или частичной формы происходят видоизменения, соче тания, комбинации» [26, с. 841].
Обсудив вопрос о формах водородных (RX—RX4) и кислородных соединений (R2O—RO4), а также их взаимосвязи (H„R— HnRO4), Менделеев высказал правило четности: «Если элемент R дает высшую форму RXn, то часто существуют низшие формы RXn_2, RXn_4 и вообще такие, которые отличаются от формы RXn на четное число X» [26, с. 842].
Далее Менделеев рассматривает одно из важных свойств окислов промежуточных степеней окисления — их способность к распаду на соединения с высшей и низшей степенями окисления (т. е. к реакциям диспропорционирования).
Памятуя о роли, которую Менделеев отводил коренным свойствам атома (элемента) в понимании свойств соединений, следует обратить внимание и на такое его замечание: «...важное качество элемента познается не по низшим, а по высшей степени окисления» [26, с. 842].
В качестве примера Менделеев приводит соединения хлора и марганца и пишет: «Конечно, в низшей форме масса элемента R, соединенного с Хп, преобладает, а потому в низшей форме качества R выражаются яснее, чем в соединениях того же R с большим количеством Х-ов. Это особенно ясно из того, что все высшие формы имеют кислотный характер... Здесь, несмотря на разность элементов, присутствие большей относительной массы кислорода влияет на свойство образующегося окисла» [26, с. 843]. В подтверждение тезиса о преобладании качества R в низших формах Менделеев приводит пример сходства Т12О с К2О. Таким образом, выдвигается тезис о потере индивидуальности элемента при переходе к высшим формам соединений, что становится особенно
372
ПЕРИОДИЧЕСКИЙ ЗАКОН
важным при интерпретации короткой формы системы, которая строится на высших формах соединений.
Рассмотрев вопрос о формах соединений, Менделеев переходит к вопросу о формулировке периодического закона, предварительно подчеркнув роль атомного веса как коренного свойства атома, в зависимости от которого должны быть рассмотрены все другие свойства.
Приведя в таблице три группы (галогены, щелочные и щелочноземельные металлы — «в этих трех группах видна сущность дела»), Менделеев продолжает: «...если все элементы расположить по величине их атомного веса, то получится периодическое повторение свойств. Это выражается законом периодичности: свойства простых тел, также формы и свойства соединений элементов находятся в периодической зависимости... от величины атомных весов элементов» [26, с. 847].
Этим, по нашему мнению, как бы завершается одна часть главы — тот ее раздел, который был связан с описанием (или раскрытием, хотя более строго это дано позже) взаимосвязи свойств простых и сложных веществ с коренными свойствами атома.
Далее следует изложение системы представлений, раскрывающей содержание периодического закона как бы с другой стороны, основанной на использовании периодической системы. Речь идет о рассмотрении, наряду с общими, специфических и индивидуальных свойств элементов.
Центральными в этой системе представлений, сложившейся в эти годы, оказались представление о свойствах элементов, стоящих в четных и нечетных рядах (два закона изменения свойств пли два типа элемент-аналогии, начало рассмотрению которых положил Ж. Б. Дюма), а также понятие о месте элемента в систем ме. Если последняя уже рассматривалась в ряде статей по периодическому закону (1871), то обсуждение специфических закономерностей изменения свойств элементов, стоящих 1) в четных и 2) в нечетных рядах системы, в столь полном виде впервые дается Менделеевым в третьем издании «Основ химии».
«Четные и нечетные ряды одинаковых групп,—пишет Менделеев,— представляют одинаковые формы, но по свойствам различаются... Совокупность двух рядов, четного и рядом стоящего нечетного, дает, таким образом, один период... Элементы двух первых рядов, обладающих наименьшим атомным весом, вследствие этого самого обстоятельства хотя имеют общие свойства группы, но при этом и много особых, нередко резко выраженных, свойств. Так, фтор... отличается многим от других галоидов, литий... от щелочных металлов и т. д. Эти легчайшие элементы можно назвать типическими. Сюда относятся Н, Li, Be, В, С, N, О, F» [26, с. 847-848].
Далее следует важное с методологической точки зрения раскрытие сущности учения о периодичности: Менделеев формули
373
ГЛАВА ВОСЬМАЯ
рует семь положений, относящихся к формам и свойствам кислородных п водородных соединений, металлоорганических соединений, понятию элемент-аналогпи и разностям в атомных весах месту элемента в системе, способу определения атомных весов ц других свойств через место элемента в системе, сопоставляя при этом описываемые свойства с принадлежностью данного элемента к той (четные ряды) или иной (нечетные ряды) совокупности элементов.
Наконец, как это отмечалось ранее, одним из важных положений, особенно четко сформулированных в новой, 27-й, главе, является положение о месте элемента в системе: «Каждый элемент по периодической системе имеет место, определяемое группою (означаем римскою цифрою) и рядом (цифра арабская), в которых находится. Они указывают величину атомного веса, аналогию, свойства и форму высшего окисла, водородного и др. соединений, словом, главные количественные и качественные признаки элемента, хотя затем и остается еще цельш ряд подробностей или индивидуальностей (подчеркнуто нами.—4. Af.), причину которых, по смыслу всего учения, лежащего в основе системы, должно искать в небольших разностях величины атомного веса» [26, с. 849—850].
Менделеев раскрывает этот тезис на примере вычисления атомного веса элемента, окруженного четырьмя соседями по группе и ряду (взят селен) и на примере предсказания свойств экасили-ция. В дальнейшем он неоднократно обращается к рассмотрению принципов сопоставления элементов, например элементов, стоящих не в соседних группах, а в ближайших четных или нечетных и т. д. [27] (см. подробнее [28]).
Известно, что в ряде статей, опубликованных в 1869—1872 гг., Менделеев затрагивал важные для понимания свойств элементов вопросы: изменение объемов атомов при образовании соединений разных классов, теплоемкость простых и сложных веществ, полимеризацию и т. п. Эти вопросы особенно интересовали Менделеева по двум причинам: 1) они давали путь для понимания «природы» разных элементов, тесно связанной с учением о периодичности, 2) давали объяснение реакционной способности («энергичности») одних веществ и пассивности других.
Понимая роль химической динамики, учения о химическом процессе в формировании системы химических знаний, Менделеев хотел к этой области приложить те принципы, которые он положил в основу периодической системы (масса и форма соединения). Этим объясняется то обстоятельство, что его ученики занимались .изучением «пределов» протекания химических реакций замещения (Г. Г. Густавсоп и А. Л. Потылицын) и изучением зависимости скорости химических реакций от массы реагирующих веществ (Ю. Е. Богуский, Н. Н. Каяндер).
Последовавшие в 1875 г. открытие галлия и в 1879 г. открытие скандия имели важное значение для утверждения периодического
ПЕРИОДИЧЕСКИЙ ЗАКОН
закона и менделеевских предсказаний. Работы, выполненные химиками разных стран по изучению периодичности свойств элементов, также способствовали утверждению новой системы понятий.
Весной 1880 г. в «Журнале Немецкого химического общества» была опубликована статья Менделеева «К истории периодического закола» [28]. В историческом плане цель статьи связана с намерением русского ученого разрешить приоритетные вопросы, возникшие после признания периодического закона. В этом отношении статья является прямым продолжением ранее опубликованной статьи 1871 г. [29] и связана с оценкой вклада ученых, в том числе Лотара Мейера, в развитие учения о периодичности. В логическом плане это выступление Менделеева должно быть рассмотрело как важная веха в формировании учения о периодичности. Действительно, анализ третьего издания «Основ химии» (1877), сравнение его с четвертым изданием (1881—1882) показывают, что двум выступлениям в иностранной печати (1879 —в Moniteur scientifi-que...» и 1880 г.— в «Berichte») Менделеев отводил особую роль. Это не просто пересказ или повторение ранее высказанных мыслей. Во-первых, Менделеев здесь впервые вводит понятие «учение о периодичности» [3, с. 404], во-вторых, дает краткую сводку о развитии представлений о коренных свойствах элементов, в-третьих, впервые отмечает роль изучения периодичности изменения физических свойств (температуры плавления, магнитные свойства), в связи с чем отмечены особые заслуги английского химика Т. Карнелли, являвшегося учеником Г. Роско, в-четвертых, подчеркивается, что из всех химиков, занимавшихся «сличением величин атомных весов элементов», Менделеев наиболее обязан двум: Э. Ленсену и Ж. Б. Дюма. Наконец, Менделеев дает новую форму (таблицу) выражения закона, которую считал «за наилучшее и полнейшее выражение гармонии элементов, или периодического закона, и в то же время за наиболее удобную в типографском отношении» таблицу [3, с. 405].
УТВЕРЖДЕНИЕ ПЕРИОДИЧЕСКОГО ЗАКОНА. ОТКРЫТИЕ ПРЕДСКАЗАННЫХ МЕНДЕЛЕЕВЫМ ЭЛЕМЕНТОВ
Утверждение периодического закона в пауке представляет яркий пример того, как происходит восприятие научных открытий. Совершив свое замечательное открытие в начале 1869 г., Менделеев в течение последующих лет упорно работал над тем, чтобы привести в соответствие с периодическим законом возможно большее число фактов. Эта работа требовала от ученого огромного мужества, так как идея периодичности не сразу встретила сочувствие в научных кругах. «Ваша система,— писал один из корреспондентов Менделеева из Бонна,— считается здесь [Кекуле] остроумной спекуляцией, не дающей, однако, прямых выводов».
375
ГЛАВА ВОСЬМАЯ
Не было недостатка и в иронических и даже откровенно враждебных высказываниях. Именно этот кропотливый и многолетний труд, опирающийся на глубокую убежденность в правильности и общности открытого Закона составляет существо научного под-вига Менделеева, о котором говорил Ф. Энгельс16.
Постепенно, однако, ученые разных стран начинают понимать огромную значимость открытия Менделеева. Хотя мотивы, по которым они становятся последователями взглядов Менделеева, во многом различны, тем не менее всех их можно поставить в один ряд людей, которых Менделеев называл «укрепителями» или «утвердителями» периодического закона. Автор периодической системы очень ценил этих ученых, старался всячески поддержать их работу, не упуская возможности указать на их заслуги, в особенности при подготовке новых изданий своего фундаментального труда «Основы химии».
В первом ряду «укрепителей» стоят, разумеется, ученые, открывшие новые элементы, предсказанные Менделеевым: в 1875 г. П. Э. Лекок де Буабодран открыл галлий (экаалюминий), в 1879 г. Л. Ф. Нилсон — скандий (экабор), в 1886 г. К. А. Винклер —германий (экасилиций).
Открытие галлия
Судьба открытия каждого из перечисленных выше элементов была очень различна. Наиболее драматичным оказалось, как это, впрочем, и следовало ожидать, открытие экаалюминпя — галлия. Французский ученый П. Э. Лекок де Буабодран (1838—1912), прекрасный химик-аналитик, был одним пз пионеров использования в химической практике спектрального анализа. Темой его исследований были активные металлы: щелочные, щелочноземельные и трехвалептпые — типа алюминия, индия и таллия. Благодаря способности этих элементов переходить в пар легко было изучать их свойства, исследуя спектры их паров. Лекок де Буабодран установил вполне четкую закономерность изменения спектров с ростом атомного веса: чем тяжелее металл, тем более сдвигается его спектр в длинноволновую область. «Так, спектр рубидия аналогичен спектру калия, но только сдвинут в красную область»,— писал он в статье, опубликованной в 1865 г. [30, с. 446].
«Буабодран был фактически первым, кто начал рассматривать эту проблему, так сказать, на атомном уровне, поскольку он анализировал характер изменения двух свойств элементов — атомного веса и длин воли спектральных линий» [31, с. 20].
В той же статье 1865 г. Лекок де Буабодран отмечал, что «...закономерный ход спектральных линий будет полезен для установления с помощью простого спектрального анализа не только существования новых тел, но и их свойств» [30, с. 446—447].
16 Энгельс Ф. Диалектика природы. М.: Политиздат, 1953, с. 42—43.
376
ПЕРИОДИЧЕСКИЙ ЗАКОН
Такая же, как и при изучении щелочных металлов, закономерность наблюдалась и для трехвалентных элементов, с той лишь разницей, что между алюминием и индием был явный разрыв. Это обстоятельство и побудило Лекока де Буабодрана искать новый элемент. В 1868 г. он приступил к поискам, выбрав в качестве объекта цинковую обманку, в которой еще в 1863 г. также спектральным путем был открыт индий.
Одновременно он продолжал работу над критериями применения спектров сходных по химическим свойствам элементов, опубликовав в 1870 и 1871 гг. статьи [32, 33], которые обратили внимание Менделеева, сделавшего соответствующее добавление при подготовке второго издания «Основ химии» [34, с. 85—86].
27 августа 1875 г. П. Э. Лекок де Буабодран сделал свое замечательное открытие, а 20 сентября того же года опубликовал о нем сообщение [35]. «Исключительно малое количество вещества не позволило мне отделить новое тело от избытка цинка, который является его спутником» [35, с. 493]. Тем не менее он заметил появление новых спектральных линий и зарегистрировал новый элемент. Его выделением и изучением физико-химических свойств Лекок де Буабодран занимался последующие три месяца в лаборатории А. Вюрца. В результате в ноябре 1875 г. оп выделил новый элемент, который был назван галлием, а в феврале 1876 г.— после получения в достаточном количестве и соответствующей очистки — смог определить некоторые его физические свойства и сопоставить их с теоретическими предсказаниями [36, с. 86—89]. Удельный вес металла, найденный при работе с образцом весом 64 мг, оказался равным 4,7 при 15° С.
Как только в Петербурге был получен французский журнал с сообщением Лекок де Буабодрана, Д. И. Менделеев, ознакомившись с этим открытием, сообщил о новом элементе на заседании Русского физического общества (4 ноября) и Русского химического общества (6 ноября). 5 ноября 1875 г. Менделеев отправил Ж. Б. Дюма свою «Заметку по поводу открытия галлия» [37] с сопроводительным письмом, в котором писал: «Если мнение о идентичности галлия с одним из предсказанных элементов оправдается, то моя заметка может представлять, на мой взгляд, некоторый интерес». В своей статье Менделеев показал, как на основе периодического закона могут быть вычислены свойства элемента. Так, например, удельный вес металла должен быть равен 5,9.
Существенно отметить, что предсказания свойств галлия Менделеев сделал па основе широкого сопоставления свойств элементов как по вертикали (Al, —, Tn, Т1), так и по горизонтали (Си, Zn, —, —, As, Se), а также с учетом диагональных свойств. Третья группа вообще обладает переходными свойствами, и одна интерполяция по вертикальному ряду А1—In—Т1 представляется недостаточной. Она может привести к неверным заключениям, о чем, кстати говоря, знал и Лекок де Буабодран.
377
ГЛАВА ВОСЬМАЯ
Таблица 1. Свойства галлия
Предсказаны Менделеевым (1871)
Определены Лекок де Буабодраном (1875—1876)
Экаалюминий (Еа)
Атомный вес около 68
Простое тело должно быть низкоплавко
Удельный вес близок к 5,9 Атомный объем 11,5
Не должен окисляться в воздухе
Должен разлагать воду при краснокалильном жаре
Формулы соединений: ЕаС13, Еа2О3, Ea2(SO4)3
Должен образовывать квасцы Ea2(SO4)3 • Me2SO4 • 24Н2О, но труднее, чем А1
Окись Еа2О3 должна легко восстанавливаться и давать металл, более летучий, чем А1, а потому можно ожидать, что Еа будет открыт при помощи спектрального анализа ЕаС13 — летуч
Галлий (Ga)
Атомный вес 69,72
Точка плавления свободного галлия 30 °C
Удельный вес 5,904 (тв.); 6,095 (ж.)
Атомный объем 11,7
Слегка окисляется только при красном калении
Разлагает воду при высокой температуре
Формулы соединений: GaCl3, Ga2O8, Ga2(SO4)3
Образует квасцы NH4Ga(SO4)2-12H2O
GaO3 восстанавливается из окиси прокаливанием в токе воздуха; открыт при помощи спектрального анализа
GaCl3 — т. кип. 215—220 °C
Французский ученый повторил свои опыты и действительно подтвердил число, соответствующее значению удельного веса, данному Менделеевым. Это обстоятельство произвело огромное впечатление на широкую научную общественность во всем мире. Периодический закон, которому до этого верили очень немногие, приобрел неожиданно характер точного закона естествознания. Открытие Менделеева стали сравнивать с предсказанием существования планеты Нептун по отклонению орбиты уже известной планеты Уран.
И все же в более поздних работах (1877 и 1881) Лекок де Буа-бодран писал, что, не зная об открытии Менделеева, он самостоятельно пришел к заключению о существовании элемента в ряду алюминий—индий и что знакомство с предсказаниями Менделеева только бы затруднило выделение этого металла, потому что химические свойства галлия (в частности, отношение его солей к аммиаку и соде) не занимают срединного положения между свойствами алюминия и индия. Кроме того, по мнению французского ученого, температура плавления галлия (30° С) также оказалась неожиданно низкой [38].
Определение свойств галлия, проведенное вскоре, подтвердило правильность основных предсказаний Менделеева (табл. 1), кото
378
ПЕРИОДИЧЕСКИЙ ЗАКОН
рый позже писал: «Если бы не было указаний периодической закономерности, так бы и считалось долго, что галлии имеет удельный вес 4,7, как было определено сперва, потому что ничто бы не указывало на неверность определения, ничто бы не побуждало проверять трудное получение и выделение галлия» [39, с. 262]. В ряде других работ Менделеев показал, какие соображения о характере изменения свойств элементов им были положены в основу предсказания свойств галлия, в частности его низкой температуры плавления. Именно об этом писал Менделеев в марте 1879 г. редактору журнала «Le Monileur scientifique» доктору Кепевиллю, решившему ознакомить французских читателей со знаменитой статьей русского ученого «Периодическая законность химических элементов», которая была уже ранее переведена на немецкий язык (1872). В этом письме, которое было предпослано статье, сказано: «Открытие металла галлия, сделанное Лекок де Буабодраном, которого теперь я имею честь считать в числе моих друзей, может рассматриваться как утверждение периодического закона и считаться одной из блестящих страниц в летописях пауки» [40, с. 393].
Открытие скандия
Открытие экабора — скандия знаменательно тремя существенными обстоятельствами, связанными с содержанием учения о периодичности:
1. Если галлий, открытый в 1875 г., относится к числу р-эле-ментов и элементов естественных групп, составивших в таблице Менделеева главные подгруппы, то скандий был первым d-элемен-том, открытым после 1869 г.
2. И галлий, и скандий относились к элементам третьей группы, формирование которой особенно затянулось [41].
3. И галлий, и скандий являются не только редкими, но и рассеянными элементами. Однако если галлий был открыт при помощи чувствительного физического метода — спектрального анализа, то скандий обнаружен химическим путем.
Первое обстоятельство было важно для формирования учения о периодичности с точки зрения развития представлений о двух типах элементов или двух типах сходства, отмечаемых еще в работах Ж. Б. Дюма.
Второе обстоятельство имеет прямое отношение к развитию представлений о структуре периодической системы и формам графического выражения периодического закона.
Наконец, третье обстоятельство оказалось важным для последующих открытий не известных еще элементов.
В опубликованной Менделеевым в марте 1869 г. таблице «Опыт системы элементов, основанной на их атомном весе и химическом сходстве» было отмечено место для будущего элемента: «?=45». Позже о свойствах «экабора» Менделеев писал: «...пра-
379
ГЛАВА ВОСЬМАЯ
Таблица 2. Свойства скандия
Предсказаны Менделеевым (1871)
Определены Нилсоном (1879—1880)
Эка бор (ЕЬ)
Атомный вес 44
Окись должна иметь формулу ЕЬ2О3 Гидроокись нерастворима в щелочах Сульфат малорастворим
Должен давать соль, вряд ли изоморфную с квасцами
Скандий (Sc)
Атомный вес 44,1
Формула окиси Sc2O3
Гидроокись нерастворима в щелочах
Sc2 (SO4)3 малорастворим
K3Sc(SO4)3 кристаллизуется в призмах, а не в октаэдрах, как квасцы
вильное предугадывание свойств могло случиться именно только при допущении того изменения в атомных весах церитовых и гадолинитовых элементов, которое было одним из первых пунктов применения периодической системы элементов к фактическому запасу химии» [42].
Именно в этом — в определении правильных атомных весов редких элементов — состояла выдающаяся роль прогнозов Менделеева.
В марте 1879 г., т. е. спустя 10 лет после первых предсказаний Менделеева и пять лет после открытия галлия — первого из предсказанных Менделеевым элементов, появилась публикация шведского химика Л. Ф. Нилсона (1840—1899) «О скандии — новом редком металле», помещенная в шведском, немецком и французском журналах [43, 44]. Однако первоначальное представление о скандии было у автора далеким от действительности. Он предполагал, что этот элемент с атомным весом 160—180 занимает место между оловом и торием.
Правильное место скандия в системе сумел определить другой шведский химик —П. Т. Клеве (1840—1905). В статье, представленной Парижской Академии наук в августе того же года, Клеве верно указал валентность скандия, равную трем, и тождество свойств этого элемента со свойствами экабора [45]. Тогда же Клеве известил письмом Менделеева, с которым познакомился в 1877 г. в Упсале на 400-летнем юбилее Университета, о своих наблюдениях: «Имею честь сообщить Вам, что Ваш элемент экабор выделен. Это - скандий, открытый Нилсоном весной этого года...» [46}. В ответном письме Менделеев с удовлетворением писал: «Возвратившись в Петербург, я нашел Ваше интересное письмо, которое меня особенно обрадовало, потому что напомнило то внимание, какое Вы обратили на мой закон периодичности и его следствия... Признаюсь Вам, что я не ждал, что на моем веку придется видеть такое оправдание, какое дают следствия периодическому закону...» [47].
380
ЙЕРИОДИЧЕСКЙЙ ЗАКОЙ
- — — ! .1. . — 1 П«|4
В 1879—1880 гг. в шведском и немецком журналах появляются большие статьи Клеве [48] и Нилсона [49], посвященные описанию свойств скандия и его соединений. Из этих статей можно заключить, что основная трудность в признании формулы окиси скандия ЕЬ2О.? состояла в понижении основности Sc2O3 по сравнению с Y2O3, тогда как для исследованных окислов редкоземельных элементов наблюдалось понижение основности с увеличением молекулярного веса.
Сопоставление свойств экабора и скандия дано в табл. 2.
Это сопоставление дало основание Нилсону писать: «...не остается никакого сомнения, что в скандии открыт экабор... так подтверждаются самым наглядным образом мысли русского химика, позволившие не только предвидеть существование названного простого тела, но и наперед дать его важнейшие свойства» (цит. по [50]).
Открытие германия
Весьма поучительная история произошла с открытием германия, когда, после получения некоторых его соединений, встал вопрос о положении его в системе.
Вначале немецкий химик К. Винклер (1838—1904), открывший экасилиций — германий в аргиродите, думал, что этот элемент является металлоидом, стоящим по свойствам близко к мышьяку и сурьме. На эту мысль наводило поведение сернистых соединений германия, аналогичное поведению сульфидов элементов V группы [51, 52].
Это заблуждение смог разъяснить профессор химии в Бреславе В. фон Рихтер, который 25 февраля 1886 г. сообщил Винклеру, что «...германий, название которого Вы должны сохранить... как фактический его отец, есть предсказанный Менделеевым элемент экасилиций, Es = 73, паипизший гомолог олова, стоящий в первом большом периоде между Ga(69,8) и As (79,9)... Экасилиций — элемент, которого мы ждем с величайшим напряжением, и во всяком случае ближайшее исследование германия явится самым определенным experimentum crucis для периодической системы» (цит. по [53]).
В письме к Винклеру от 14(26) февраля 1886 г. Менделеев писал: «...растворимость хлористого германия в воде и характерный белый цвет сернистого германия указывают на иное положение германия, вследствие чего желательно попытаться определить его как экакадмий.
...Большая летучесть самого германия и большая летучесть его соединений не позволяют считать его за экасилиций... хотя другие свойства довольно близки к свойствам [последнего]» [54].
Таким образом, Менделеев правильно оценил более металлический характер нового элемента (экакадмия или экасилиция),
381
ГЛАВА ВОСЬМАЯ
но из-за недостатка данных не мог сделать окончательного вывода хотя и считал, что он более похож на экасилиций.
Заслуживает внимания и ответное письмо К. Винклера от 5 марта 1886 г., которое мы приводим с некоторыми сокращениями: «По составу аргиродита я вычислил, что атомный вес германия должен был быть много ниже такового сурьмы и что, таким образом, элемент не может стоять между сурьмой и висмутом, как я сперва предполагал. Я забыл Вам сказать, что предположение о том, что есть предсказанный Вами экасилиций, высказано не мною, а В. фон Рихтером в Бреславе, и он об этом сообщит в „Berichte der deutschen chemischen Gesellschaft44. В письме ко мне, почти одновременно с ним, это же предположение высказал и Лотар Мейер в Тюбингене.
Первое мое сообщение о германии содержало столь недостаточные данные, что более или менее правильный вывод о том, какое место занимает элемент, едва ли было возможно сделать, и потому Вам... он напоминал экакадмий. До сих пор мне еще не удалось установить атомный и удельный веса нового вещества, и потому вопрос о том, какое место занимает оно в периодической системе, должен оставаться открытым до тех пор, пока это не будет точно установлено. Но все мои наблюдения, сделанные за это время, указывают на то, что германий стоит ближе к олову, чем сурьме, т. е. он принадлежит к группе четырехвалентных элементов, и все говорит за то, что это действительно экасилиций.
Теперь же, когда я работаю с большим количеством чистого материала, мне удалось удалить чрезвычайно мешающие и загрязняющие примеси — мышьяк и сурьму, и я нашел, что летучесть германия не так велика. Расплавить мне его не удалось; далее я установил, что существуют не один, а два сульфита, из которых высший, вероятно GeS2, в осажденном, прокаленном и возогнанном состоянии — белого цвета. Низший же сульфид, GeS, напротив, имеет темную окраску и по своей летучести и поведению при нагревании может быть легко принят за трехсернистую сурьму. Последнее обстоятельство всегда и вводило в заблуждение.
Хлорид германия — бесцветная жидкость, которая кипит около 100° или даже ниже, совершенно так, как Вы предсказывали для хлорида экасилиция. Иодид — оранжево-красный, плавкий и летучий. Вчера мне удалось получить германий в кристаллическом виде. Хотя кристаллы и видны невооруженным глазом, но все они малы; под микроскопом видно, что это — прекрасные октаэдры. Таким образом, германий, подобно углероду и кремнию, принадлежит к правильной системе. Однако здесь пока не исключена .возможность ошибки...» [55].
Вот почему Менделеев, повторяя уже не раз высказывавшуюся им мысль, вспоминает об истории открытия германия: «Разнообразные периодические отношения принадлежат элементам, а не
382
ПЕРИОДИЧЕСКИЙ ЗАКОН
Таблица 3. Свойства германия
Предсказаны Менделеевым (1871)
Определены Винклером (1886)
Экасилиций (Es)
Атомный вес около 72
Удельный вес около 5,5
Атомный объем около 13
Темно-серый тугоплавкий металл
Окись будет иметь состав EsO2 Кислоты почти не будут действовать
С щелочами будет реагировать легче, чем с кислотами
Хлорид экасилиция — ЕьС14, точка кипения, вероятно, несколько ниже 100 °C, уд. вес около 1,9 (0°С), уд. объем около 113
Германий (Ge)
Атомный вес 72,3
Удельный вес 5,469
Атомный объем 13,2
Серовато-белый металл с т. пл. 959 °C
Окись имеет состав GeO2
С НС1 не реагирует, с HNO3 образует осадок гидрата окиси
При кипячении с щелочами постепенно переходит в раствор
Хлорид германия — GeCl4, т. кип. 86 °C, уд. вес 1,887 (18 °C), уд. объем 113,3
простым телам, и это весьма важно заметить, потому что периодический закон относится к элементам, так как им свойствен атомный вес, а простым телам, как и сложным, частичный (молекулярный.— А. М.) вес. Физические свойства определяются преимущественно свойствами частиц (молекул.—Л. 71/.) и только посредственно зависят от свойств атомов, образующих частицы. По этой-то причине периоды, ясно и совершенно резко выраженные, напр., в формах соединений, уже до некоторой степени усложняются в физических свойствах. Так, напр., кроме maxima и minima, отвечающих периодам и группам, являются новые частные maxima и minima; так, в температуре плавления германия является местный maximum, который, однако, предвиделся периодическим законом при определении его свойств (экасилиция)» [56, с. 392].
В начале 1887 г. Винклер опубликовал данные об определении многих других свойств германия [57].
Сопоставление свойств экасилиция (Es) и германия (Ge) дано в табл. 3 *.
❖
В ироцессе формирования учения о периодичности начался пересмотр некоторых сложившихся к тому времени понятий («элемент», «простое вещество», «валентность»). В рамках учения
* Более подробно история открытия элементов, предсказанных Д. И. Менделеевым, изложена в книге: Кедров Б. М. Прогнозы Д. И. Менделеева в атомистике. Т. 1. Неизвестные элементы. М.: Атомиздат, 1977. 263 с.— Прим. отв. ред.
383
ГЛАВА ВОСЬМАЯ
Титульный лист пятого издания книги Д. И. Менделеева «Основы химии» (1889)
о периодичности сформировались новые понятия: «периодичность», «система элементов» «периоды», «ряды», «группы элементов», «место элемента в системе», было развито представление об особенностях изменения свойств определенных совокупностей элементов, относящихся к тем или иным фрагментам системы, дано новое определение понятия «элемент».
«...Теперь часто смешивают понятия простого тела с понятием об элементе,— писал Д. И. Менделеев,— а между тем, чтобы избегнуть путаницы в химических представлениях, эти понятия должно строго отличать друг от друга. Простое тело — есть нечто материальное; это металл или металлоид, обладающий данными физическими свойствами... Понятию простого тела отвечает молекула... Оно может существовать в изомерных и полимерных видоизменениях и от химически сложного тела отличаться лишь однородностью материальных частиц, его образующих. Элементы суть материальные составные части простых и химически сложных тел, которые обусловливают их* физическое и химическое отношение... Углерод — элемент, но уголь, графит и алмаз — простые тела» [58, с. 199-200].
Периодический закон дал толчок к накоплению экспериментального материала, к обоснованию таких количественно измеримых свойств, как атомный объем (В. Траубе), изоморфизм (Г. Чер-мак, А. Арцруни), формы соединений (Ф. М. Флавицкий), валентность (Л. Мейер), а также к изучению зависимости различных свойств от положения элемента в системе или от атомных весов. Он привел также к уточнению классификации соединений (например, отличие перекисей от окислов, аналогия аммиакатов с гидратами), к синтезу новых соединений.
В рассматриваемый период была показана периодичность изменения большого числа физико-химических свойств: удельных объемов простых тел и соединений, коэффициентов линейного расширения (Т. Карнелли), теплот образования и плавления (Ю. Томсен, Лаури), сжижаемости простых тел (Т. Ричардс),
384
ПЕРИОДИЧЕСКИЙ ЗАКОН
электропроводности металлов (В. И. Селезнев), проводимости расплавленных солей (В. А. Горстман), подвижности ионов (Ф. Коль-рауш, В. Гитторф), спектральных и магнитных свойств (П. Лекок де Буабодран, В. Хартли, Дж. Чамичан, Л. Эррера, Бахметьев, Б. Паскаль, Э. Ведекинд, Ж. Урбен, С. Мейер), окраски ионов (А. Ладенбург и др.) и т. п. Наряду с открытием элементов, предсказанных Менделеевым, результаты изучения физико-химических свойств элементов явились дальнейшим подтверждением периодического закона [59].
Исследования в плане периодического закона тесно смыкались с исследованиями в области физической химии: изучение химического сродства, химического равновесия, изучение растворов и сплавов. Первые работы в этом направлении были осуществлены учениками Д. И. Менделеева — Г. Г. Густавсоном и А. Л. Поты-лицыным (1869—1872), показавшими, что предел протекания химических реакций замещения зависит от положения элементов в системе. Такие процессы, как реакции замещения галогенов, изученные Густавсоном (1869—1877) и Потылицыным (1870— 1880), показали, по мнению Менделеева, «ясную зависимость хода разложения не от каких-либо неизвестных особенностей элементов (от сродства и т. п.), а от их массы и места в системе» [26, с. 852; см. также 56, с. 90].
Одновременно были предприняты попытки установить зависимость и других физико-химических характеристик, например скоростей химических реакции, от молекулярных весов реагирующих веществ (II. II. Каяндер, Ю. Е. Богуский). Однако в данном случае связь оказалась более сложной. Другие работы этой школы характеризуют стремление Д. И. Менделеева, а затем Д. П. Коновалова выяснить применимость периодического закона для возможно большего числа физико-химических свойств и явлений. Это — изучение форм соединений кристаллогидратов, аммиакатов (Менделеев, Лучак, 1870-е годы), комплексообразования (Менделеев, 1870-е годы, Е. В. Бирон, 1902—1903), металлических соединений (А. А. Байков, 1902), изучение поведения веществ в растворе: растворимость солей (Менделеев, 1880-е годы), измерение плотности растворов как метод определения атомного веса (Менделеев, В. Я. Бурдаков, 1880-е годы) (подробнее см. [60]).
В рассматриваемый период были установлены также следующие основные закономерности химического поведения элементов в соответствии с их положением в периодической системе: 1. Особый характер элементов малых периодов, их большая индивидуальность. Одной из особенностей их оказалось сильное диагональное сходство. 2. Увеличение сходства соседних элементов в больших периодах (особенно триады VIII группы и редкоземельные элементы) . 3. Большие отличия по сравнению с аналогами элементов шестого периода. 4. Отличия в закономерностях изменения свойств элементов четных и нечетных рядов или главных и дополнитель-
15 Всеобщая история химии
385
ГЛАВА ВОСЬМАЯ
Пер!одическая система элементовъ по группамъ и рядамъ.
I 'HtlfJ ГРУППЫ ЭЛЕМЕНТОВЪ:
0 I II III IV V VI VII VIII
Водо-родъ. н 1,008 Ли-Т1Й. 7,03 На-ipi й. Na 23,05 Берил-л!й. Ве 9.1 Маржи. Мд 24,36 Борт,- В 11,0 Алю-нижй. AI 27,1 Угле-родь. с 12,0 Крем-nifi. Si 28,2 Азоть. N 14,01 Фос-форъ. р 31,0 Кисло-родъ. 0 16,00 Cipa S 32,06 Фторъ. F 19,0 Хлоръ 01 35,45 1
1 2 3 Гел1й. He 4,0 Неонъ Ne 19,9
4 5 Ap-гонъ. Аг 38 Ка- Л1Й. К 39,15 М1дь Си 63,6 Каль-цш. Са 40.1 Цинкъ. Zn 65.4 Скан- Д1Й. Sc 44,1 Гал-Л1Й. Ga 70,0 Ти-танъ. Ti 48.1 Гер-Yanifi. Ge 72,5 Вана- Д1Й. V 51,2 Мышь* якъ. As 75 Хромь. Сг 52,1 Селенг. Se 79,2 Мар-танецъ. Мп 55,0 Бромь Вг 79,95 Же- Ко- Ник-л!зо.бальтъ.кель. Fe Со Ni (Си) 55,9 59 59
6 7 Крип-тонъ. Кг 81,8 Ру-бид!й. Rb 85.5 Серебро. Ад 107,93 Строи-Ц1Й. Sr 87,6 Над xiii. Cd _ 112,4 Ит-Tpiw. Y 89,0 Ин-Д1Й. Jn 115,0 Цир К0Н1Й. Zr 90,6 Олово. Sn 119,0 Hio-rtiii. Nb 94,0 Сурьма. Sb 120,2 Молиб-денъ. Мо 96,0 Тел-луръ. Те 127 1одъ J 127 Ру- Ро- Нал-тенш. Д1Й лад!Й. Ru Rh Pd (Ag) 101,7 103,0 106,5
8 9 Ксе-нонъ. Хе 128 Це-з!й. Cs 132,9 Ба-piii. Ba 137,4 Лантань. La 138,9 Це-pifi. Се 140,2 — —— - - - »
10 11 — Золото. Аи 197.2 Ртуть. Нд 200 0 Иттер-б!Й. Yb 173 Ta-iift. Tl 204.1 Сви-нець. РЬ 206,9 Тан-таль. Та 183 Виснуть. Bi 208,5 Вольф-рамъ. W 184 Ос- Ири- Пла- М1й. Д1й. тина. Os Jr Pt (Au) 191 193 194,8
12 —— — Радш. Rd 225 — Topiii. Th 232,5 — У ранг и 238.5
В ы с ш i е солеобразные окислы. R R50 RO R203 RO2 R205 RO3 R207 RO* &ucuiia газеобразныя водородный соединен! я; RH4 RH3 RH2 RH
Д Менйелпевъ
386
ПЕРИОДИЧЕСКИЙ ЗАКОН
ных подгрупп. 5. Способность элементов середины системы (III— VI группы), особенно второго и третьего периодов, образовывать цепи. 6. Отличие форм интерметаллических соединений от обычных форм бинарных соединений.
Изучение периодичности химических свойств привело к широкому распространению периодического закона в химии, к углублению основных понятий химии (амфотерность, химическая активность и др.). Вместе с тем оно поставило вопрос об изучении связи физико-химических свойств не только с массой, но и с другими свойствами. В этом огромное самостоятельное значение работ по изучению изменения различных свойств и их взаимосвязи. Появилась, как никогда раньше, необходимость физического истолкования вновь открытых свойств.
В первые годы после открытия периодического закона многие химики считали, что в группах периодической системы физикохимические свойства элементов, в том числе атомные веса, изменяются монотонно. Однако в 1887—1888 гг. А. II. Базаров в России и II. Г. Ридберг в Швеции указали на случаи немонотонного изменения величин атомных весов в группах и рядах системы. Одновременно были накоплены сведения о некоторых «отступлениях» от монотонного изменения и других свойств. Эти отступления пытались связать с отступлением в значениях атомных весов. Так, Менделеев писал: «...необходимо [видеть] различать у элементов общие их свойства, находящиеся в периодической зависимости от атомных весов (напр., способность давать известные формы окисления — это свойство само по себе периодично), и индивидуальные свойства, зависящие от упомянутых выше отступлений (в изменениях атомных весов.—А. 71/.) [61, с. 67—68].
Такой характер изменения свойств соединений Менделеев сознавал уже в процессе открытия периодического закона: «...в соединениях, составленных аналогически, нередко замечается сходство в величине объемов и такое отношение объемов, которое нисколько пе предугадывается и нисколько не согласуется с такими объемами, какие имеют входящие элементы в отдельном состоянии. Оттого и становится попятным, что в системе, предложенной нами и основанной на величине атомных весов и сходстве в химическом характере, заключаются указанные выше отступления от того простого порядка, которого можно было бы ожидать» [3, с. 48].
Изучение различных физико-химических свойств показало, что чем более сложным (т. е. зависящим от многих других свойств или условий) оказывалось данное свойство, тем более сильные отличия наблюдались при сравнении характера его изменения по группам или периодам системы. Такой вывод, в частности, можно было сделать при изучении теплот образования соединений. После того как Т. Карнеллп и Лаури констатировали периодический характер изменения теплот образования, был установлен ряд правил, харак
387 15*
ГЛАВА ВОСЬМАЯ
теризующих различные стороны изменения этой величины (правила Бейли).
А. Ш. Вюрц в следующих словах отразил итоги решения этой проблемы за 15 лет, последовавших после открытия периодического закона: «...следует признать, что изменения... свойств далеки от того, чтобы прогрессировать правильно и одинаковым образом во всех группах. Иногда эти изменения быстры... иногда очень постепенны... Словом, если справедливо, что свойства тел вообще периодически изменяются с возрастанием атомного веса, то закон этих изменений ускользает от нас и, во всяком случае, не должен быть прост...» [62].
Уже в 1880-е годы предпринимались попытки изучить количественно зависимость одного из физико-химических свойств не только от атомного веса элементов, но и от других основных свойств. Так, А. Л. Потылицын установил формулу для вычисления процента замещения хлора на бром в хлоридах металлов: 4Л/72я2, где М — атомный вес металла, а п — его валентность. Активность простых тел и соединений (т. е. их способность вступать в дальнейшие взаимодействия) пытались связать с величиной атомных объемов [63]. Эти работы свидетельствовали о стремлении дать количественную интерпретацию химическим свойствам.
Одновременно разрабатывался термодинамический подход к оценке неуловимого «химического сродства».
Невозможность установления строгой количественной зависимости между изучаемыми свойствами, распространяющейся на все элементы или свойства, привели, в частности, к широкому развитию в химии метода аналогии, или сравнения. Менделеев по этому поводу писал: «Проверка одного пути другими составляет обычный прием, вполне необходимый, потому что явления диссоциации, полимеризации и т. п. могут усложнять отдельные определения — по каждому способу».
Изучение различных физических, механических, кристаллографических и химических свойств указало на их зависимость от скрытых внутренних свойств атомов. Изучение образования соединений в растворах и расплавах, комплексных соединений и изучение кристаллического состояния неминуемо вело к отысканию причин появления этих различных типов структур.
Изучение характера изменения многочисленных свойств ясно приводило к выводу: «...периодическая изменяемость простых и сложных тел подчиняется некоторому высшему закону, природу которого, и тем более причину — ныне еще нет средства охватить. По всей вероятности, она кроется в основных началах внутренней ’ механики атомов и частиц» [2, с. 384].
ЗАКЛЮЧЕНИЕ
На рубеже XVIII и XIX веков, как на перевале, видна была длинная извилистая дорога блужданий химической мысли, дорога упорного поиска решения глобальных проблем: что такое химический элемент и химическое соединение, химическое сродство и химическая реакция, вода и воздух, дыхание человека и окисление металла?
Виден был путь создания первых обобщений разрозненных фактов, путь, отмеченный вехами открытия новых элементов и газов атмосферы. Полученный Дж. Пристли и К. Шееле кислород в 70-х годах XVIII в. зажег костер химической революции.
К концу XVIII в. была установлена истина фундаментального значения, что вода и угольная кислота — основа существования живого мира на Земле. В системе
Вода
Газы
Жизнь
к тому времени был выяснен состав воды п состав атмосферы, но весь цикл предстояло еще изучить.
Из теории Лавуазье следовал вывод, что все реакции окисления «уносят» с собой кислород. Однако анализы состава атмосферы показали, что количество свободного кислорода планеты остается неизменным. Неясно было, откуда же берется свободный кислород взамен прочно связанного в химических соединениях. Этот вопрос также ждал своего решения.
В начале XIX в. перед учеными открывалась невиданная ранее перспектива развития химических исследований, освещенная восходящими лучами атомно-молекулярного учения. Химиков-аналитиков ждала неизведанная область — химия вновь открытых элементов и их соединений. Фнзико-химиков (так мы назовем Дж. Дальтона, А. Авогадро, П. Дюлонга, Ж. Гей-Люссака, Т. Гротгуса, Г. Дэви) интересовал мир атомов и молекул, электрохимических явлений. «Придет время,— писал Я. Берцелиус в 1828 г.,— когда гипотезы об атомных частицах и общее взаимодействие тел превратятся в прекрасно обоснованную теорию, и тогда химия станет на прочный путь» 4.
1 Цит. по кн.: Жизнь науки. Антология вступлений к классике естествознания. М.: Наука, 1973, с. 241.
389
ЗАКЛЮЧЕНИЕ
Чем мощнее становилась научная теория, чем сильнее выступали ее предсказательные функции, тем глубже вливалась химия в русло внутренней логики своего развития. Ее течение в этом русле убыстрялось с каждым годом.
Из «внешних» факторов, воздействующих на развитие химии, в первой половине XIX в. на первый план выступают запросы развивающегося производства. В конце XVIII — начале XIX в. создались благоприятные условия для сближения и более тесного взаимодействия науки и производства. Развиваются новые формы этого взаимодействия. Ученые-химики (Ж.-Л. Гей-Люссак, Г. Дэви, Н. Клеман, Т. Грэм и др.) выступают в качестве консультантов химических предприятий и принимают непосредственное участие в разработке производственных процессов (производство искусственной соды по методу Леблана, сернокислотное производство и др.). Правительства и промышленные фирмы различных стран начинают щедрее ассигновать средства на создание новых химических лабораторий, кафедр технической химии, политехнических школ 2.
Следствием этого явилось увеличение числа внедрений химических открытий в химические производства. С каждым десятилетием XIX в. росло число химических фабрик и заводов. Рождалась химическая индустрия.
Характеризуя развитие химии в начале XIX в., русский химик А. А. Воскресенский писал: «Наука едва успевала дать отчет в открытиях, уже вслед за этими открытиями являлся целый ряд фабрик, которые были обязаны им своим существованием. ITT ев -рель еще не совсем окончил свои ученые исследования о жировых существах, а уже, основываясь на немногих данных, какие он сообщил публике, мы успели усовершенствовать мыльные заводы, нашли новое средство выгодным образом сбывать наши отечественные произведения, приготовляя стеариновую кпслоту»3.
«Кто спрашивает у научного исследования, зачем оно нужно, тот ничего не понимает в науке,— писал Я. Берцелиус в 1839 г,— Всякое новое положительное знание, как ни кажется оно ненужным в обыденной жизни, приводит к новым знаниям, с которыми оно связано, и способствует развитию понятий, приносит результаты, важность которых в большинстве случаев не может быть предсказана у колыбели исследования. В науке нет ничего незначительного, что не заслуживало бы быть исследованным и познанным, и то, что с виду казалось незначительным, будучи исследовано, имеет значение, которое с течением времени может стать очень большим» 4.
2 Первая политехническая школа была создана в 1794 г. во Франции.
3 Воскресенский А. А. Об успехах химии в новейшее время.— Жури. Мин. народи, проев., 1840, ч. 26, отд. II, с. 149 и сл.
4 Berzelius J.— Jalires-Bericlite. 1839, Jg. 18, S. 1—2.
390
ЗАКЛЮЧЕНИЕ
В первой половине XIX в. утверждение химии как науки сопровождалось процессом дифференциации химии на самостоятельные направления (аналитическая, неорганическая и органическая химия), а также процессом возникновения пограничных дисциплин.
Для развития химии первой половины XIX в. характерно ее взаимодействие с минералогией (создание химической минералогии), физиологией (создание физиологической химии), физикой (создание электрохимии).
В области прямого контакта физического и химического знания в первые два десятилетия XIX в. открываются «золотые россыпи» новых теоретических представлений, которые обогащают все естествознание новым видением «электрофицированной» материи (учение о ионах).
В результате дифференциации и интеграции знании непрерывно менялись контуры химии: они расширялись и углублялись. Возникли новые области исследований. Иной стала инструментальная вооруженность химии.
Конец XX III и начало XIX в. характерны тем, что в это время образовалась когорта ученых 'нового типа. Они соединили в себе порыв научного поиска, уважение к экспериментальным данным и непримиримость к ошибочным концепциям. Химик-профессионал, педагог, пропагандист новых научных идей выходил на арену общественной жизни и начинал играть в пей заметную роль. Ученый становится активным участником процесса преобразования материальных условий жизни общества.
Один из первых, кто в полную меру осознал высокий общественный долг ученого, был А. Л. Лавуазье, научная деятельность которого была направлена на решение важнейших теоретических и прикладных задач, возникших на почве запросов практики.
«Испытатель природы в тиши своей лаборатории и своего кабинета,— говорил Лавуазье,— также может исполнять патриотические обязанности; он может надеяться уменьшить своими работами тяжесть бедствий, угнетающих человеческий род, увеличить его радости и счастье. И если ему, благодаря открытым им новым путям, удастся способствовать тому, чтобы продлить среднюю жизнь человека на несколько лет, даже на несколько дней, он может рассчитывать на славное звание благодетеля человечества» 5.
Успешное решение жизненно важных практических задач подняло престиж ученых и привлекло к науке широкое внимание общественности.
Характерная черта в деятельности химиков первой половины XIX в. заключалась в том, что они в редких случаях работали только в одной области. Яркий пример тому — деятельность Бер-
5 Цит. по статье: Погодин С. А. Антуан Лоран Лавуазье — основатель химии Нового времени.— Успехи химии, 1943, т. 12, вып. 5, с. 332.
391
ЗАКЛЮЧЕНИЕ
целиуса, Гей-Люссака, Дэви и многих других. Но с углублением исследований в различных направлениях энциклопедизм уступает место специализации,
К середине XIX столетия в полный голос заявляют о себе ученые — специалисты в определенной области химии, для которых характерна систематическая разработка того или иного направления химической науки. Они создают первые научные школы химиков, организуют химические лаборатории, издают химические журналы и учебные руководства; начинают выходить специализированные международные журналы6. Ежегодные обзоры Я. Берцелиуса, научные журналы, издаваемые Ю. Либихом, И. Погген-дорфом, Ж. Б. Дюма, служат новым мощным фактором воздействия на развитие химии.
Развитию химии в XIX в. во многом способствовали диксуссии по кардинальным научным проблемам. Научная дискуссия. — как важный фактор научного прогресса — приобрела современную форму именно в XIX в. Столкновение различных гипотез, обсуждение спорных вопросов в научных журналах, участие в дискуссии ученых различных школ и поколений, привлечение эксперимента как беспристрастного судьи,— все это присутствовало в научных дискуссиях и обогащало химию новыми идеями и представлениями.
Живой обмен результатами научных исследований проходил на заседаниях научных обществ и конгрессов. Научные конгрессы способствовали преодолению разрыва между успешной работой специалиста в узкой области и утратой понимания им общих путей развития данной науки. Научные общества призваны были объединить специалистов той или иной области науки для коллективного обсуждения актуальных научных проблем. Так, в 1841 г. организуется Лондонское химическое общество7, в 1857 г.— Парижское химическое общество, в 1867 г.— Берлинское химическое общество, в 1868 г.— Русское химическое общество. Каждое из этих обществ издавало свой журнал8, где были сосредоточены все оригинальные исследования ученых.
6 В 1828 г. О. Эрдман основал «Journal fur technische und Okonomische Chemie» (Журнал технической и экономической химии); с 1834 г.— «Journal fur praktische Chemie» (Журнал практической химии); в 1862 г. К. Фрезениус основал «Zeitschrift fur analytische Chemie» (Журнал аналитической химии).
7 В Уставе Лондонского химического общества, утвержденном в 1848 г.т организация именовалась так: «Химическое общество продвижения химической науки, тесно связанное с процветанием промышленников Соединенного королевства, многие из которых [промышленников] зависят от применения химических принципов и открытий, что должно способствовать их выгодному развитию, экономическому применению промышлен-• ных источников и санитарному состоянию общества».
8 С 1849 г. начал издаваться «Journal of the Chemical Society of London»; c 1863 r.— «Bulletin de la Societe Chimique de Paris»; c 1868 r.— «Berichte der Deutschen chemischen Gesellschaft»; c 1869 r.— «Журнал Русского химического общества».
392
ЗАКЛЮЧЕНИЕ
К середине XIX в. химия подошла с впечатляющими результатами. В области неорганической химии насчитывалось уже более 50 открытых химических элементов, изучением которых занимались многие видные химики. Сложились все предпосылки для: открытия периодического закона.
В области физической химии определились и плодотворно развивались самостоятельные направления: электрохимия, термохимия, учение о растворах9.
С накоплением сведений о веществе все яснее становилось понимание того, что изучение свойств химических соединений и их состава — это не предел химического знания, есть более важная задача науки — она состоит в отыскании зависимости свойств веществ от их состава и строения. Исследования в этом направлении подготовили необходимые предпосылки для создания теории химического строения органических соединений.
К середине XIX в. химия стала ярко демонстрировать свою» практическую значимость. На промышленных выставках уже широко были представлены разнообразные продукты химических производств.
Рост народонаселения, концентрация его в крупных городах остро поставили вопрос об увеличении продовольствия. Речь шла уже не о поддержании плодородия почв, а об его увеличении. В связи с этим возникла необходимость научного ведения сельского хзояйства. На этом фоне в 30—40-е годы XIX в. возникает агрохимия как наука, изучающая взаимоотношения между растениями, почвой и удобрениями. Количественные методы анализа,, кислородная теория Лавуазье, закон сохранения массы вещества — все это легло в основу изучения обмена веществ между растениями и окружающей средой.
В 1840 г. Ю. Либих впервые четко высказал идею о сознательном регулировании обмена веществ между человеком и природой.. Возвратить взятые из нее минеральные вещества, не только сохранить, но и увеличить плодородие почвы путем внесения в нее искусственных химических удобрений — это основные требования, которые выдвинул Либих в своей книге «Органическая химия в приложении к земледелию и физиологии» (1840). «Придет время,— писал Ю. Либих,— когда каждое поле, сообразно с растением, которое на нем будут разводить, будет удобряться свойственным удобрением, приготовленным на химических заводах» 10. По словам К. Маркса, «выяснение отрицательной стороны современного зем
9 Более подробно см.: Всеобщая история химии. История учения о химическом процессе. М.: Наука, 1981. 447 с.
10 Либих Ю. Химия в приложении к земледелию и физиологии. М.; Л.: Сель-хозгиз, 1936, с. 18; см. также: Либих Ю. Искусственные удобрения или: туки. СПб., 1856.
393
ЗАКЛЮЧЕНИЕ
леделия, с точки зрения естествознания, представляет собой одну из бессмертных заслуг Либиха» п.
Отвечая на социально-экономические потребности восходяще!! промышленной буржуазии, Ю. Либих больше чем кто-либо из химиков 30—40-х годов XIX в. содействовал установлению тесных связей между химической наукой и химической промышленностью. Либих убедительно показал необходимость фундаментального химического образования для лиц, предназначающих себя для работы в промышленности, сельском хозяйстве и фармации.
В 30—40-е годы XIX в. создаются первые школы химиков — новые организационные формы, которые обеспечивали передачу эстафеты научных поколений от учителя к ученикам, от ученых одной страны к ученым другой страны.
Без тесного научного сотрудничества, без участия ученых различных национальностей в разработке научных проблем уже пе представлялся научный прогресс.
11 Маркс К. Капитал. М., 1969, т. 1, с. 515.
ЛИТЕРАТУРА
Глава первая
1. Кузнецов Б. Г. Идеи и образы Возрождения: (Наука XIV—XVI вв. в свете современной науки). М.: Наука, 1979, с. 212—221.
2. Boas Hall М. Robert Boyle on Natural Phylosophy: An Essay with Selections from his Writings. Bloomington: Indiana Univ, press, 1966. 406 p.
3. Gibbs F. W. Peter Shaw and the Revival of Chemistry.— Ann. Sci., 1951, vol. 7, p. 221—237.
4. Bowen W. Industrial Society in England towards the End of the Eighteenth Century. N. Y.: Harcourt Brace and Co. 1925. 300 p.
5. Reuleaux F. Theoretische Kinematik. Grundziige einer Theorie des Maschi-nenwesens. Braunschweig: Kunst und Welt, 1875. 622 S.
6. Лейкин Э. Г. Система механического производства и ее место в истории цивилизации: (Об одной научной проблеме, поставленной и решенной К. Марксом).— В кн.: Механика и цивилизация XVII—XIX вв./Под ред. А. Т. Григорьяна, Б. Г. Кузнецова. М.: Наука, 1979, с. 383—446.
7. Методология историко-научных исследований: Реф. сб. М.: ИНИОН АН СССР, 1978. 269 с.
<8. Лекторский В. А. Субъект, объект, познание. М.: Наука. 1980, 359 с.
9. Кузнецов В. II. Тенденции развития химии: (История химии и ее роль в прогнозировании развития химических наук). Новое в жизни, пауке, технике. М.: Знание, 1976. 64 с. (Серия «Химия». № 3).
10. Антология мировой философии. В 4-х т. Т. 1. Философия древности и средневековья / Под ред. В. В. Соколова. М.: Мысль, 1969, ч. 2, с. 830—831.
И. Зубов В. П. Развитие атомистических представлений до начала XIX века. М.: Наука, 1965. 371 с.
12. Ахутин А. В. История принципов физического эксперимента (от античности до XVII в.). М.: Наука, 1976. 292 с.
13. Boas Hall М. Robert Boyle and Seventeenth-century Chemistry. Cambridge: Univ, press, 1958. 240 p.
14. Metzger II. Les doctrines chimiques en France du debut du XVIle a la fin du XVIII-e siecle. P.: Press. Univ., 1923. 536 p.
15. Metzger H. Les concepts scientifiques. P.: Libr. Felix Alcan, 1926. 356 p.
16. a) Boyle B. The Works of the Honourable Robert Boyle, to which is Prefixed the Life of the Author: 5 vol. / Ed. T. Birch. L.: A. Millar, 1744. б) Второе издание в 6-ти т., 1772. в) Имеется также современное факсимильное издание шеститомника 1772 г. (Hildesheim: 01ms, 1965—1966). Кроме того, в XVII—XVIII вв. появилось множество изданий сочинений Бойля на латинском языке.
17. Всеобщая история химии: Возникновение и развитие химии с древнейших времен до XVII века/Под ред. Ю. И. Соловьева. М.: Наука, 1980. 399 с.
18. Helmont van I. В. Ortus medicinae. Id est Initia physicae inauditae. Pro-gressus medicinae novus in Morborum ultionem, ad Vitam Longam / Edente Authoris filio, Francisco Mercurio Van Helmont, cum ejus Praefatione ex Belgico translata. Amsterdam: Louis Elzevier, 1648. 800 p.
19. Hooykaas R. The Experimental Origin of Chemical Atomic and Molecular Theory before Boyle.— In: Chymia. Philadelphia: Univ. Penna press, 1949, vol. 2, p. 65—80.
20. Jungii J. Doxoscopiae physicae minores, sive Isagoge Physica Doxoscopiae. Hamburgi: Sumtibus Johannis Naumann, 1662, Pt 2, Sect. 1, cap. 12, sec-tiuncula 18.
395
ЛИТЕРАТУРА
21. Stahlii G. E. Fundamenta Chymiae Dogmatico-Rationalis et Experimentalis quae planam ac plenam viam ad Theoriam et Praxin Artis hujus tam Vul’ gatiocinia et dextras Enchirises sternunt. Norimbergae: Impeusis B. Guolfg Maur. Endteri Filiarum, 1732. 199 p.
22. Stahlii G. E. Fundamenta Chymiae Dogmaticae et Experimentalis, et qui-dem turn communioris physicae mecanieae pharmaceuticae ac medicae tuni sublimioris sic dictae hermeticae atque alchymicae. Norimbergae: Sumpti-bus Wolfgang i Mauritii, 1723. 255 p. (Трактат был подготовлен к печати учеником Шталя И. С. Карлом (J. S. Carl, 1675—1757), им же было написано и предисловие к нему, содержащее резкую критику корпускулярных теорий Бойля и Лемери). Есть также английское издание: Stahl G. Е. Phylosophical Principles of Universal Chemistry / Drawn from the Collegium Jenense of Dr. G. E. Stahl by Peter Shaw. L.: Osborn and Longman, Paternoster-Row, 1730. 424 p. П. Шоу опустил при переводе последний, алхимический, раздел трактата и приложение к нему. Цитирование в основном тексте настоящей главы приводится нами по латинскому оригиналу.
23. Kargon R. Н. Atomism in England from Hariot to Newton. Oxford: Clarendon press. 1966. 168 p.
24. Бэкон Ф. Соч. В 2-х т. М.: Мысль, 1977—1978.
25. Kepler J. Gesammelte Werke: Bd. 1—22/Hrsg. von M. Caspas, W. Van Dyck und F. Hammer. Munich: R. Oldenbourg, 1937—1968.
26. Спекторский E. Проблема социальной физики в XVII столетии. Т. 1. Новое мировоззрение и новая теория науки. Варшава, 1910. 563 с.
27. Декарт Р. Избранные произведения / Пер. с фр. И. Латинского; под ред. В. В. Соколова. М.: Госполитиздат, 1950. 711 с.
28. Декарт Р. Рассуждение о методе. С приложениями: «Диоптрика», «Метеоры», «Геометрия» / Ред., пер., статьи и коммент. Г. Г. Слюсарева, А. П. Юшкевича. М.: Изд-во АН СССР, 1953. 656 с.
29. Stahlii G. Е. Fundamenta Chymiae Dogmatico-Rationalis et Experimentalis. Pars III. Norimbergae, 1747. 508 p.
30. Stahlii G. E„ Chymia Rationalis et Experimentalis, oder Griindliche der Na-tur und Vernunfft gemasse und mit Experimenten erwiesene Einleitung zur Chymie. Leipzig, 1720. 520 S.
31. Gassend P. Opera omnia: Vol. I—VI. Lugduni, 1658.
32. Гассенди П. Соч.: В 2-х т./Пер. с лат., общ. ред. и вступ. статья Е. П. Ситковского. М.: Мысль, 1966—1968.
33. Быховский Б. Э. Гассенди. М.: Мысль, 1974. 202 с.
34. Jacob J. R. The Ideological Origins of Robert Boyle’s Natural Philosophy.— J. Europ. Stud., 1972, vol. 2, p. 1—21.
35. Jacob J. R. Robert Boyle and Subversive Religion in the early Restoration.— Albion, 1974, vol. 6, N 4, p. 275—293.
36. Jacob J. R. Boyle’s Atomism and the Restoration Assault on Pagan Naturalism.— Social Stud. Sci., 1978, vol. 8, p. 211—233.
37. a) Rossi P. Hermetism, Rationality and the Scientific Revolution.— In: Reason, Experiment and Mysticism in the Scientific Revolutioon / Ed. M. L. Righini et al. N. Y.: Sci., Hist. PubL, 1975, p. 247—273. 6) Science and Society, 1600—1900/Ed. P. Mathias. L.: Cambridge Univ, press, 1972. 166 p.
38. Royal Society, Boyle Papers, XXVI, ff. 162—163. Рукопись впервые опубликована в статье: Westfall R. S. Unpublished Boyle Papers Relating to Scientific Method. Pt I.— Ann. Sci., 1956, vol. 12, N 1, p. 63—79; Pt IL— Ibid., N 2, p. 103—117 (особенно p. Ill—113).
39. Maddtson R. E. W. The Life of the Honourable Robert Boyle. L.: Taylor and Fransis ltd, 1969. 332 p.
40. Gassend P. a) Antimadversiones in decimum librum Diogenis Laertii, qui est de vita, moribus placitisque Epicuri 3 vols. Lugduni, 1649. (Предисловие датировано 1646 г.) 6) Syntagma philosophiae Epicuri, cum refutatio-nibus dogma turn quae contra fidem christianam ab eo asserta sunt, appo-
396
ЛИТЕРАТУРА
sitis per Petrum Gassendum.— In: Opera omnia. Vol. III. Lugduni, p. 1—94.
41. Charleton W. Pliysiologia Epicuro-Gassendo-Charltoniana: Or a Fabrick of Science Natural, upon the Hypothesis of Atoms, Founded by Epicurus, Repaired by Petrus Gassendus, Augmented by Walter Charleton, Dr. in Medicine, and Physician to the Late Charles, Monarch of Great Britain / Indexes and introduction by R. H. Kargon. Reprint of London Edition of 1654. The Sources of Science, N 31. N. Y.; L.: Johnson Repr. Corp., 1966. 394 p.
42. Hobbes T. The English Works of Thomas Hobbes of Malmesbury: 11 vol. / Now first collect, and ed. by Sir W. Molesworth. L.: J. Bohn, 1839—1845.
43. Cavendish M. Poems and Fancie. L., 1653. 356 p.
44. Гегель Г. Ф. Соч.: В 14-ти т./Под ред. А. Деборина. М.: Партиздат, 1932. Т. 9. Лекции по истории философии. Кн. 1. 314 с.
45. Ньютон II. Лекции по оптике/Пер. с англ., коммент, и ред. С. И. Вавилова. М.: Пзд-во АН СССР, 1946. 295 с.
46. The Oxford English Dictionary: 12 vol. Oxford: The Clarendon press, 1933. 47. Погодин С. А. Бойль.— В кн.: БСЭ. 3-е изд. Т. 3, 1970, с. 468, колонка 1391. 48. Boyle R. Of the Incalescence of Quicksilver with Gold, Generously Imparted by B. R.— Philos. Trans., 1675/1976, vol. 10, p. 515—533.
49. Джу a M. История химии. 2-е изд. / Пер. с пт. Г. В. Быкова; под ред. С. А. Погодина. М.: Мир, 1975. 477 с.
50. More L. Т. The Life and Works of the Honourable Robert Boyle. L. etc.: Oxford Univ, press, 1944. 313 p.
51. Рабинович В. Л. Алхимия как феномен средневековой культуры. М.: Наука, 1979. 392 с.
52. Wohlwill Е. Joachim Jungius und die Erneuerung Atomistischer Lehren in 17. Jahrhandert. Hamburg: L. Friedrischen und Co, 1887. 356 S.
53. Кедров Б. M. Энгельс о развитии химии. 2-е доп. изд. М.: Наука, 1979. 496 с.
54. More L. Т. Boyle as Alchemist.— J. Hist. Ideas, 1941, vol. 2, p. 61—76.
55. Kopp H. Geschichte der Chemie. Theil 2. Braunschweig: Friedrich Vieweg und Sohn, 1844, S. 275. См. также его Beitrage zur Geschichte der Chemie. Stuck 3. Braunschweig: Friedrich Vieweg und Sohn, 1875, S. 165, 182.
56. Partington J. R. A History of Chemistry: 4 vol. L.: Macmillan and Co ltd, 1961, vol. 2, p. 486—549 (особенно c. 496, 500-502).
57. Фигуровский H. А. Очерк общей истории химии: От древнейших времен до начала XIX в. М.: Наука, 1969. 455 с.
58. Мейер сон Э. Тождественность и действительность: Опыт теории естествознания как введения в метафизику / Пер. с фр. под общ. ред. Д. М. Койгена. СПб.: Шиповник, 1912. 498 с.
59. Кун Т. Структура научных революций / Пер. с англ. И. 3. Налетова; общ. ред. и предисл. С. Р. Микулинского, Л. А. Марковой. М.: Прогресс, 1975.
60. Kuhn Т. S. Robert Boyle and Structural Chemistry in the Seventeenth Century.— Isis, 1952, vol. 43, N 131, p. 12—36.
61. Ланжевен Л. M. В. Ломоносов и P. Бойль: Корпускулярная теория материи и механистическая концепция мира / Пер. М. Г. Новлянской; ред. и примеч. С. А. Погодина.— В кн.: Ломоносов. Сборник статей и материалов. Л.: Наука, 1977, вып. 7, с. 30—57.
62. Соловьев Ю. И. Представления Р. Бойля о химических элементах.— Химия в школе, 1981, № 1, с. 13—15.
63. Вихалемм Р. А. Понятие научности и социальная детерминация формирования науки (на материале истории химии).— В кн.: Наука в социальных, гносеологических и ценностных аспектах: Сб. статей / Под ред. Л. А. Баженова, М. Д. Ахундова. М.: Наука, 1980, с. 345—358.
64. Boas М. An Early Version of Boyle’s Sceptical Chymist.— Isis, 1954, vol. 45, pt 2, N 140, p. 153—168.
65. Boyle R. The Philosophical Works of the Honourable Robert Boyle Esq. 3 vol. / Abridged, methodized and disposed under the general heads of physics, statics, pneumatics, natural history, chemistry and medicine by Peter Shaw. L., 1725.
397
ЛИТЕРАТУРА
66. Clave Е. de. Nouvelles luniiere philosophique des vrais principes et elements de nature, et qualite d’iceux. P.; Olivier de Verennes, 1641. 410 p.
67. Shaw P. A New Method of Chemistry. 3rd ed. correct. L.: T. and T. Longman, 1741, vol. 1. 246 p. "b
68. Holms F. L. Analysis by Fire and Solvent Extractions: The Metamorphosis of a Tradition.— Isis, 1970, vol. 62, pt 2, N 212, p. 129—147.
69. Debus A. G. Fire Analysis and the Elements in the Sixteenth and the Seventeenth Centuries.— Ann. Sci., 1967, vol. 23, p. 127—147.
Глава вторая
1.
9
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
. 18.
19.
20.
Le Fevre N. Traile de la chymie. 2nd ed. P., 1669. vol. 1. 389 p.
Погодин С. A. M. В. Ломоносов и химия XVIII в.— Вопросы истории естествознания и техники, 1962, вып. 12, с. 28—43.
Вайте A. Chymie experimentale et raisonnee. P., 1773, vol. 1. 482 p.
Чугаев Л. А. Открытие кислорода и теория горения в связи с философскими учениями Древнего мира. Иг.: Науч, хим.-техн. изд-во, 1919. 80 с. Glaser Ch. Traite de la chimie. 4th ed. Brysselles, 1676.
Lemery N. Cours de chymie. lOe ed. P., 1713.
Bohn I. Dissertationes chymico-physicae, chimiae finein instrumenta et ope-rationes frequentiores explicantes cum Indice Rerum et Verborum / Qvibus accesfit Ejusdem Tractatus olim editus de Aeris in sublinaria influxu. Lip-siae, 1685.
Barnero J. Chymia philosophica perfecte delineata, docte enucleata et feli-citer desiderata munc vera omnibus philatris conseerata cum brevised accu-rata et fundamentalis salium Doctrina. Medicamentis etiam fine igne culi-nari facile parabilibus nec non exercitio chymiae Appendicis loco locuple-tata. Norimbergae, 1689.
Stahl G. E. Fundamenta chymiae dogmaticae et experimentalis. Norimbergae, 1723.
Teichmeyer H. F. Institutiones chemiae dogmaticae et experimentalis in quibus chymicorum principia, instrumenta, operationes et producta succinc-ta methodo tracuntur in usurn auditorii sui cum figuris aeneis et indicibus. lenae, 1729.
Juncker I. Conspectus chemiae theoretico-practicae in forma tabularum... Halae; Magdeburgae, 1730. 1086 p. См. также Kangro H. loachim Jungius* experimente und Gedanken zur Begrundung der Chemie als WissenschafL Wiesbaden: Franz Steiner Verl., 1968. 479 S.
Boerhaave H. Elementa Chemiae. T. 1. Lipsiae: Lugduni Batavorum, 1732. Цит. no [13, c. 244].
Фигуровский II. А. Очерк общей истории химии от древнейших времен до начала XIX в. М.: Наука, 1969. 455 с.
Соловьев Ю. II. Очерки по истории физической химии. М.: Наука, 1964.
340 с.
Погодин С. А. Диссертации М. В. Ломоносова и И. Г. Пича о селитре.— В кн.: Ломоносов. Сборник статей и материалов. Л.: Наука, 1977, т. 7, с. 9—29.
Ломоносов М. В. Поли. собр. соч. Т. 2. Труды по физике и химии 1747— 1752 гг. М.; Л.: Изд-во АН СССР, 1951. 756 с.
Фигуровский Н. А. Предмет и задачи химии в определениях М. В. Ломоносова.— Вопросы истории естествознания и техники, 1962, вып. 12, с. 22—27.
Ludolf II. Einleitung in die Chymie... Erfurt, 1752.
Metzger H. Newton, Stahl, Boerhaave et la doctrine chimique. P., 1930. 332 p.
Stahl G. E. Zymotechnia fundamentalis seu Fermentationis Theoria gene-ralis... Halae, 1697. Переп. в кн.: Stahl G. E. Opusculum chijmico-physi-co-medicum... Halae; Magdeburgae, 1715. См. также [60, c. 665—671].
398
ЛИТЕРАТУРА
21. Мейер Э. История химии от древнейших времен до настоящих дней / Пер. со 2-го нем. изд. М. А. Розенталя с пред. Д. И. Менделеева. СПб., 1899. XVI, 513 с.
22. Соловьев Ю. II. Эволюция основных теоретических проблем химии. М.: Наука, 1976. 376 с.
23. De la Metrle J. Abrege de la theorie chimique tire des propres Ecrits de-м. Boerhaave. P., 1741. 160 p.
24. Hoffmann F. De calcinationis ac reductionis fundamento et causis. Observationuni physico-chymicarum selectiorum: Libri 3. Halae, 1722, p. 323—327. См. также [13, c. 243; 60, c. 691—700].
25. Neumann C. Chymia medico-dogmatico-experimentalis: Bd. 1—4. Zullichau, 1749—1754. См. также [60, c. 702—706].
26. Pott H. Chymische Untersuchungen, welche furnehmlich von der Litho-geognosie oder Erkenntniss und Beobachtung der gemeinen einfacheren Steinen und Erden infleichen von Feuer und Licht handeln. Potsdamm, 1746. 431 S.
27. Eller I. F. Physikalisch-niedicinisch Abhandlungen. Berlin, 1764, Bd. 1, S. 254—273, 364—381.
28. Эллер И. Господина Эллера Опыт о начале и рождении металлов / Пер. с нем. языка Горного училища студентом Андреем Пикароном. СПб., 1780.
29. Gerlac Н. Some French Antecedents of the Chemical Revolution.— In: Chymia. Philadelphia: Univ, of Penna press, 1959, vol. 5, p. 73—112.
30. Marggraf A. S. Chemische Schriften. 2 Teil. Berlin, 1767.
31. Chemische Annalen fur die Freunde der Naturlehre, Arzneygelehrtheit, Haushaltungskunst und Manufakturen. Helmstadt, 1784—1803; Die neues-ten Entdeckungen in der Chemie. Leipzig, 1781—1783; Beytrage zur Erwei-terung der Chemie. Helmstadt; Leipzig, 1786—1799; Neues chemisches Ar-chiv. Weimar, 1798.
32. Gren A. C. Systematisches Handbuch der gesamten Chemie. Bd. 1—4. Halle, 1787—1790; 2 Aufl. Halle, 1794—1796.
33. Джу a M. История химии/Пер. с ит. Г. В. Быкова; под ред. С. А. Погодина. М.: Мир. 1975. 477 с.
34. Rappaport R. Rouelle and Stahl — the Phlogistic Revolution in France.— In: Chymia. Philadelphia: Univ, of Penna press, 1961, vol. 7, p. 73—102.
35. Rappaport R. G.-F. Rouelle. An Eighteenth-Century Chemist and Teacher.— In: Chymia. Philadelphia: Univ, of Penna press, 1960, vol. 6, p. 68—101.
36. Macquer P. J. Dictionnaire de chymie. 2 ed. P., 1778, Vol. 1.
37. Coleby L. J. M. The Chemical Studies of P. J. Macquer. L., 1938. 132 p.
38. Блох M. А. Торберн Бергман.— В кн.: Академику В. И. Вернадскому к пятидесятилетию научной и педагогической деятельности. М.: Изд-во АН СССР, 1936, т. 2, с. 1239—1265.
39. Partington J. R. A History of Chemistry. L., 1962. Vol. 3.
40. Ломоносов M. Б. Поли. собр. соч. Т. 1. Труды по физике и химии 1738— 1746. М.; Л.: Изд-во АН СССР, 1950. 619 с.
41. Saechtling Ch. Die Entwicklung der Phlogiston Theorie und der Sauerstoff-theorie bis Lavoisier. Mischcolz, 1933. 54 S.
42. Lavoisier A. L. Reflexions sur le phlogistique.— In: Oeuvres. P., 1862, vol. II, p. 623—655.
43. Ньютон И. Оптика или трактат об отражениях, преломлениях, изгибаниях и цветах света. 2-е изд. М.: Гос. изд-во тех.-теорет. лит-ры, 1954.
44. Lindeboom G. A. Herman Boerhaave. The Man and his Work. Amsterdam; London, 1968. XX, 452 p.
45. Макер П. Ж. Господина Макера начальные основания умозрительной химии, составляющей часть первую / Пер. Косма Флоринского. СПб.: имп. Акад. наук. 1774. 428 с.
46. Меишуткин Б. Н. О корпускулярной философии М. В. Ломоносова.— В кн.: Ломоносовский сборник. СПб., 1911, с. 151—156.
399
ЛИТЕРАТУРА
47. Меншуткин Б. Н. Труды М. В. Ломоносова по физике и химии. М • Л . Изд-во АН СССР, 1936. 537 с.
48. Neville R. J. Maquer and the First Chemical Dictionary, 1766. A Bicente-nial Tribute.— J. Chem. Educ., 1966, sept., vol. 43, N 9, p. 486—491.
49. Fourcroy A. Systeme des connaissances chimiques. P., 1801, vol. 1. Histoire de la chymie, sect. 1, art. 3.
50. Cassebaum H., Kauffman G. The Analytical Concept of a Chemical Element in the Work of Bergman and Scheele.— Ann. Sci., 1976, vol. 33, N 5 p. 447—456.
51. Baume A. Manuel de chimie, ou expose des operations de la chymie et de leurs produits. P., 1766.
52. Валъден И. П. Теории растворов в их исторической последовательности / Пер. Н. П. Страхова; под ред. М. А. Блох. Пг., 1921. 197 с.
53. Шептунова 3. II. Химическое соединение и химический индивид (очерк развития представлений). М.: Наука, 1972. 214 с.
54. Mauskopf S. Criteria for Chemical Combination in the Late Eighteenth Century. Histoire de la chimie depuis le XVIII siecle.— In: Actes de Xlle Congr. intern, d’hist, des sci. Paris, 1968. P., 1971, vol. 6, p. 63—67.
55. Weeler T. S., Partington J. R. The Life and Work of William Higgins. Chemist (1763—1825). L. etc., 1960. 173 p.
56. Higgins W. A Comparative View of the Phlogistic and Antiphlogistic Theories. 2nd ed. L., 1791. 316 p.
57. Kopp H. Geschichte der Chemie. Th. 2. Braunschweig, 1844.
58. Scali ger J. C. Exotericarum exercitationum. Francofurti, 1582, liber 15, sect. 8.
59. Зубов В. П. Развитие атомистических представлений до начала XIX в. М.: Наука, 1965. 371 с.
60. Partington J. R. A History of Chemistry. L., 1961, vol. 2. XXIV+795 p.
61. Bloch E. Das chemische Affinitatsproblem geschichtlich betrachtet.— Isis, 1926, vol. 25, p. 119—157.
62. Люблинская А. Д. К вопросу о влиянии Ньютона па французскую науку (спор ныотонианцев с картезианцами).— В кн.: И. Ньютон/Под ред. С. И. Вавилова. М.; Л.: Пзд-во АН СССР, 1943, с. 361—391.
63. Sivin N. William Lewis (1708—1781) as a chemist.— In: Chymia. Philadelphia: Univ. Penna, press, 1962, vol. 8, p. 63—88.
64. Karsten С. I. B. Revision der chemischen Affinitiitslehre mit bestandiger Rucksicht auf Berthollet’s neue Theore. Leipzig, 1803.
65. Bergmann T. Traite des Affinites Chimiques, ou Attraction Electives. P., 1788.
66. Partington J. R. Joseph Black’s «Lectures on the Elements of Chemistry».— In: Chymia. Philadelphia: Univ. Penna press, 1960, vol. 6, p. 27—67.
67. Wenzel C. F. Lehre von der Verwandschaft der Korper. Dresden, 1777.
68. Farber E. Early Studies Concerning Time in Chemical Reactions.— In: Chymia. Philadelphia: Univ. Penna press, 1961, vol. 7, p. 135—152.
69. Szabadvary F. The Birth of Stochiometry.— J. Chem. Educ., 1962, vol. 39, N 5, p. 267.
70. Ладенбург А. Лекции по истории развития химии от Лавуазье до наших дней. Одесса: Матезис, 1917. 360 с.
71. Kirwan R. De la force des acides et de la proportion des substances qui composent les seis neutres.— Ann. chim., 1792, vol. 14, p. 138.
72. Berthollet C. L. Essai de statique chimique. P., 1803. T. 1.
73. Holmes F. L. From Elective Affinities to Chemical Equilibria: Berthollet’s Law of Mass Action.— In: Chymia. Philadelphia: Univ. Penna press, 1962, vol. 8, p. 105—145.
74. Петров Л. П. Исследования газов атмосферы: Дис. ... канд. хим. наук. М.: ИНЕТ АН СССР, 1974.
75. Turner Н. D. R. Hooke and the Theories of Combustion.— Centaurus, 1956, N 4, p. 297—310.
400
ЛИТЕРАТУРА
76. Partington J. R. John Mayow (1641—1679).— Isis, 1956. vol. 47, p. 217—230.
77. Mayow J. Medico-physical Works. Oxford, 1907.
78. Hales S. Vegetable Staticks. L. etc.: Amer. Elsevier publ. co., 1969. XXXII+ +214 c.
79. Hartog P. J. The Newer Views of Priestley and Lavoisier.— Ann. Sci., 1941, vol. 5, N 1, p. 1—56.
80. Priestley J. Observations on Different Kinds of Air.— Philos. Trans. (L.), 1772, vol. 62, p. 147-264.
81. Priestley J. Autobiography of Joseph Priestley (1733—1804). Teaneck etc.: Facrleigh Dickinson Univ, press, 1970. 159 p.
82. Scheele K. W. Chemische Abhandlung von der Luft und Feuer. Upsala; Leipzig, 1777.
83. Partington J. R. The Discovery of Oxygen.— J. Chem. Educ., 1962, vol. 39, N 3, p. 123—125.
84. Соловьев Ю. II. История химии. M.: Просвещение, 1982, с. 90.
85. Scheele К. W. Bruna boken. Stockholm, 1961. 411 s.
Глава третья
1. Fayet J. La Revolution fran^aise et la science. 1789—1795. P., 1960. 498 p.
2. Le Chatelier II. De la methode dans les sciences experimentales. P., 1936. 319 c.
3. Daumas M. Lavoisier theoricien et experimentateur. P., 1955. 180 p.
4. Fourcroy A. Systeme des connaissances chimiques. P., 1801, vol. I, sect. I, art. III.
5. Lavoisier A. L. Sur la destruction du diamant par le feu (1772). Premier memoire.— Oeuvres. P., 1862, vol. II, p. 38—64. Deuxieme memoire.— Ibid., p. 64-88.
6. Дорфман Я. Г. Лавуазье. 2-е перераб. изд. М.: Изд-во АН СССР, 1962. 327 с.
7. Lavoisier A. L. Details historiques sur la cause de 1’augmentation de poids, qu’acquierent les substances metalliques lorsqu’on les chauffe pendant leur exposition a 1’air.— Oeuvres. P., 1862, vol. II, p. 99—103.
8. Velluz L. Sur 1’ouverture par 1’Academie des Sciences le 5 mai 1773, du premier pli cachete depose par Antoine — Laurent de Lavoisier.— C. r. Acad, sci. (P.), Ser. C, 1973, vol. 276, N 26, p. 149—152.
9. Lavoisier A. L. Memoire sur la calcination de Petain dans les vaisseaux fer-mes et sur la cause de 1’augmentation du poids qu’acquiert ce metal pendant cette operation (1774).— Oeuvres. P., 1862, vol. II, p. 105—120.
10. Lavoisier A. L. Opusqules physiques et chimiques.— Oeuvres. P., 1864, vol. I, p. 439—655.
11. Ahlers W. С. P. J. Macquer et le rapport sur les Opusqules physiques et chimiques de Lavoisier.— In: Actes de Xlle Congr. intern, d’hist. sci. Paris, 1968. P., 1971, vol. 6, p. 5—9.
12. Lavoisier A. L. Memoire sur la nature du principe que se combine avec les metaux pendant leur calcination et qui en augmente le poids (1775).— Oeuvres. P., 1862, vol. IT, p. 122—128.
13. Лавуазье А. Л. Мемуары. О природе вещества... / Под ред. с биографией А. Лавуазье и пред. М. А. Блох. 2-е изд. Л.: Ленингр. обл. изд-во, 1931. 79 с.
14. Lavoisier A. L. Memoire sur 1’existence de 1’air dans 1’acide nitreux et sur les moyens de decomposer et de recomposer cet acide (1776).— Oeuvres. P., 1862, vol. II, p. 129-138.
15. Lavoisier A. L. Considerations generales sur la nature des acides et sur le principes dont ils sont composes (1778).— Ibid., p. 248—260.
16. Crosland M. Lavoisier’s Theory of Acidity.— Isis, 1973, vol. 64, N 30, p. 306—325.
16 Всеобщая история химии
401
ЛИТЕРАТУРА
17. Lavoisier A. L. Experiences sur la respiration des animaux.— Oeuvres P 1862, vol. II, p. 174—183. ’ *’
18. Culotta Ch. A. Respiration and the Lavoisier Tradition: Theory and Modi-fications, 1777—1850. Philadelphia, 1972. 41 p.
19. Lavoisier A. L. Memoire sur la dissolution du morcure dans Facide vitriolL que, et sur la resolution de cet acide en acide sulphureux aeriforme et en air eminemment respirable (1777).—Oeuvres. P., 1862, vol. II, p. 194—198.
20. Lavoisier A. L. Experiences sur la combinaison de Falun avec les matieres charbonneuses et sur les alterations qui arrivent a Fair dans lequel on fait bruler du pyrophore (1777).—Ibid., p. 199—208.
21. Lavoisier A. L. Memoire sur la vitriolisation des pyrites martialcs (1777) — Ibid., p. 209—211.
22. Lavoisier A. L. De la combinaison de la matiere du feu avec les fluides evaporables et de la formation des fluides elastiques aeriformes.— Ibid p. 212-224.
23. Lavoisier A. L. Memoire sur la combustion et general (1777).— Ibid p. 225—233.
24. Crosland M. Le heritiers de Lavoisier. P.: Univ, de Paris, Palais de la Decou-verte, 1967. 45 p.
25. Partington J. R. A History of Chemistry. L., 1961. Vol. 2. XXIV, 795 p.
26. Ломоносов M. В. Поли. собр. соч. T. 2. Труды по физике и химии 1747— 1752. М.; Л.: Изд-во АН СССР, 1951.
27. Lavoisier A. L. Traite elementaire de chimie. Vol. 1—2. P., 1789.
28. Ломоносов M. В. Поли. собр. соч. T. 1. Труды по физике и химии 1738— 1746. М.; Л.: Изд-во АН СССР, 1950.
29. Lavoisier A. L. Consideration generales sur la dissolution des metaux dans les acides (1782).— Oeuvres. P., 1862, vol. II, p. 509—527.
30. Lavoisier A. L. Memoire sur 1’affinite du principe oxygine avec les diffe-rentes substances auxquelles il est susceptible de s’unir (1782).— Ibid., p. 546—556.
31. Lavoisier A. L. Memoire sur 1’union du principe oxygine avec le fer (1782).—Ibid., p. 557.
32. Lavoisier A. L., Laplace P. de. Memoire sur la chaleur (1780).— Ibid., p. 283—333.
33. Delacre M. Histoire de la chimie. P., 1920. 632 p.
34. Daumas M., Duveen D. Lavoisier’s Relatively Unknown Large Scale Decomposition and Synthesis of Water, february 27 and 28, 1785.— In: Chymia. Philadelphia: Univ. Penna press, 1959, vol. 5, p. 113—129.
35. Lavoisier A. L., Meusnier L Memoire ou 1’on prouve, par la decomposition de 1’eau que ce fluide n’est point unesustance simple, et qu’il у a plusieurs moyens d’obtenir en grand Fair inflammable qui entrc comme principe con-stituant (1781).— Oeuvres. P., 1862, vol. II, p. 360—373.
35a. Berthe lot M. La revolution chimique. Lavoisier. P., 1902.
36. Lavoisier A. L. Reflexiones sur la phlogistique pour servir de suit a la theo-rie de la combustion et la calcination, publiee en 1777 (1783).— Ibid., p. 623—655.
36a. Погодин С. А. Антуан Лоран Лавуазье — основатель химии Нового времени.— Успехи химии, 1943, т. 12, вып. 5, с. 329—358.
37. Partington L R. Berthollet and the Antiphlogistic Theory.— In: Chymia Philadelphia: Univ. Penna press, 1959, vol. 5, p. 130—137.
38. Nomenclature chimique ou synonimie ancienne et moderne. P., 1789.
39. Кедров Б. M. Эволюция понятия элемента в химии. М.: Изд-во АПН РСФСР, 1956. 360 с.
. 40, Менделеев Д. И. Памятник Лавуазье.— Новое время [газ.], 1895, 23 мая.
ЛИТЁРЛТУРЛ
Глава четвертая
1. Баталин А. X. Аналитическая химия и пути ее развития (История возникновения и развития основных методов и направлений аналитической химии).— В кн.: Тр. Оренбургского сельскохоз. ин-та. Т. 12. Работы по химии. Оренбург, 1961. 388 с.
2. Szabadvary F. Geschichte der analytischen Chemie. Bp.: Akad. kiado, 1966. 410 S.
3. Kopp H. Geschichte der Chemie. Braunschweig, 1844. Th. 2. 436 S.
4. Madsen E. R. The Development of Titrimetric Analysis till 1806. Copenhagen: G.E.C. Gad., 1958. 238 p.
5. Лыс T. И. История развития классических методов количественного анализа: Дис. ...канд. хим. наук. М.: ИИЕиТ АН СССР, 1974. 259 с.
6. Boyle R. Philosophical Works. Vol. I—III. L., 1725, vol. I, p. 58.
7. a) Lampadius W. A. Handbuch zur Chemischen Analyse. Freyberg, 1801. б) См. также: Richter J. Lampadius und Freiberg. Freibergs Hiittenwesen in Zeitalter der industriellen Revolution (1800—1870). Freiberg (Sachs), 1972. 156 S. (Из серии: Veroffentlichungen in wissenschaftlichen Informa-tionzentrums der Bergakademie Freiberg, N 54).
8. Всеобщая история химии. Возникновение и развитие химии с древнейших времен до XVII в. М.: Наука, 1980. 399 с.
9. Цукерман А. М., Гельман Н. Э., Володина М. А., Давыдова С. Л. Некоторые проблемы органического анализа.— Журн. анал. хим. 1978, т. 23, № 11, с. 2258—2266.
10. Lehman J. G. Probier-Kun st. В., 1761. LXXXVI+318 S. Рус. пер.: Леман И. Г. Пробирное искусство. СПб., 1772. 538+2 с.
11. Еганян А. М. Из истории алгебры. Ереван: Луйс, 1977. 176 с.
12. Марк Витрувий Поллион. Об архитектуре / Пер. с лат. Л.: ОГИЗ, 1936. 343 с.
13. The Elder Plinys Chapter on Chemical Subjects. Pt 1. L., 1929. 249 p. Лат. и англ, текст. Ранний рус. текст: Кайя Плиния Секунда Естественная история ископаемых тел, переложенная на российский язык в азбучном порядке и примечаниями дополненная трудами В. Севергина. СПб., 1819. 364 с.
14. Дильс Г. Античная техника/Пер. с нем. М.; Л.: ОНТИ, 1934. 215 с.
15. a) De La Pirotechnia. Libri. X. DOVE AMPIAMEN re si tratta non solo di ogni sorte & dinersita di minere, ma anchora quan to si ricerica intorno a la prattica di quelle cose di quelche gitto de metalli come d’ogni altra cosa simile a questa / Composti per il S. Vanoccio Birinquccio Sennese. Veneto MDXL. Более доступны переводы: 6) De La Pirotechnia. Ten books in which are fully treated not only every kind and sort of mineral but also all that is necessary for the practice of those things belonging to the arts of smelting or casting metals and all related subjects / Composed by Signor Vannoccio Biringuccio of Siena. N. Y., 1943. XXVI, 474 p. Transl. from I tai. в) Biringuccio V. Biriguccios Pirotechnia / Ubersetzt und erliiutert von dr. Otto Johannsen. Braunschweig, 1925. XVI+544 S. г) На русском языке подробное изложение содержания см. в примечаниях ред. руд. пер. кн. Г. Фестера «История химической техники» проф. М. А. Блох. См. [17, с. 259—261].
16. a) Agricolae Georgii. De re metallica. Libri XII. Basiliae, 1556. б) Факсимильное немецкое издание: Agricola Georg. Zwolf Bucher vom Berg- und Huttenwesen. Dusseldorf, 1953. 564 S. в) Рус. пер.: Агрикола Георгий. О горном деле и металлургии. М.: Изд-во АН СССР, 1962. 599 с.
17. Фестер Г. История химической техники/Пер. с нем. Харьков: ОНТИ, 1938. 304 с.
18. Beierlin Р. R. Lazarus Ercker, Bergmann, Huttenman und Munzmeister im 16 Jahrhundert. B., 1955. 143 S. (Из серии: Freiberger Forschungshefte, N D12).
403
16*
ЛИТЕРАТУРА
19. a) Beschreibung Allesfurnemistcn mineralischen Ertzt und Berckwerck ar-ten wie diselbigen und eine jede in sonderheit ihrcr Natur und Eigenschaft nach auf alle Metall probiert... / Durch Lazarus Erckern. Pr., 1574. б) Апгл. nep.: Ercker L. Lazarus Ercker’s Treatise of Ores and Assaying. Chicago* 1951. XXXIV+360 p.
20. Бергман T. Пробирное искусство или способ разлагать металлические руды мокрым путем/Пер. с нем. СПб., 1801. 108 с. Оригинальный лат. текст: De Docimasia minerarum humida см. [21, с. 399—454].
21. Torbcrni Bergman Opuscula Physica et Che mica, pleraque seorsim antea edita, jam ab auctore collecta, revisa et aucta: Vol. I—II. Upsaliae MDCCLXXX.
22. Соловьев Ю. Куринной В. II. Якоб Берцелиус: Жизнь и деятельность. 2-е изд. М.: Наука, 1980. 320 с.
23. Страдынъ Я. П., Страдынъ П. И. Шведский ученый XVIII в. Урбан Перне и его связи с Прибалтикой.— В кн.: Из истории медицины. Рига, 1957 вып. 1, с. 57—62.
24. Берцелиус Я. О употреблении паяльной трубки/Пер. с нем. СПб., 1831. 395 с.
25. Berzelius J. Liirbok i kemien af Dr. J. Jac. Berzelius. Trede delen. Stockholm, 1818, vol. II, p. 473—493.
26. Генкель II. Ф. Руководство к химическому рудословшо /Пер. с нем. СПб. 1775. 248 с.
27. Кронштедт А. Ф. Опыт Кронштедтова рудословия / Пер. с нем. СПб., 1775. 412 с.
28 Шлаттер И. А. Описание при монетном дворе потребного искусства, в двух главных частях состоящее, из которых первая часть содержит описание монетного дела и как употребляемые к тому металлы пробовать и перечищать, тако ж какие потребности к такому делу надобны и как оные приготовлять. СПб., 1739. 100 с.
29. Шлаттер И. А. Задачи, касающиеся до монетпого искусства, сочиненные для обучения определенных при монетных дворах Коллегии и титулярных юнкеров и прочих учеников статским советником и монетной канцелярии главным судьею Иваном Шлаттером в 1754 г.: Ч. 1—3. СПб., 1754— 1758. Ч. 1. О пробирной науке. СПб., 1754. 80 с.
30. Ломоносов М. В. Первые основания металлургии или рудных дел. СПб., 1763.— В кн.: Ломоносов М. В. Поли. собр. соч. Т. 5. Труды по металлургии и горному делу. М.; Л.: Изд-во АН СССР, 1954.
31. Bergman Т. Torbern Bergman’s Foreign Correspondence. Stockholm; Uppsala, 1965, vol. 1. LVI, 466 p.
32. Bergman T. De analyse aquarum. Cm. [21, vol. I, p. 68—128].
33. Bergman T. De praecipitatis metallics. Cm. [21, vol. II, p. 349—398].
34. Bergman T. De tubo ferruminatorio. Cm. [21, vol. II, p. 455—505].
35. Sage B. G. Analyse Chimique et Concordance des Trois Reignes: Vol. 1—3. P., 1786.
36. Sage B. G. Analyse des Bles. P., 1776. 118 p.
37. Sala A. Hydrealogia. Darinen wie man alleley Wasser, Olipeten und bren-nende spiritus. Rostok, 1633. 8°, 320 S.
38. Wenzel C. F. Lehre von der Verwandschaft der Korper. Dresden, 1777. 492 S.
39. Берцелиус И. Я. Руководство по разложению неорганических тел/Пер. с фр. СПб., 1833. 222 с.
40. Раскин А. А., Б ар ваковский В. П. Оборудование химической лаборатории Ломоносова.— В кн.: Ломоносовский сборник. М.; Л.: Изд-во АН СССР, 1951, вып. 3, с. 124—205.
41. Ihde A. J. The Development of Modern Chemistry. N. Y.; L., 1964. 851 p.
42. Kirwan B. An Essay on the Analysis of Mineral Waters. L., 1799. См. также нем. nep.: Richard Kirwan1s Esq. Physisch-chemische Schriften / Aus dem englischer iibcrsetzt von Lorenz von Crell: Bd. 1—3. Berlin; Stettin, 1796—1802.
43. Dann G. E. Martin Heinrich Klaproth (1743—1817). B.: Akad.-Verl., 1958. X+171 S.
404
ЛИТЕРАТУРА
44. Klaproth М, N. Beit rage zur Chemischen Kentniss der Mineralkorper: Bd. 1—4. Posen; Berlin, 1795—1803; Berlin; Leipzig, 1810. Bd. 5; B.; Stettin, 1815. Bd. 6.
45. Фигуровский II. А. Открытие элементов и происхождение их названий. М.: Наука, 1970. 207 с.
46. Крицман В. А. О работах Лавуазье по органической химии.— В кп.: Очерки по истории органической химии. М.: Паука, 1977, с. 5—20.
47. Ostwald W. Lebenslinen. В., 1926, Bd. 1, S. 222. Цит. по [22, с. 198].
48. Gay-Lussac. Р., 1852. 52 р.
49. a) Tenard L. Л Traite de chimie elementaire, theoretique et pratique. Vol. 1—5. 5e ed. P., 1813—1816; Paris; Bruxells, 1827. XXXIII+480 p. б) Рус пер.: Тенар Л. Основания химического разложения минеральных тел/Пер. с 5-го фр. изд. СПб., 1829. 1V+256 с.
50. Pfaff С. Н. Handbuch der analytischen Chemie. Bd. 1. Propadeutischer Theil oder Lehre von den Reagentien. Haupttheil 1. Analytische Chemie der anorganische Korper. Altona, 1821, XXIV+464 S.; Bd. 2. Die Fortsetzung der Ersten Haupttheilen der analytischen Chemie der anorganische Korper und den zweiten Haupttheil — die analytische Chemie der organischen Korper. Altona, 1822. XXII+696 S.
51. Страдынъ Я. П. Теодор Гротгус. 1785—1822. М.: Наука, 1966. 184 с.
52. Ilarkort Е. Die Probierkunst mit dem Lothrohre. Freiberg, 1827.
53. Гаркорд Э. Руководство к испытаниям посредством паяльной трубки, или наставление, как при пособии паяльной трубки определять с точностью содержания металлов в минералах и продуктах их. Соч. Эдуарда Гаркорда / Пер. с нем. СПб., 1829. Х+63 с.
54. Plattner С. F. Die Probierkunst mit dem Lotrohre. Freiberg, 1835. 6te AufL: Carl Friendrich Plattner’s Probierkunst mit dem Lotrohre. Eine vollstandige Anleitung zu qualitativen und quantitativen Lotroht-Untersuchungen/Be-arbeitet von Dr. F. Koibeck. Leipzig, 1897. XVI+488 S.; 7te AufL: Leipzig, 1927. XVI+500 S.
55. Berzelius J. J. Essai sur la theorie de proportions chimiques et sur 1’influen-ce chimique de I’electricite. P., 1819. 122 p.
56. Rose H. Handbuch der analytischen Chemie von Heinrich Rose. Ite AufL: B., 1829. VII+620 S.; 3te AufL: Die Lehre von den qualitativen chemisch-analytischen Untersuchungen. B., 1833, Bd. 1. 657 S. Die Lehre von den quantitativen chemisch-analytischen Untersuchungen. B., 1834, Bd. 2. IV+819 S.
57. Розе Г. Аналитическая химия Генриха Розе, переведенная с немецкого с третьего издания и частично переделанная в таблицы корпуса горных инженеров капитаном II. Евреиновым. СПб., 1837. 308 с.
58. Fresenius С. R. Geschichte des chemische Laboratorium in Wiesbaden. Wiesbaden, 1873.
59. Fresenius C. R. Anleitung zur qualitativen chemischen Analyse, oder die Lehre von den operationen, von den Reagentien und von dem Verhalten der bekannteren Korper zu Reagentien, sowie systematisches Verfaren zur Auffindung der in der Pharmacie haufige Korper in einfachen und zusam-mengesetzten Verbindungen. Ite AufL: Bonn, 1841; 5te AufL: Braunschweig, 1847. XX+293 S.
60. Фрезениус К. P. Аналитическая качественная химия или учение о производствах, реагенциях... / Первый рус. пер. с 5-го нем. изд. СПб., 1848; Фрезениус К. Р. Руководство к качественному химическому анализу. Т. 1, 2 / Пер. с 9-го нем. изд. М., 1858.
61. Fresenius Н. Zur Erinerung an R. Fresenius. Wiesbaden, 1898.
62. Fresenius C. R. Anleitung zur quantitativen chemischen Analyse. Ite AufL Braunschweig, 1845.
63 Фрезениус К. P. Минеральный количественный анализ/Пер. с 6-го нем. изд. СПб., 1875. ХХ+711 с.
64. Zeitschrift fhr analytische Chemie / Herausgegeben von Dr. C. Remigius Fresenius Herz. Nass. Gen Hofrathe, Direktor des Chemisches Laboratoriums
405
Литература
zu Wiesbaden und Professor der Chemie, Physik und Technologie am land-wirtschaftliches Institute dasselbst. Erster Jahrgang. Wiesbaden, 1862. C 1947 г. выходит под названием: Fresenius Zeitschrift fur analytische Chemie.
65. Gay-Lussac. Instruction sur 1’essai du chlorure de chaux.— Ann. chim. et phys., 1824, vol. 26, p. 162—175.
66. Jounniaux A. Les origines fran$aises de la shimie analytique. P., 1938. 59 p. (Ser. Actualites scientifiques et industrielles; N 707).
67. Gay-Lussac. Instruction sur 1’essai des potasses du commerce.— Ann. chim. et phys., 1828, vol. 39, p. 337. См. также [66, p. 11—51].
68. Gay-Lussac. Instruction sur 1’essai des matieres d’argent par la voie hu-mide. Cm. [70, c. 137—244].
69. a) Mohr F. Lehrbuch der chemisch-analytischen Titriermethode. Nach eige-nen Versuchen und systematise!! dargestellt. Fiir Chemiker, Artze und Far-maceuten, Berg- und Iluttenmanner, Fabrikanten, Agronomen, Metallurgen, Miinzbeamte u.s.w. in zwei Abtheilungen. Ite Abth.: Braunschweig, 1855.’ XVI, 396 S.; 2te Abth.: Braunschweig, 1856. VIII+272 S. 6) Pyc. nep.: Mop Ф. Руководство к химическому анализу — мерою (метода титрования). СПб., 1859. 599 с.
70. Vauquelin L. Manuel complet de 1’essayeur: Par Vaquelin. Suivi de 1’inst-ruction de M. Gay-Lussac sur 1’essai des maiteres d’argent par la voie hu-mide et des dispositions du laboratoire de la monnaie de Paris. Nouv. (VII) ed. P., 1836. 248 p.
71. Тредвел Ф. Курс аналитической химии. Т. II. Количественный анализ. Кн. 2. Объемный и газовый анализ / Пер. с нем. М.; Л.: Госиздат, 1927. 329 с.
72. Bunsen R. Jodometrie.— Lieb. Ann., 1853, Bd. 86, S. 265—274.
73. Marqueritte F. Sur un nouveau procede de dosage du fer par la voie humi-de.—Ann. chim. et phys., 1846. Ser. 3, vol. 18, p. 244—256. См. также [66, p. 28—35].
74. Schwarz K. Praktische Anleitung zu MaaBanalysen (Titrier-Methode). Be-sonders in ihren Anwendung auf die Bestimmung des technischen Werthers der chemischen Handelsproducte: Potasche, Soda, Ammoniak, Chlorkalk, Jod, Brom, Braunstein, Sauren, Arsen, Chrom, Eisen, Kupfer, Zink, Zinn, Blei, Silber, Indigo u. a. Ite Aufl.: Braunschweig, 1850. 100 S.; 2te Aufl.: Braunschweig, 1853. 150 S.
Глава пятая
1. Меншуткин H. А. Очерк развития химических воззрений. СПб., 1888.
2. Gmelins Handbuch der anorganischen Chemie. 8-e Aufl. В., Bd. 1, 1926.
3. Partington J. R. A History of Chemistry: Vol. 1—4. L., 1961—1970.
4. The Encyclopedia of the Chemical Elements I Ed. C. A. Hampel. N. Y.; L., 1968. 849 p.
5. Mellor J. W. A Comprehensive Treatise on Inorganic and Theoretical Chemistry: Vol. 1—16. L., 1922—1938.
6. Weeks M. E. Discovery of Elements. 5th ed. N. Y., 1945. XIV+578 p.
7. Фигуровский H. А. Открытие элементов и происхождение их названий. М.: Паука, 1970. 207 с.
8. Альтшулер С. В. и др. Открытие химических элементов. М.: Просвещение, 1980. 174 с.
9. Трифонов Д. Н., Кривомазов А. Н., Лисневский Ю. И. Химические элементы и нуклиды. Специфика открытий. М.: Атомиздат, 1980. 156 с.
, 10. Lavoasier A. Traite elementaire de Chimie. P., 1801, vol. 1. XLIV+386 p.
И. Дэви Гемфри. О некоторых химических действиях электричества. М.; Л.: Гостехтеоретиздат, 1933. 160 с.
12. Gay-Lussac. Untersuchungen uber das Jod (1814). Leipzig, 1889. 52 S. (Ostwald’s Klassiker der exakten Wissenschaften, N 4).
406
1
ЛИТЕРАТУРА
13. Либих Ю. Письма о химии/Пер. с нем. под род. П. П. Алексеева. Т. 1, 2. СПб, 1861.
14. Трифонов Д. Н. Редкоземельные элементы и их место в периодической системе. М.: Наука, 1966. 192 с.
15. Dalton J. A New System of Chemical Philisophy. Manschestcr, 1810. Pt IL
16. Менделеев Д. И. Периодическая законность химических элементов.— В кн.: Менделеев Д. И. Периодический закон. М.: Изд-во АП СССР, 1958, с. 102—176. (Классики науки.)
17. Berzelius J. Lehrbuch der Chemie. 3le Aufl. Dresden; Leipzig, 1835, Bd. V.
18. Фигуровский H. А. Очерк общей истории химии. Развитие классической химии в XIX столетии. М.: Наука, 1979. 477 с.
19. Prout W. On the Relation between the Specific Gravities of Bodies in their Gaseous State and the Weights of their Atoms.— Ann. Philos, 1815, vol. 6, p. 321—330.
20. Праут В. Об отношении между удельными весами тел в их газообразном состоянии п весами их атомов.— Успехи химии, 1940, т. 9, вып. 2/3, с. 285—292. Исправление ошибки в статье «Об отношении между удельными весами тел в их газообразном состоянии и весами их атомов».— Там же, с. 293—294.
21. Dobereiner J. W. Darstellung der Verhaltnisszahlen der irdischen Elemente zu chemischen Verbindungen. Jena, Iste Aufl. 1816; 2te Aufl, 1823.
22. Dobereiner J. W.— Ann. Phys. (Gilbert), 1817, Bd. 57, S. 331.
23. Dobereiner J. W.~ Pogg. Ann, 1829, Bd. 15, S. 301—307; перепечатано: Dobereiner J. W., Pettenkofer M. Abhandlungcn uber die Anfiinge des na-turlichen Systemes der chemischen Elemente, nebst einer geschichtlichen Ubersicht der Weiterentwicklung der Lehre von der Triaden der Elemente) Hrsg. von Lothar Meyer. Leipzig, 1895. (Ostwald’s Klassiker der exacten Wissenschaften; N 66).
24. Рабинович E., Тило Э. Периодическая система элементов: История и теория. М.; Л.: Гостехтеоретиздат, 1933. 409 с.
25. Van Spronsen J. W. The Periodic System of Chemical Elements: A History of the First Hundred Years. Amsterdam etc, 1969. 368 p.
26. Gmelin L. Handbuch der Chemie. 4te Aufl. Heidelberg, 1843, Bd. 1, S. 48.
27. Partington J. R. A History of Chemistry. L, 1964. Vol. 1.
28. Lenssen E. Uber die Gruppirung der Elemente nach ihrer chemisch-physi-kalischen Charakter.— Ann. Chem. (J. Lieligs), 1857, Bd. 103, S. 121—131.
29. Менделеев Д. И. К истории периодического закона.— В кн.: Менделеев Д. И. Избр. соч. Т. 2. Л.: Изд-во АН СССР, 1934, с. 282—289.
29а. Менделеев Д. И. Периодическая законность химических элементов: Фарадеевское чтение проф. Д. Менделеева в Лондонском химическом обществе, 23 мая / 4 июня 1889 года.— Там же, с. 347—366.
30. Odling W. On the Natural Grouping of the Elements.— Philos. Mag, 1857. Ser. 4, vol. 13, p. 423—439, 480—497.
31. Odling W. A Manual of Chemistry. L, 1861. Vol. 1.
32. Odling W. On the Proportional Numbers of the Elements.— Quart. J. Sci, 1864, vol. 1, p. 642-648.
33. Odling W. A Course of Practical Chemistry. L, 1865; 3rd ed. L, 1868.
34. Watts H. A Dictionary of Chemistry. L, 1868. Vol. 3.
35. Newlands J. A. R. On Relation among the Equivalents.— Chem. News, 1863, vol. 7, p. 70-72.
36. Newlands J. A. R. Relations between Equivalents.— Ibid, 1864, vol. 10, p 59______go, 94_95,
37. Newlands J. A. R. On the Law Octaves.— Ibid, 1865, vol. 12, p. 83; 1866, vol. 13, p. 113, 114, 130.
38. Newlands J. A. R. On the Discovery of the Periodic Law and on Relations among the Elements. L, 1884. VI11+39 p.
39. Smeaton W. Centenary of the Law of Octaves.— J. Roy. Inst. Chem, 1964, vol. 88, p. 271-274.
407
ЛИТЕРАТУРА
40.
41.
42.
43.
44.
Chancourtois А. Е. В. de. Memoire sur un classement naturel des corns simples ou radicaux appele vis tellurique.— Compt. rend., 1862, vol p. 757—761, 840—843, 967—971. * °4’
Chancourtois A. E. B. de. Vis teleurique, classement naturel des corps simples ou radicaux, obtenu au moyen d’un systeme de classification helicoidai et numerique. P.: Mallet-Bachelier, 1863. 21 p.; Tableau du classement naturel des corps simples, dit vis tellurique.— Ibid., 1863, vol. 56, p. 253, 479 Чугаев Л. А. Периодическая система химических элементов. СПб. IQm’ XVII1+258 с.
Meyer L. Die modernen Theorien der Chemie und ihre Bedeutung fur die chemische Statik. Breslau, 1864.
Mak-Donald D. A History of Platinum. L., 1960. 254 p.
Глава шестая
1. Кедров Б. М. Три аспекта атомистики. II. Учение Дальтона: Исторический аспект. М.: Наука, 1969, с. 14—32, 36—43.
2. Henry W. Ch. Memoirs of the Life and Scientific Researches of John Dalton. L., 1854; Кедров Б. M. Атомистика Дальтона. M.; Л.: Госхимиздат, 1949. 312 с.; John Dalton and the Progress of Science. Manchester, 1968' 352 p.
3. Дальтон Дж. Сборник избранных работ по атомистике (1802—1810) / Пер. с англ. А. Л. Либермана; ред. и прим. Б. М. Кедрова. Л.: THTHXJI, 1940. 243 с.
4. Thomson Т. A. System of Chemistry of Inorganic Bodies. 3 rd ed. Edinburgh, 1807, vol. 3, p. 424.
5. Dalton J. A New System of Chemical Philosophy. Manchester, 1810, pt 2, p. 236.
6. Smith B. Memoir of John Dalton and History of the Atomic Theory up to his Time. L., 1856.
7. Boscoe H. E.f Harden A. A New View of the Origin of Dalton’s Atomic Theory; A Contribution to Chemical History, together with Letters and Documents Concerning the Life and Labours of John Dalton. L., 1896.
8. Вюрц А. История химических доктрин от Лавуазье и до настоящего времени. СПб., 1869, с. 17.
9. Канниццаро С. Исторический обзор применения атомистической теории к химии и систем формул, выражающих строение соединений. Увив. изв. (Киев), 1873, № 1, с. 15.
10. Демокрит в его фрагментах и свидетельствах древности: Сборник статей. М.: Гос. соц.-экон. изд-во, 1935. 382 с.
И. Guerlac Н. The Background to Dalton’s Atomic Theory.— In: John Dalton and the Progress of Science. Manchester, 1968, p. 57—91.
12. Die Grundlagen der Atomtheorie. Leipzig, 1921, S. 12 (Ostwald’s Klassiker; N 3).
13. Ладенбург А. Лекции по истории развития химии от Лавуазье до нашего времени. Одесса, 1917. VIII+690 с.
14. Герц Г. Очерк истории развития основных воззрений химии. Л., 1924, с. 59—60.
15. Мейер Э. История химии от древнейших времен до настоящих дней. СПб., 1899, с. 156—158.
16. Jac. Berzelius Bref. 2. Lettres detachees (1809—1847) / Ed. H. G. Soderbaum. •Uppsala: Almquist, 1920, sect. VII, vol. 3. 320 p.
17. Hisinger W., Berzelius J. Versuche fiber die Wirkung der elektrischen Saule auf Salze und einige von ihren Basen.— Neues allg. J. Chem. (Hgn. von A. F. Gehlen), 1803, Bd. 1. S. 115—149.
18. Дмитриев И. С.— В кн.: Соловьев IO. II., Курипион В. И. Якоб Берцелиус» Жизнь и деятельность. 2-е изд. М.: Наука, 1980, с, 93—162,
408
ЛйтературА
19. Менделеев Д. И. Основы химии. 8-е изд. СПб., 1906. 816 с.
20. Berzelius J. J. a) Forsok rorande de bestamde proportioner, hvari den cor-ganiska naturens bestandsdelar finnas forenade.— Afhandl. fys., kemi och miner., 1810, Bd. 3, s. 162—276. б) Перепечатка с добавлениями: Versuch, die bestimmten und einfachen Verhaltnisse aufzuf inden, nach welchen die Bestandtheile der unorganischen Natur mit einander verbunden sind.— Ann. Phys. (Hgn. von L. W. Gilbert), 1811, Bd. 37, S. 249—334, 415—472. в) Фр. nep.: Suite des experiences sur les proportions determinees, d’apres les-quelles les elemens de la nature inorganique s’unissent.— Ann. chim., 1811, t. 79, p. 113—142, 233—264; 1811, t. 80, p. 5-37.
21. Berzelius J. J. Fortsetzung des Versuchs, die bestimmten ein fachen Verhaltnisse aufzufinden, nach welchem die Bestandtheile der unorganischer Natur mit einander verbunden sind. (Erste, Zweite, Dritte).— Ann. Phys. (Hgn. von L. W. Gilbert), 1811, Bd. 38, S. 161—226; 1812, Bd. 40, S. 162— 208, 235—320.
22. Wollaston W. H. On Super-acid and Sub-acid Salts.— Philos. Trans. Roy. Soc., 1808, vol. 98, p. 96—102; J. Natur. Philos., Chem. and the Arts (Ed. W. Nicholson), 1808, vol. 21, p. 164—169.
23. Berzelius J. J. Om de bestamda proportioner hvari den oorganiska naturens bestandsdelar finnas forenade.— Kongl. vetenskapsakad. handlinger, 1811, s. 169—197.
24. Berzelius J. J. Essay on the Cause of Chemical Proportion, and on some Circumstances Relating to them; Together with a Short and Easy Method of Expressing them.— Ann. Philos., 1813, vol. 2, p. 443—454; 1814, vol. 3, p. 51-62, 93—106, 244—257, 353—364.
25. Berzelius J. J. Memoire sur la composition des Acides phosphorique et phosphoreux ainsi que de leurs combinaisons avec des bases salines.— Ann. chim. et phys., 1816. Ser. 2, vol. 2, p. 151—176, 217—240, 329—339.
26. Berzelius J. J. Untersuchungen iiber die Zusammcnstetzung der Phosphor-saure, der phosphorigen Saure und ihrer Salze.— Ann. Phys. (Hgn. von L. W. Gilbert). N. F„ 1816, Bd. 53, S. 393—446; Bd. 54, S. 31—55.
27. Lundgren A. Berzelius och den kamiska atomteorin. Uppsala: Almquist and Wiksell, 1979. 210 s.
28. Melhado E. M. Jac. Berzelius: Foundations and Development of his Chemistry. Princeton: Univ, press, 1977. 645 p.
29. Berzelius J. J. Ueber das Verhaltniss der Sauerstoffmengen etc. zu einander, welche die Korper auf verschiedenen Oxydationsstufen aufnahmen, etc. (Aus einem Schreiben and den Akademiker Gehlen vom 14 Aug. 1810).— J. Chem. und Phys. (Hgn. von J. S. C. Schweigger), 1811, Bd. 1, S. 257—262.
30. Berzelius J. J. a) Forsok till ett riittfardigande at de theoretisk chemiska asigter, pa hvilka den systematiska Uppstallningen i mitt forsoket till en forbattring af den chemiska nomenclaturen sig.— Kongl. vetenkapsacade-miens nyahandlinger, 1812, D. 33, s. 166—198, 233—244; 1813, D. 34, s. 39—62, 175—215. б) Нем. nep.: Versuch, die chemischen Ansichten, welche die systematische Aufstellung der Korper, in meinem Versuch einer Verbesserung der chemischen Nomenclatur begriinden, zu rechtfertigen.— J. Chem. und Phys. (Hgn. von J. S. C. Schweigger), 1812, Bd. 6, S. 119—144, 144—176, 284—322; 1813, Bd. 7, S. 43—78.
31. Дмитриев И, С. Обоснование и развитие химической атомистики.— В кн.: Соловьев Ю. И., Куринной В. И. Якоб Берцелиус: Жизнь и деятельность. М.: Наука, 1980, с. 44—75.
32. Thomson Т. On the Compounds of Sulphur and Oxygen.—J. Nat. Philos. Chem. and the Arts (Ed. W. Nicholson), 1803, vol. 6, p. 92—109.
33. Bucholz C. F. Versuche iiber Thomson’s schwefelhaltige Salzsaure; zur nii-heren Kenntniss dieser mcrkwurdigen Verbindung.—Neues allg. J. Chem. (Hgh. von A. F. Gehlen), 1810, Bd. 9, S. 172—188.
34. Berthollet A. B. Memoire sur combinaison du soufre avec 1’oxigene et 1’aci-de muriatique.— Mem. Soc. d’Arcueil, 1807, vol. 1, p. 161—179.
409
ЛИТЕРАТУРА
35. Berzelius J. J. Experiment on the Nature of Asote, of Hydrogen, and of Ammonia, and of the Degrees of Oxidation of which Azone is Susceptible.— Ann. Philos., 1813, vol. 2, p. 276—284, 357—368.
36. Lavoisier A. L. Traite elementaire de chimie, presente dans un ordre nouveau et d’apres les decouvertes modernes. P., 1789. Vol. 1.
37. Berzelius J. J. Essai sur la theorie des proportions chimiques et de 1’in-fluence chimique de I’electricite. P., 1819.
38. Jac. Berzelius Bref. Correspondance entre Berzelius et A. Marcet (1812— 1822) / Ed. H. G. Soderbaum. Uppsala: Almquist, 1914, sect. Ill, vol. 1. 198 p.
39. Berzelius J. J. Nouvelles recherches sur les proportions chimiques.— Ann. chim. et phys., 1817, vol. 5, p. 174—181.
40. Berzelius J. J. a) Forsok att narmare bestamma atskilliga oorganiska krop-pars sammansattning, till vinnande af en narmare utveckling af laran om de kemiska proportionerna.— Afhandl. fyk., kemi och miner., 1818, D. 5, s. 379—520. б) Нем. nep.: Versuche die Zusammensetzung verschiedener unorganischer Korper naher zu bestimmen, zur genaueren Entwickelung der Lehre von den chemischen Proportionen.— J. Chem. und Phys. (Hgn. von J. S. C. Schwegger), 1818, Bd. 23, S. 98—122, 129—202, 279—299.
41. Berzelius J. J. Forsok att genom anviindandet af den electrokemiska theo-rien och de kemiska proportionerna gundliigga ett rent vetenskapligt system for mineralogien. Stockholm, 1814. 104 s.
42. Фаерштейн M. Г. История учения о молекуле в химии (до 1860 г.). М.: Изд-во АИ СССР, 1961, с. 131—148.
42а. Berzelius J. J. Versuch fiber die Theorie der chemischen Proprotionen. Dresden, 1820. 200 S.
43. Berzelius J. Gleichformige Zusammenlegen der Atome bringt gleiche Crystalform hervor.— Jahres-Berichte, Tubingen, 1823, Jg. 2, S. 41—43.
44. Berzelius J. Uber die Bestimmung der relativen Anzahl von einfachen Ato-men in chemischen Verbindungen.— Pogg. Ann., 1826, Bd. 7, S. 397—416; Bd. 8, S. 1—24.
45. Meldrum A. Avogadro and Dalton: The Standing in Chemistry of their Hy-pothese. Edinburg: W. F. Clay, 1904. 113 p.
46. Berzelius J. Traite de chimie. 2e ed. P.: F. Didot, 1847, vol. 4, 615 p.
47. Berzelius J. Atomgewichte der einfachen Korper.— Jahres-Berichte, Tubingen, 1828, Jg. 7, S. 79-91.
48. Kopp H. Die Entwicklung der Chemie in der meueren Zeit. Munchen: R. 01-denbourg, 1873. 854 S.
49. Меншуткин H. А. Очерк развития химических воззрении. СПб., 1888. 394 с.
50. Delacre М. Histoire de la chimie. P.: Gauthier-Villars, 1920. 632 p.
51. Berzelius J. Lehrbuch der Chemie. 3te Aufl. Dresden; Leipzig: Arnold, 1835, Bd. 5. 484 S.
52. Berzelius J. Verhaltnisse zwischen Atomen und Volumen.— Jahres-Berichte, Tubingen, 1834, Jg. 13, S. 59—63.
Глава седьмая
1. Crosland M. P. Gay-Lussac, Scientists and Bourgeois. Cambridge: Univ, press, 1978. 333 p.
2. Gay-Lussac J. L. Lemons de physique de la Faculte des Sciences de Paris, recueillies et redigees par M. Grosselin: En 2 vol. P., 1828. Vol. 1.
3. Crosland M. P. The Library of Gay-Lussac.— Ambix, 1981, vol. 28, pt 3, N 3, p. 158—170.
^.'Gay-Lussac J. L. Memoire sur 1’iode.— Ann. chim. ou rec. mem. concern, chim. et arts qui en depend., 1814, vol. 91, p. 5—160. Перепечатка: Ann. Philos., 1815, vol. 5, p. 101—109, 207—214, 296—302, 401—413; vol. 6, p. 124— 132; 183—198 (цитата в основном тексте дана по английскому варианту статьи).
410
ЛИТЕРАТУРА
5. Дмитриев И. С. Открытие Гей-Люссака и его влияние на теоретические взгляды Берцелиуса.— В кн.: Соловьев Ю. И., Куринной В. И. Якоб Берцелиус: Жизнь и деятельность. 2-е изд. М.: Наука, 1980, с. 63—71.
6. Gay-Lussac J. L. Memoire sur la combination des substances gazeuses, les unes avec les autres.— Mem. phys. et chim. Soc. Arcueil, 1809, vol. 2, p. 207—234.
7. Фаерштейн M. Г. История учения о молекуле в химии (до 1860 г.). М.: Изд-во АН СССР, 1961. 368 с.
8. Rocke A. J. Gay-Lussac and Dumas: Adherents of the Avogadro — Ampere Hypothesis? — Isis, 1978, vol. 69, N 249, p. 595—600.
9. Dumas J.-B. Sur quelques points de la theorie atomistique.— Ann. chim. et phys., 1826. Ser. 2, vol. 33, p. 337—391.
10. a) Gay-Lussac J. L., Becquerel A. C. Le rapport a 1’Academie des Science sur le travail de M. Gaudin: «Recherches sur les atomes» (5 Nov. 1832).— Proces-verb. seances Acad, tenues depuis fondation Inst, jusqu’au mois d’Aout 1835, 1922, vol. 10, p. 142—145. б) Этот отзыв был также опубликован в кн.: Gaudin М. A. L’Architecture du monde des atomes. P.: Gauthier-Villars, 1873, а также в кн.: Actes du colloque Gay-Lussac (11—13 decemb-re 1978). Palaiseau: Ecole polytechn., 1980, p. 149—163.
Основные идеи Годен изложил затем в следующих двух статьях: в) Gaudin М. A. Recherches sur la structure intime des corps inorganique definis, et considerations generales sur le role que jouent leurs dernieres particules dans les principaux phenomenes de la nature.— Ann. chim. et phys., 1833, vol. 52, p. 113—133; 6) Gaudin M. A. Note sur quelques pro-prietes des atomes.— Bibl. Univ. Sci., Belles-Lettres et Arts (Geneve). Sci. et Arts, 1833, vol. 52, p. 124—111.
11. Gay-Lussac J. L., Thenard L, Memoire sur 1’acide fluorique.— Ann. chim. ou rec. mem. concern, chim. et arts qui en depend., 1809. Ser. 1, vol. 69, p. 204—224.
12. Wollaston W. H. On Super-acid and Sub-acid Salts.— Philos. Trans. Roy. Soc., 1808, vol. 98, p. 96—102.
13. Thomson Th. On Oxalic Acid.— Ibid., p. 63—95.
14. Дальтон Дж. Сборник работ по атомистике (1802—1810) / Пер. с англ. А. Л. Либермана; ред. и прим. Б. М. Кедрова. Л.: Госхимиздат, 1940. 243 с.
15. Avogadro A. Essai d’une maniere de determiner les masses relatives des molecules elementaires des corps, et les proportions selon lesquelles elles entrent dans ces combinaisons.— J. phys. chim. et hist. Natur. (Ed. J. G. de Lametherie), 1811, vol. 73, p. 58—76.
16. Cole T. (jr.). Dalton, Mixed Gases, and the Origin of the Chemical Atomic Theory.— Ambix, 1978, vol. 25, pt 2, N 2, p. 117—130.
17. Brooke J. H. Avogadro’s Hypothesis and its Fate: a Case-study in the Failure of Case-studies.— Hist. Sci., 1981, vol. 19, p. 235—273.
18. Fisher N. W. Avogadro, the Chemists...— Ibid., 1982, vol. 20, N 48, p. 77.
19. Fricke M. The Rejection of Avogadro’s Hypotheses.— In: Method and Appraisal in the Physical Sciences/Ed. C. Howson. Cambridge: Univ, press, 1976, p. 277—307.
20. Mundy B. W. Avogadro on the Degree of Submolecularity of Molecules.— In: Chymia. Philadelphia: Univ, of Penna press, 1967, vol. 12, p. 151—155.
21. Bonner J. K. Amedeo Avogadro: a Reassessment of his Research and its Place in Early Nineteenth Century Science: Ph. D. Diss. Baltimore: John Hopkins Univ., 1974. 356 p.
22. Fox R. The Caloric Theory of Gases. From Lavoisier to Regnault. Oxford: Clarendon press, 1971. 378 p.
23. Fox R. The Rise and Fall of Laplacian Physics.— In: Historical Studies in the Physical Sciences/Ed. R. McCormmach. Princeton: Univ, press, 1975, Sixth Annual Vol., p. 89—136.
24. Coley N. G. The Physico-chemical Studies of Amedeo Avogadro.— Ann. Sci., 1964 (publ. Nov. 1965), vol. 20, N 3, p. 195—210. См. также: Crosland M. Л
411
ЛИТЕРАТУРА
Avogadro.— In: Dictionary of Scientific Biography / Ed. С. G. Gillispie. 1970 vol. 1, p. 346-347.
25. Morselli M. A. The Manuscript of Avogadro’s «Essai d’une maniere de determiner les masses relatives des molecules elementaires».— Ambix, 1980 vol. 27, pt 3, N 3, p. 147—172.
26. Быков Г. В. Амедео Авогадро: Очерк жизни и деятельности. М.: Паука 1970. 184 с.
27. Avogadro A. Memoire sur les masses relatives des molecules des corps simples ou densites presumees des gaz, et sur la constitution de quel-ques-uns de leurs composes, pour servir de suite а Г«Essai» sur le meme sujet.— J. phys., chim. et hist. Natur. (Ed. par J. C. de Lametherie), 1814, vol. 78, p. 131—156.
28. Avogadro A. Memoria sui calorici specifici dei corpi solidi e liquidi.— Mem. Soc. I tai. Sci. residente Modena, 1832, vol. 20, p. 451—486. Перепечатка: Ann. chim. et phys., 1833. Ser. 2, vol. 55, p. 80—111 (ссылка в основном тексте дана на французский перевод статьи).
29. Mollet J. De la constitution in time des gaz et de leur capacite pour le ca-lorique.— J. phys., chim., et hist. Natur. (Ed. par J. C. de Lametherie), 1820, vol. 90, p. 13—32.
30. Avogadro A. Nouvelles considerations sur la theorie des proportions deter-minees dans les combinaisons, et sur la determination des masses des molecules des corps.— Mem. Reale Accad. Sci. Torino, 1821, vol. 26, p. 1—162.
31-32. Dumas J. B. Traite de Chimie Appliquee aux Arts. P.: Bechet Jeune, 1828, vol. 1. 420 p.
33. Dumas J. B. Dissertation sur la densite de la vapeur de quelques corps simples.— Ann. chim. et phys., 1832. Ser. 2, vol. 50, p. 170—178.
34. Baudrimont A. E. Introduction a 1’etude de la chimie par la theorie atomi-que. P., 1833.
35. Baudrimont A. E. Traite de chimie generale et experimentale: 2 vol. P., 1844—1846.
36. Dumas J. B. Legons sur la philosophic chimique. P., 1837, p. 224, 227.
37. Соловьев Ю. И., Куринной В. И. Якоб Берцелиус. М.: Наука, 1980. 319 с.
38. Berzelius J. J. Versuch uber die Theorie der chemischen Proportionen. Dresden, 1820.
39. Фаерштейн M. Г. Шарль Жерар (1816—1856). M.: Паука, 1968. 163 с.
40. Жерар Ш. Введение к изучению химии по унитарной системе Шарля Жерара. СПб., 1859.
41. Фаерштейн М. Г. О роли Д. И. Менделеева в утверждении закона Авогадро.— Тр. ИИЕиТ, 1955, т. 6, с. 68—85.
42. Менделеев Д. И. Соч. Т. 25. М.; Л.: Изд-во АН СССР, 1952. 385 с.
43. Быков Г. В., Крицман В. А. Станислао Канниццаро: Очерк жизни и деятельности. М.: Наука, 1972. 214 с.
44. Канниццаро С, Исторический обзор применения атомистической теории к химии и систем формул, выражающих строение соединений.— Унив. изв. (Киев), 1873, № 1/6.
45. Фаерштейн М. Г. К столетию Первого Международного конгресса химиков в Карлсруэ.— Вопросы истории естествознания и техники, 1960, вып. 10, с. 24—34.
46. Frankland Е. Uber eine Reihe organischer Korper, welche Metalle enthal-ten.— Ann. Chem. Pharm., 1853, Bd. 85, S. 329.
47. Гъелът Э. История органической химии с древнейших времен до настоящего времени. Харьков; Киев: ОНТИ НКТП, 1937, с. 135.
48. Palmer W. A History of the Concept of Valency to 1930. Cambridge: Univ, press, 1965. VII+177 p.
49. Rassel C. The History of Valency. Oxford, 1971. 373 p.
50. Быков Г. В. Август Кекуле. М.: Наука, 1965, с. 47.
51. Kekule A. Uber die Constitution des Knallquecksilbers.— Ann. Chem. Pharm,, 1857, Bd. 101, S. 200—213.
412
ЛИТЕРАТУРА
52. Anschutz R. August Kekule: Leben und Werken. B., 1929, Bd. 1. 708 S.
53. Столетие теории химического строения: Сборник статей. М.: Изд-во АП СССР, 1961. 148 с.
54. Congres des chimistes a Carlsrue. Moniteur Scientifique de Quesneville. P., 1860, vol. 2.
55. Kekule A. Ueber die Constitution des Mesitylens.— Ztschr. Chem., 1867, [2], Bd. 3, S. 214-218.
56. Wichelhaus H. Ueber die Verbindungen des Phosphors.— Liebig’s Ann., 1868, Suppl., Bd. 6, S. 257—280.
57. Меншуткин Б. II. Курс общей (неорганической) химии. М.: Гос. техн, изд-во, 1924. 373 с.
58. Friend J. N. The Theory of Valency. 2nd ed. L., 1915. 192 p.
59. Kekule A. Lehrbuch der organischen Chemie. Erlangen: Enke, 1866, Bd. II. 744 S.
60. Быков Г. В. История классической теории химического строения. М.*. Изд-во АН СССР, 1960. 312 с.
61. Wurtz A. Cours de philosophie chimique. P., 1864. 80 p.
62. Delavaud Ch. Sur 1’atomicite.— Bull. Soc. chim. (Paris), 1865, vol. 4, p. 421—429.
63. Blomstrand C. Die Chemie der Jetztzeit. Heidelberg, 1869. 462 S.
64. Frankland E. Contributions to the Notation of Organic and Inorganic Compounds.— J. Chem. Soc., 1866, vol. 19, p. 372—395.
Глава восьмая
1. Менделеев Д. II. Основы химии. М.; Л.: ГНТИ, 1947, т. 1. 624 с.
2. Менделеев Д. И. Периодический закон. Дополнительные материалы. М.: Изд-во АН СССР, 1960, 711 с. (Классики науки.)
3. Менделеев Д. И. Периодический закон. М.: Изд-во АН СССР, 1958. 830 с. (Классики науки.)
4. Кедров Б. М. О механицизме в химии.— Успехи химии, 1952, т. 21, с. 969-996.
5. Менделеев Д. И. Органическая химия. Соч. Т. 8. М.; Л.: Изд-во АН СССР, 1948. 664 с.
6. Бернал Д. Наука в истории общества. М.: Изд-во иностр, лит., 1956, с. 166.
7. Менделеев Д. И. Учение о промышленности. СПб., 1901, т. I, ч. 1, с. 199.
8. Менделеев Д. И. Научный архив. Т. 1. Периодический закон. М.; Л.: Изд-во АН СССР, 1953. 866 с.
9. Менделеев Д. И. Соч. Т. 1. М.; Л.: Изд-во АН СССР, 1937. 348 с.
10. Graham Т., Otto F. J. Ausfiihrliches Lehrbuch der Chemie. Braunschweig, 1847, Bd. 1, S. 609—650.
И. Менделеев Д. И. Соч. T. 25. М.; Л.: Изд-во АН СССР, 1954. 804 с.
12. Щукарев С. А., Добротин Р. Б. Первые научные работы Д. И. Менделеева как этап на пути к открытию периодического закона.— Вестн. ЛГУ, 1954, № 2, с. 165—177.
13. Менделеев Д. И. Соч. Т. 15. М.; Л.: Изд-во АН СССР, 1949. 647 с.
14. Менделеев Д. И. Соч. Т. 2. М.; Л.: Изд-во АН СССР, 1937.
15. Dumas J. В.— Compt. rend., 1854, vol. 39, р. 1037—1069.
16. Cooke J. P.— Amer. L, 1854, vol. 14, p. 211.
17. Научный архив Д. И. Менделеева при ЛГУ 11-А-17 1-1. Лекции 1857 г.
18. Schtrecker A. Theorien und Experimente fur Bestimmung der Atomgewicht der Elemente. Braunschweig, 1859, S. 18.
19. Научный архив Д. И. Менделеева при ЛГУ. Библиотека Д. И. Менделеева 1-16-3.
20. Nordenskiold N. Ueber die atomisches Mineralsystem und das examinations System der Mineralien. B., 1849.
413
ЛИТЕРАТУРА
21. Менделеев Д. И. Основы химии. 1-е изд.— Соч. Т. 14. МЛ.: Изд-во АН СССР, 1949, 943 с.
22. Архив Д. И. Менделеева. Т. 1. Л.: Изд-во ЛГУ, 1951.
23. Меншуткин Б. Н. Химия и пути ее развития. М.; Л.: Изд-во АП СССР 1937. 351 с.
23а. Менделеев Д. И. Соч. Т. 13. М.; Л.: Изд-во АН СССР, 1949. 850 с.
24. Кедров Б. М. День одного великого открытия. М.: Соцэкгиз, 1958. 580 с.
25. Менделеев Д. И. Новые материалы по истории открытия периодического закона. М.; Л.: Изд-во АН СССР, 1950. 145 с.
26. Менделеев Д. И. Основы химии. 3-е изд. СПб., 1877. ХП+1434 с.
27. Добротин Р. Б., Макареня А. А. Прогнозирование свойств скандия и германия в работах Д. И. Менделеева.— В кн.: Прогнозирование в учении о периодичности. М.: Наука, 1976, с. 53—70.
28. Mendelejefj D. I. Zur Geschichte des periodischen Gesetzes.— Ber. Deutsch, chem. Gesellsch., 1880, Bd. 13, S. 1796—1804.
29. Mendel]eff D. I. Die periodische Gestzmassigkeit der chemischen Ele-mente.— Ann. Chem. Pharm., 1871, Suppl., Bd. 8, S. 133—229.
30. Lecoq de Boisbaudran P. E. Sur la constitution des spectres lumineux.— Compt. rend., 1869, vol. 69, p. 445—451.
31. Дмитриев И. С. Теоретические исследования П. Э. Лекок де Буабодрана по классификации химических элементов и систематике спектров.— В кн.: Учение о периодичности. История и современность. М.: Наука, 1981, с. 19—36.
32. Lecoq de Boisbaudran Р. Е. Sur la constitution des spectres lumineux.— Compt. rend., 1870, vol. 70, p. 144—164.
33. Lecoq de Boisbaundran P. E. Sur la constitution des spectres lumineux.— Compt. rend., 1871, vol. 73, p. 658—660.
34. Менделеев Д. 11. Основы химии. 2-е изд. СПб., 1873, ч. 2. 356 с.
35. Lecoq de Boisbaudran Р. Е. Caracteres chimiques et spectroscopques d’un nouveau metal, le gallium, decouvert dans une blende de la mine de Pierre-fitte, valee d’Argeles (Pyrenees).—Compt. rend., 1875, vol. 81, p. 493—495.
36. Lecoq de Boisbaudran P. E. Sur les physiques du gallium.— Compt. rend., 1876, vol. 83, p. 611—613.
37. Менделеев Д. И. Заметка по поводу открытия галлия.— В кн.: Периодический закон. М.: Изд-во АН СССР, 1958, с. 198—202. (Классики науки.)
38. Кедров Б. М. Открытие галлия — первое химическое открытие нового типа (Д. И. Менделеев и П. Э. Лекок де Буабодран).— В кн.: Прогнозирование в учении о периодичности. М.: Наука, 1976, с. 5—19.
39. Менделеев Д. И. Периодическая законность химических элементов.— В кн.: Периодический закон. М.: Изд-во АН СССР, 1958, с. 237—273. (Классики науки.)
40. Менделеев Д. И. {Письмо о периодическом законе.} Господину доктору Кеневиллю.— Там же, с. 391—396.
41. Макареня А. А. Д. И. Менделеев и физико-химические науки. 2-е изд. М.: Энергоиздат, 1982, с. 117—125.
42. Менделеев Д. И. Основы химии. 5-е изд. СПб., 1889, с. 513.
43. Nilson L. F. От Scandium en ny jordmetall.— Kongl. Vensk. Akad. For-handl., 1879, N 3, p. 47.
44. Nilson L. F. Ueber Scandium, ein neues Erdmetall.— Ber. Deutsch, chem. Gesellsch., 1879, Bd. 12, S. 554; Sur la Scandium, d’element nouveau.— Compt. rend., 1879, vol. 89, p. 645.
45. Cleve P. T. Sur la Scandium.— Ibid., p. 419.
46. Научный архив Д. И. Менделеева при ЛГУ: Письмо П. Т. Клеве Д. И. Менделееву от 30 августа 1879 г.
47. Там же: Письмо Д. И. Менделеева П. Т. Клеве (черновик).
48. Cleve Р. От Scandium.— Kongl. Vensk. Akad. ForhandL, 1879, N 7, p. 3.
49. Nilson L. F. Ueber das Atomgewicht und einige charakteristische Verbin-dungen des Scandium.— Ber. Deutsch, chem, Gesellsch., 1880, Bd. 13, S. 1439,
414
ЛИТЁРаТУРА
•- ГЖ-.-Г*'
50. Семишии В. И. Периодическая система химических элементов Д. IL Менделеева. М.: Химия, 1972, с. 33.
51. Winkler Cl.— J. prakt. Chem., 1886, Bd. 34, S. 117.
52. Winkler Cl. Germanium, ein neues nichtmetallisches Element.— Ber, Deutsch, chem. Gesellsch., 1886, Bd. 9, S. 210—211.
53. Лисснер А. Связи Д. И. Менделеева с Горной академией во Фрейберге.— Вопросы истории естествознания и техники, 1957, вып. 5, с. 51.
54. Научный архив Д. И. Менделеева при ЛГУ: Письмо Д. И. Менделеева Кл. Винклеру от 14 февраля 1886 г. (черновик).
55. Научный архив Д. И. Менделеева при ЛГУ: Письмо Кл. А. Винклера Д. И. Менделееву от 5 марта 1886 г. 2-й альбом писем, док. 91.
56. Менделеев Д. И. Основы химии. М.; Л.: ГНТИ, 1947, т. 2. 708 с.
57. Winkler Cl. Mitteilungen iiber das Germanium.—J. prakt. Chem., 1887, Bd. 34, S. 177—209.
58. Менделеев Д. II. Избранные лекции но химии. М.: Высшая школа, 1968. 224 с. См. также [3, с. 102, 237—241].
59. Чугаев Л. А. Периодическая система химических элементов: Пособие при изучении химии. СПб.: Образование, 1913. См. также: Чугаев Л. А. Избранные труды. М.: Изд-во АН СССР, 1962, т. 3, с. 54—208.
60. Макареня А. А. Д. И. Менделеев и физико-химические науки. 2-е изд. М.: Эпергоиздат, 1982. 256 с.; Кедров Б. М. Прогнозы Д. И. Менделеева в атомистике. Т. 2. Атомные веса и периодичность. М.: Атомиздат, 1978. 197 с.; Кедров Б. М. Прогнозы Д. И. Менделеева в атомистике. За гранью системы элементов. М.: Атомиздат, 1979. 184 с.
61. Менделеев Д. И. Новые материалы по истории открытия периодического закона. М.; Л.: Изд-во АН СССР, 1950. 148 с. См. также [3, с. 159].
62. Вюрц А. Гипотезы и развитие атомов/Пер. с фр. Киев, 1889, с. 115.
63. Потылицын А. Л. О способах измерения химического сродства. СПб.. 1880. См. также [1, с. 345].
ИСТОЧНИКИ ИЛЛЮСТРАЦИИ
Приводимые в книге иллюстрации заимствованы из следующих источников.
Рисунок на стр. 164 — из книги: Rancke-Madsen Е. The Development of Titri-metric Analysis till 1806. Copenhagen, 1958.
Рисунок на стр. 165 — из книги: Vanquelin L. Manuel complet de 1’essayer P., 1836.
Рисунок на стр. 186 — из статьи: Kirchhoff G., Bunsen R.— Pogg. Ann. Phys, und Chem., 1860, Bd. 110, N 6.
Рисунок на стр. 187 — из книги: Szabadvary F. Geschichte der analytischen Chemie. Bp., 1966.
Рисунок на стр. 201 — из книги: Mak-Donald D. A History of Platinum. L. 1960.
Рисунок на стр. 251, 260 — из книги: Crosland М. Р. Hysterical Studies in the Language of Chemistry. L., 1962.
Рисунок на стр. 331 — из книги: Soloviev J. I. L’Evoluzion del pensiero chi-mico dal’ 600 ai giorni nostre. Milano, 1976.
Рисунки на первой вклейке:
Химическая лаборатория М. В. Ломоносова — из ЖРХО, 1912, т. 36, вып. 6, отд. 2.
Приборы Лавуазье — из книги: Lavoasier A. Traite elementaire de chimie.
P., 1801. T. 1, 2. Рисунки выполнены женой ученого Мари Лавуазье.
Оборудование пробирной лаборатории и пробирные весы Лемана — из книги: Леман, И. Г. Пробирное искусство. СПб., 1772.
Весы Бойля — из книги: Szabadvary F. Geschichte...
Весы Лавуазье — из книги: Duamas М. Lavoisier theoricien et experimenta-teur. P., 1955.
Рисунки па второй вклейке:
Промывание золы для капеллей. Пробирные весы Эркера — из книги: Егс-ker’s L. Treatise on Ores and Assaying. Chicago, 1951.
Приборы Лсмери — из книги: Lemery N. Cours de chymie. Leyde, 1716.
Приборы Бергмана — из книги: Torberni Bergman Opuscula Phisica et Che-mica. Upsalial, 1780, vol. II.
Титриметрические приборы для объемного анализа — из книги: Ranke Madsen Е. The Development...
Приборы Дальтона — из книги: John Dalton and the Progress of Science. N. Y., 1968.
Прибор Гей-Люссака и Тепара — из книги: Gay Lussak J. L., Thenard L. J. Recherches physico-chimiques. P., 1810. T. 2.
Лабораторные приборы Берцелиуса — из книги: Szabadvary F. Geschichte...
Рисунок па первом форзаце — из книги: Соловьев Ю. И. История химии. М.: Просвещение, 1976.
Рисунок па втором форзаце — из книги: Soloviev J. I. L’Evoluzion...
ПРИЛОЖЕНИЕ
Ё Приложения включены фрагменты работ основателей химии Нового времени. В текст современной истории химии мы включили голоса ученых минувших эпох, которые определили развитие химии своего времени. Текст трактатов ученых конца XVII — начала XVIII вв. любопытен манерой изложения, приемами доказательств выдвинутой гипотезы, экзотической химической терминологией. В публикуемых впервые на русском языке фрагментах из работ Р. Бойля и Г. Э. Шталя обсуждаются животрепещущие проблемы химии XVII—XVIII вв.: атомно-корпускулярные представления, участие флогистона в химических превращениях, его свойства.
В классической статье Антуана Лорана Лавуазье «О горении вообще» изложены основы кислородной теории.
Впервые на русском языке публикуется фрагмент из «Учебника химии» Якоба Берцелиуса «Опыт теории химических пропорций и химического действия электричества», в котором знаменитый шведский ученый изложил историю атомной теории и стехиометрических законов.
Приложения органически дополняют основной текст данной книги.
Приложение I
ФРАГМЕНТ ИЗ ТРАКТАТА Р. БОЙЛЯ ПО ФИЛОСОФИИ НАУКИ (1657)
Все, что необходимо для хорошей гипотезы, суть:
1. Чтобы она была понятной (intelligible).
2. Чтобы она пе принимала и не предполагала ничего невозможного, непонятного, абсурдного или явно ложного.
3. Чтобы она была согласована сама с собой.
4. Чтобы опа была пригодной и достаточной для объяснения явлений, особенно главных.
5. Чтобы она была, по крайней мере, согласована с остальными явлениями, особенно с теми, к которым она относится, и не противоречила бы любым другим явлениям природы или очевидным физическим истинам.
Условиями и свойствами (qualityes) превосходной гипотезы являются следующие:
1. Чтобы она пе была необоснованной (precarious), но имела бы достаточные основания в природе самой вещи или, по крайней мере, была хорошо представлена некоторыми вспомогательными доказательствами.
2. Чтобы она была простейшей из всех хороших гипотез, которые мы в состоянии построить и, по меньшей мере, пе содержала бы в себе ничего лишнего или неуместного.
3. Чтобы она была единственной гипотезой, которая может объяснить данные явления, или, по крайней мере, чтобы она объясняла их так же хорошо.
4. Чтобы она давала возможность искусному натуралисту предсказывать будущие явления по тому, согласуются они с нею или не согла-
Примечание. Опубликован в кп.: Boas Ilall М. Robert Boyle on Natural Philosophy. An Essay with Selections from his Writings. Bloomington: Indiana University Press, 1965, p. 134—135. (Перевод с английского И. С. Дмитриева.)
419
ПРИЛОЖЕНИЕ
суются, и особенно исход таких экспериментов, гбторые специально предназначены для рассмотрения этой гипотезы, а т^кже [предсказывать] вещи, которые должны или не должны быть ее следствием.
Приложение II
ФРАГМЕНТ ИЗ ТРАКТАТА Р. БОЙЛЯ «ПРОИСХОЖДЕНИЕ ФОРМ И КАЧЕСТВ СОГЛАСНО КОРПУСКУЛЯРНОЙ ФИЛОСОФИИ»
(1666)
<...>1. В мире имеется великое множество частиц материи, каждая из которых слишком мала, чтобы, взятая отдельно, быть доступной для ощущения. Будучи целой и неделимой, такая частица непременно должна иметь определенную форму и быть очень твердой. Так что хотя она и может быть разделена мысленно или божественным всемогуществом, однако в действительности, она столь незначительна и тверда, что природа едва ли способна когда-либо разделить ее, и в этом смысле подобные частицы могут быть названы minima или prima naturalia.
2. Имеется также множество корпускул, которые составлены путем объединения нескольких minima naturalia. Объем (bulk) этих корпускул столь мал, а сцепление (adhesion) в них столь плотно и столь прочно, что каждое из этих небольших первичных образований и скоплений-частиц (primitive coneretions or clusters), если так можно выразиться, по отдельности не доступно для органов чувств. Хотя по своей природе эти образования не являются абсолютно неделимыми на составляющие их prima naturalia или на другие малые фрагменты, однако в силу названных причин они крайне редко разлагаются или разламываются и, как правило, они остаются целыми в подавляющем большинстве чувственно воспринимаемых тел, принимая различные формы, или личины (disguises). Как мы уже видели... даже наиболее массивные и сложные корпускулы часто наделены такой постоянной структурой. (К примеру, ртуть из плавкого и тягучего тела может быть превращена в красный порошок или же в летучий дым или же принять многие другие формы, но при этом она всегда будет оставаться истинной и способной к восстановлению ртутью). Они (особо прочные корпускулы.— И. Д.) являются как бы семенами или непосредственными началами многих видов природных тел, таких, как земля, вода, соль и т. д., и, будучи неощутимыми в отдельности, в соединении с другими становятся способными воздействовать на наши органы чувств.
3. Каждая minima naturalia, как и каждый кластер, имеет определенный объем и форму. Когда кластеры присоединяются друг к другу, то получающаяся вследствие их определенного расположения и когезии кор-
Примечание. Boyle R. The Works: Vol. I—V / Ed. by T. Birch. L.: A. Millar, 1744, vol. II, p. 470—471. (Перевод с английского И. G. Дмитриева.)
420
ФРАГМЕНТЫ ИЗ ТРАКТАТОВ Р. БОЙЛЯ
пускула всегда будет иметь иные размеры а часто и иную форму, чем исходные частицы. Кроме того, скорость одной из них или обеих соединяющихся частиц может изменяться по направлению, величине или как-то иначе. Аналогичная картина наблюдается и при разъединении составляющих кластер корпускул, а также при разламывании малых масс. Во всех случаях меняются размеры частиц, а зачастую и их форма, которая становится конгруэнтной форме пор одних тел (а возможно, и некоторых органов чувств) и не конгруэнтной форме пор других тел. Поэтому в разных ситуациях одна и та же корпускула будет вести себя по-разному.
4. Если при образовании видимого тела из множества невидимых корпускул последние (полностью или частично) приходят в движение, то это движение, независимо от его источника, само по себе может привести к большим изменениям и к появлению у тела новых свойств... Движение способно сделать многое, даже когда оно не вызывает каких-либо видимых изменений в теле. Но часто оно производит заметные изменения в структуре тела, ибо движущиеся части всегда стремятся передать, хотя бы в некоторой степени, свое движение тем частям, которые до того или пребывали в покое, или двигались иным образом. Тем самым движущиеся частицы иногда разрушаются теми корпускулами, с которыми они соударяются, изменяя таким образом свой объем или форму пли же и то и другое... Отсюда следует, во-первых, что структура тела, хотя бы на некоторое время, окажется сильно трансформированной (если, конечно, она не будет очень стабильной и абсолютно неизменной), а во-вторых,— это особенно важно — поры тел или малые интервалы между частицами будут менять свою величину или форму или и то, и другое, в результате чего нарушается их соразмерность корпускулам, которые ранее подходили им по своей величине и форме. Вместе с тем поры становятся соразмерными тем корпускулам, которым первоначально они не были конгруэнтны.
Приложение III
ФРАГМЕНТ ИЗ КНИГИ Г. Э. ШТАЛЯ «ОСНОВЫ ЗИМОТЕХНИИ, ИЛИ ОБЩАЯ ТЕОРИЯ БРОЖЕНИЯ...»
(ОПЫТЫ С КУПОРОСНЫМ ВИННЫМ КАМНЕМ И С СЕРНОЙ ПЕЧЕНЬЮ. ПЕРВОЕ УПОМИНАНИЕ ТЕРМИНА ФЛОГИСТОН)
(...) Если в хорошо раскаленный вместительный тигель всыпать по одной драхме 1 виннокаменной соли и купоросного винного камня, то витт-нокаменная соль расплавится и растворит купоросный винный камень2. Если теперь вбросить уголь (в кусочках) или в виде грубого порошка, то вскоре купоросная кислота отнимет от угля субстанцию флогистон3, и через его посредство прекрасно возродится настоящая сера4. (...)
То же самое прекрасно удается с серой: если две или лучше три части щелочной соли и одну часть порошка серы внести в тигель и расплавить, получится серная печень 5. По прошествии более или менее четверти часа она без какой-либо добавки может быть превращенной в ту соль, которую получают из серного масла, приготовленного посредством колокола и виннокаменной соли, и обычно называют купоросным винным камнем6. В ней больше нет никакого следа серы или щелочи вместо красного цвета (свойственного) серной печени. Эта соль совершенно белого цвета, вместо крайне неприятного вкуса серной печени, соль эта горьковата, она не легкоплавка и не расплывается на воздухе, подобно серной печени, вследствие ее щелочности, но является наиболее трудно растворимой из всех солей после винного камня 7. В отличие от серной печени, которая не кристаллизуется, эта соль весьма склонна к образованию октаэдрических кристаллов 8. Она не легкоплавка, как серная печень, но совсем не плавится9. >
Примечание. Stahl G. Е. Zimotechnia fundamentalis sive fermentationis theoria generalis. Halae, 1697. Переведено по перепечатке в кн.: Stahl G. E. Opusculum chymico-pliysico-medicum. Halae Magdeburgicae, 1715, p. 143—145. (Перевод с латинского, редакция и комментарий профессора С. А. Погодина.)
422
ФРАГМЕНТЫ ЙЗ РАБОТ Г. ШТАЛЯ
Если эту новую соль, полученную, как сказано выше, из серной кислоты и щелочи и лишенную флогистона, обработать углем, то вскоре через четверть часа появится вновь серная печень, и так она может быть превращена сотню раз (...)
Посредством других опытов я могу показать, как быстро флогистон из жирных материй и углей входит в металлы и возвращает сожженным известям прежнюю способность плавиться, коваться и амальгамироваться (...)
Комментарий
1 Драхма — единица веса, около 4 г. 2 Виннокаменная соль — поташ, полученный прокаливанием винного камня (нечистого гидротартрата калия KHG4H4O6) на воздухе. Купоросный винный камень — сульфат калия. Как видно из другой работы Шталя («Новый опыт получения настоящей серы искусственным путем», 1697), он не мог расплавить эту соль в чистом виде, но после добавки поташа она плавилась легко [Stahl G. Е. Opusculum..., р. 312]. По данным М. Амадори (1912), сульфат калия (т. пл. 1066° С) и карбонат калия (т. пл. 890° С) образуют ряд непрерывных твердых растворов I типа (без экстремумов); сплав, содержащий 50% К2СО3 по массе, плавится при 938° С (см. кн.: Справочник по плавкости солевых систем. М.: Изд-во АН СССР, 1961, т. 1, с. 646—647). Отсюда видно, что бывшие в распоряжении Шталя угольные печи позволяли получить температуру примерно на 100° С ниже точки плавления меди (1083° С). Замечание Шталя, что добавка одной драхмы поташа к нескольким фунтам (1 фунт = 96 драхм) суль-* фата калия [Stahl G. Е. Opusculum..., р. 312] непонятна, так как сплав этой соли с 2,5% К2СОз плавится при 1060° С, всего на 6° ниже чистого K2SO4 [Справочник..., с. 647]. Можно только предполагать, что в последующих опьн тах Шталь пользовался печью, в которой можно было расплавить сульфат калия.
3 Обычно полагают, что здесь Шталь впервые применил термин
«флогистон» (см. кн.: Leicester Н. S., Klickstein Н. М. A Source Book in Chemistry. N. Y., 1952, p. 59), не объяснив, однако, его значения. Этим термином Шталь уже пользовался в главе X «Основ зимотехнии» для обозначения горючего вещества, содержащегося в винном спирте, угле, жирах и др. Это вещество Шталь назвал «substantia phlogistos» и «substantia срАо^ютои», т. е. «горючей субстанцией», «субстанцией горючего» [Stahl G. Е. Opusculum..., р. 119]. Не останавливаясь па неправильном согласовании (substantia — женского рода, phlogistos — мужского), отметим, что Шталь пишет «флогистон» через архаическую древнегреческую букву «сти», означающую двойной согласный звук «ст». Этой буквы в типографиях давно нет (ее изображение см. в кн.: Дворецкий И. X. Древнегреческо-русский словарь. М., 1958, т. 2, с. 1506, слово оть). По недоразумению, Б. Н. Меншуткин принял букву «сти» за «дзету» и полагал, что Шталь называл горючее начало «флогидзоном» [Меншуткин Б. Н. Химия и пути ее развития. М.; Л.: Изд-во АН СССР, 1937, с. 87].
4 Восстановление сульфата калия углеродом при высоких температурах не идет строго по уравнению K2SO4+4C=K2S+4CO, обычно приводимому в учебниках, но сопровождается побочными реакциями. В результате получается сульфид калия с примесью полисульфидов. Поэтому при подкислении раствора продукта реакции
423
Приложение
сульфата калия с углем Шталь наблюдал выпадение серы (см. Приложение V).
5 Щелочная соль — поташ. При его сплавлении с серой получается серная печень — печёночно-бурая масса, состоящая главным образом из пентасульфида калия K2S5 (т. пл. 206° С), тиосульфата K2S2O3 и сульфата калия.
6 Речь идет об окислении K2S5 в K2SO4 кислородом воздуха, которое Шталь наблюдал, но объяснить не мог. Серное масло, полученное посредством колокола,— кислая жидкость, приготовленная
сжиганием серы под стеклянным * колоколом, находящимся над сосудом с горячей водой, содержала серную и сернистую кислоты.
7 При 20° С растворимость (в процентах по массе) этих солей равна: 10,00% для K2SO4 и 0,57% для КНС4Н4О6.
8 Сульфат калия диморфен; при температурах ниже 588° С он кристаллизуется в формах ромбической сингонии, выше 588° С — в гексагональной.
9 См. примеч. 2. Серная печень плавится в пламени спиртовой лампы.
Приложение IV
ФРАГМЕНТЫ ИЗ КНИГИ Г. Э. ШТАЛЯ «СЛУЧАЙНЫЕ МЫСЛИ И ПОЛЕЗНЫЕ РАЗМЫШЛЕНИЯ ПО ПОВОДУ СПОРА О ТАК НАЗЫВАЕМОЙ СЕРЕ»
(СВОЙСТВА ФЛОГИСТОНА) 1
(...) Первое, что необходимо рассмотреть в связи с этим (серным.— Пер ев.) началом, это:
1. Его свойства по отношению к огню.
2. Его способность давать окраску.
3. Его тесное соединение с другими тонкими субстанциями.
4. Его поведение по отношению к воде и влаге.
5. Его удивительную способность к дроблению и разрежению.
6. Его природу как в твердом, так и в жидком состоянии.
7. Где оно встречается.
Есть основание утверждать, что по отношению к огню это серное начало является не только присущим движению огня, но, по-видимому, только его движению и свойственно. Более того, рассуждая здраво, надо признать, что само это начало — вещественный (материальный) огонь2, истинная материя огня, подлинное начало его движения во всех горючих соединениях; однако само по себе, вне соединения, оно никакого огня не образует, а рассеивается и улетучивается в впде неделимых частиц или же, в крайнем случае, просто дает тепло, которое представляет собой сильно разреженный огонь.
Примечание. Stahl G. Е. Zufallige Gedanken und nutzliche Bedenken uber den Streit von sogenannten Sulphure. Leipzig, 1716. Есть французский перевод, сделанный П. Гольбахом: Stahl G. Е. Traite du soufre. Р., 1766. Настоящий перевод сделан по выдержкам из него, опубликованным в кн.: Metzger Н. Newton, Stahl, Boerhaave. Р., 1930, р. 165—170. (Перевод с французского Н. П. Чернышевой при участии И. С. Дмитриева; редакция и комментарий С. А. Погодина.)
424
ФРАГМЕНТЫ ИЗ РАБОТ Г. ШТАЛЯ
С другой стороны, важно отметить, что эта огненная материя сама по себе, т. е. без содействия воздуха и воды, не является ни разреженной, ни летучей, но, однажды став таковой при соприкосновении со свободным воздухом или при движении огня, она делается столь тонкой и разреженной, что оказывается не доступной восприятию всех (органов) чувств, так что ее уже невозможно ни распознать, ни приблизить или собрать, особенно если это нужно сделать быстро и в большом количестве.
(...) По всей совокупности изложенного я рассудил, что для этой материи нет иного, более подходящего, названия, чем горючая материя, или начало горючести. В самом деле, поскольку доныне ее смогли найти и распознать не иначе, как только в соединениях, и потому ее невозможно ни определить, ни назвать по каким-либо свойствам, только ей и присущим, мне кажется, что нет ничего разумнее, чем наименовать эту материю по тем общим действиям, которые она производит в соединениях; вот почему я даю ей греческое название флогистон, или горючее 3.
1. Несмотря на это свойство, необходимо отметить одно важное явление, а именно: пока это (горючее) вещество находится в связанном состоянии, способном воздействовать на (наши) органы чувств, оно не может быть совершенно разрушено огнем и не может быть рассеяно, напротив, оно будет сопротивляться самым сильным воздействиям огня и не претерпит при этом никаких изменений, пока свободный воздух не увлечет его с собой.
2. Что касается цвета, то, по крайней мере a posteriori, можно доказать, что это серное начало в той или иной степени служит причиной и порождает цвета всех веществ, в состав которых оно входит и к которым присоединяется заметным образом.
3. Относительно способности этого начала тесно соединяться с другими субстанциями я приведу примеры, которые показывают, в каком тонком и раздробленном состоянии оно пребывает, к примеру, в обыкновенной сере и до какой степени это начало может рассеиваться, как можно об этом судить по некоторым окрашенным растительным субстанциям (таким, как шафран), что заслуживает быть отмеченным.
4. Что касается запаха, то существование бесконечного множества растительных пахучих субстанций дает возможность предположить, что они обязаны своим запахом также этому началу горючести. К тому же, при горении дым и запах разпосятся далеко и ощущаются обонянием и вкусом, что говорит о тонкости разделения этого начала.
5. Справедливо и то, что это начало совершенно не обладает способностью к соединению с водой; между тем, в растениях оно прочно соединено с водной частью посредством очень непрочной солеобразной субстанции, чему особенно способствует длительность времени роста растений и их полного созревания. Подтверждение тому мы усматриваем в наиболее летучих и тончайших растительных маслах и горючих спиртах, которые всегда образуются при длительной ферментации. С другой стороны, эта проблема до сего времени не решена совершенно, поскольку не удалось собрать п соединить искусственно это вещество с водной частью, хотя очень легко удалось выделить его из растительных масел4.
425
ПРИЛОЖЕНИЕ
6. Многочисленные примеры убедительно доказывают, что хотя очень трудно соединить это начало с водой и хотя оно очень легко выделяется из этого соединения, тем не менее оно очень охотно и тесно соединяется разными путями с твердыми веществами, из чего видно, что серное начало склонно переходить в твердое и сгущенное состояние. Таким образом, Бехер был прав, говоря, что это — сухое и землистое по своей природе вещество, способное к образованию твердых соединений.
7. Наконец, необходимо рассмотреть вопрос, где находится это начало. Как видно из предыдущего, все сложные тела содержат ту или иную ощутимую долю этой субстанции, причем во всех так называемых трех царствах Природы, т. е. в растениях, животных и минералах. Тела двух первых царств содержат это начало в большом изобилии, так что все части их пронизапы им и тесно соединены с ним, за исключением водной части, которая сама по себе от него тоже не свободна, поскольку заключена в этом теле; но в жирных частях этих двух царств начало горючести преобладает.
8. Среди царства минералов только в воде, обычной соли, чистой купоросной соли5, песке или в камнях этого начала либо мало, либо совсем нет. С другой стороны, каменный уголь и битум наделены им в изобилии; что же касается серы, то она наполнена этим началом не по весу, но по числу его бесконечно тонких частиц. И не меньше его находится в несовершенных и горючих металлах, а также в тех веществах, которые называют незрелыми металлическими субстанциями 6.
(ВОССТАНОВЛЕНИЕ МЕТАЛЛОВ ИЗ ОКАЛИН)
У какого химика найдем мы, что одно и то же начало горючести может переходить из царства животных и царства растений непосредственно и без изменений в царство минералов и в металлы, вызывая там всегда один и тот же эффект, а именно горючесть, что пытаются объяснить, ссылаясь на аналогию, слово, которым пользуются для прикрытия своего невежества?
(..^Остается лишь зайти к оловянщику7, когда он плавит олово на сильном огне (пылающих) углей, нагревает расплавленный металл до того, что он воспламеняет клочок бумаги8. Вскоре на поверхности олова ооразуется пленка, и как только ее снимут, незамедлительно образуется новая, которая по мере продолжения этого процесса станет более похожей на пыль или золу. Если поместить большое или малое количество этой золы в тигель и подвергнуть его сильному нагреву, но так, чтобы ни один уголек не попал в него, зола не испытает никаких изменений; если же туда добавить стекла как флюс, пленка останется все в том же состоянии золы. С другой стороны, если эта зола еще остается на поверхности расплавленпого олова, если добавить туда растительного масла, вара или растительной смолы либо сала пли же другого животного жира, перемешать все это палочкой, то эта зола вновь расплавится 9 и присоединится к остаткам олова, но таким образом, что на нем уже не будет даже малейшей пленки.
426
ФРАГМЕНТЫ ЙЗ РАБОТ Г. ШТАЛЯ
Если же взять чистый свинец, расплавить в плоском сосуде, помешивая или снимая образующуюся пленку, то мало-помалу он полностью превратится в известь пли золу. Если поместить эту золу в плоскую и открытую плошку, то она будет испускать легкий пар, но мало помалу весь свинец преобразуется в тонкий шлак или вид стекла. Если же эту золу долго нагревать на пламени дровяной печи, она сначала побелеет, затем пожелтеет и, наконец, покраснеет, станет тем, что называют суриком 10. Если взять примерно с горошину этой свинцовой извести, поместить ее в углубление в куске угля и с помощью паяльной трубки и лампы эмалировщиков11 направить <на известь) пламя, известь расплавится и превратится в стекло; теперь достаточно следить за тем моментом, когда капля этого стекла коснется края угля, тогда послышится легкое шипение, и в этот момент оно снова превратится в свинец 12.
(КРУГОВОРОТ ФЛОГИСТОНА)
Создается впечатление, что это начало переходит в растения из воздуха, как это позволяют предполагать некоторые наблюдения. В самом деле, всем известно, что масличные и смолистые растения лучше всего произрастают на засушливых и песчаных почвах; для них непригодна влажная и жирная земля, им необходимо легкое и губчатое удобрение, образовавшееся из гнилого дерева, которое только разрыхляет песок и делает его пористым. Различные породы сосен и елей, которые насыщены смолой, растут только на чистом песке или рыхлом песчанике, в котором их корни делают щели, что не удается в жирной и влажной земле. Эти деревья протягивают свои корни у самой поверхности земли, и у самых больших деревьев их очень мало; это совершенно не мешает их корням наполняться смолой, равно как их листьям 13 и шишкам. Никто не может себе представить, что эта субстанция приходит только из сухого песка, кажется более вероятным, что она переходит из этих растений в атмосферу, которая наполняется ею с помощью ферментации; последняя действует на листья, которые от этого падают и, сгнивая осенью и весной, несут в воздух, помимо масел, дров и горелого угля, значительное количество освобожденного горючего начала в его чистом, первозданном состоянии. Итак, это начало проникает в виде паров или переносится в растения с помощью непрочной солевой части, которая, как известно, лишена воздуха; к тому же, никто не знает, не содействует ли роса ускорению роста растений 14.
Комментарий
1 Учение Шталя о флогистоне рассеяно в его многочисленных сочинениях. По мнению французского историка химии Ф. Гёфера, Шталь лучше всего изложил это учение в работе «Случайные мысли и полезные размышления по
поводу спора о так называемой сере», которую он назвал «чрезвычайно редким небольшим сочинением» [Hoejer F. Histoire de la chimie. Paris, 1869, t. 2, p. 397]. Мы приводим три фрагмента из этого труда.
427
Приложение
2 Здесь, как и во многих других высказываниях, Шталь считает флогистон вещественным, материальным началом.
3 Флогистон — средний род от греческого прилагательного флогис-тос — горючий, воспламеняемый.
4 Подразумевается легкость воспламенения растительных масел, причем, по учению Шталя, флогистон выделяется в виде пламени.
5 Чистая купоросная соль — какой-то природный сульфат.
6 По представлениям алхимиков, металлы зарождаются и зреют в земных недрах. Золото и серебро считались металлами зрелыми, достигшими совершенства; прочие металлы (ртуть, медь, железо, олово, свинец) считались металлами несовершенными, не достигшими зрелости.
7 «Оловяничник, оловянщик, отливающий, работающий оловянную посуду» [Даль В. И. Толковый словарь живого великорусского языка. М.: Русский язык, 1979, т. 2, с. 671].
8 Олово плавится при 232° С; чтобы сообщить расплавленному металлу жидкотекучесть, необходимую
для хорошего заполнения литейной формы, его надо перегреть значительно выше точки плавления.
9 Так в авторском тексте. Но расплавится, конечно, не окись олова (II) SnO, а восстановленное из него олово.
10 Речь идет о превращении свинцового глёта в сурик по реакции 6РЬО+О2=2РЬзО4.
11 Лампа эмалировщиков — масляная лампа, которой пользовались для плавки эмалей и стеклодувных работ.
12 Шталь не поясняет, что, по его мнению, восстановление олова и свинца из оловянной золы и свинцового глёта происходит за счет присоединения флогистона, содержащегося в угле и других горючих материалах. Вероятно, он считал это очевидным.
13 Так в авторском тексте. Речь идет о хвое.
14 В другом месте Шталь пишет, что флогистон переходит из растений в животных и вместе с ними обоими попадает в подземный мир [Stahl G. Е. Experimenta..., р. 25].
Приложение V
ФРАГМЕНТ ИЗ КНИГИ Г. Э. ШТАЛЯ «ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ ОПЫТЫ, НАБЛЮДЕНИЯ, ЗАМЕЧАНИЯ, ЧИСЛОМ ТРИСТА»
(Синтез серы из серной кислоты и флогистона)
24. ОПЫТ 1
(...) Кислый спирт, или же так называемое купоросное масло соединяется до взаимного насыщения2 (...) Если эту соль с добавкой двадцатой части угля3 истереть в порошок и затем последовательно всыпать в раскаленный тигель, то смесь ожижается, хорошо течет и выливается. Она представляет собой красноватую массу, подобную крови. Она совершенно подобна той, которая получается сплавлением двух частей постоянной щелочи с одной частью минеральной серы4. Если эту красную
Примечание. Stahl G. Е. Experimenta, observationes, animadversiones, CGG numoro, chymicae et physicae. Berolini, 1731, p. 34—35. (Перевод с латинского, редакция и комментарий G. А. Погодина.)
428
фрагменты из работ г. шТаля
массу растворить в обыкновенной воде, профильтровать (раствор) и осадить приливанием винного уксуса, появится белый порошок5, который, будучи хорошо промыт и расплавлен, окажется настоящей обыкновенной серой, каковую добывают в рудниках. Короче говоря, она ничем, даже самым малым, не отличается от самородной серы. Отсюда всем ясно, что обыкновенная сера — смешанное тело (состоящее) из минеральной кислоты (находящейся в купоросе, а также в природной сере) и из той горючей материи, которую опа во время опыта отняла от угля и с которой соединилась 6.
Комментарий
1 Настоящий фрагмент — сделанное Шталем краткое изложение сущности его работы «Новый опыт получения истинной серы искусственным путем» [Experimentum novum vcruin sulphur arte produ-cendi. Halae, 1697; перепечатано в кн.: Stahl G. E. Opusculum..., p. 299—333]. Описываемые результаты Шталь и его последователи считали одним из самых убедительных доказательств существования флогистона.
2 Речь идет о получении сульфата калия по реакции K2CO3+H2SO4= =K2SO4+CO2+H2O.
3 В латинском тексте vigesima pars carbonis, т. е. двадцатая часть угля. Это количество угля явно недостаточно, для полного восстановления сульфата калия в сульфид требуется по уравнению (см. примеч. 2) на 100 частей сульфата 27 частей углерода. В литературе рекомендуется брать 30 частей угля на 100 частей сульфата [Graham Т., Otto F. J. Ausfiihrli-
ches Lehrbuch der Chemie. Aufl. 3. Braunschweig, 1853, Bd. 2, Abt. 2, S. 90—91], что хорошо согласуется с расчетом по уравнению. Можно только предполагать, что «двадцатой частью» Шталь назвал 20% от массы сульфата; тогда количество угля будет в небольшом избытке против требуемого уравнением K2SO4+2C=K2S+2CO2.
4 С этим согласиться нельзя. Продукт нагревания сульфата калия с углем состоит из K2S с примесью полисульфидов калия, а серпая печень — из K2S5 и K2S2O3 (см. примеч. 3 и 4 к Приложению III). Общее у обоих продуктов лишь то, что они при подкислении выделяют белую мелкодисперсную серу (о чем Шталь пишет далее) и сероводород (о чем Шталь умалчивает).
5 Здесь описано получение так называемого «серного молока» (lac sulphuris).
6 Горючая материя из угля — флогистон.
Приложение VI
А. Л. ЛАВУАЗЬЕ
О ГОРЕНИИ ВООБЩЕ
Если в физических науках опасно стремление подбирать факты под определенную систему, то не менее опасным является беспорядочное нагромождение опытных данных, которое способно лишь затемнить науку, вместо того чтобы внести в нее ясность, и сделать доступ желающим приобщиться к ней крайне затруднительным. В конце концов в награду за долгую и утомительную работу можно получить лишь полный беспорядок и путаницу. Факты, наблюдения и опыты являются материалами для построения большого здания, по, собирая их, надо избегать загромождения науки; наоборот, следует стремиться их классифицировать и отмечать то, что относится к каждому отделу и к каждой части того целого, к которому опи принадлежат.
С этой точки зрения системы в физике являются только орудиями, которые должны помочь слабости наших чувств. Это, собственно говоря, приближенные методы, направляющие нас на путь решения задачи; это гипотезы, которые, будучи последовательно изменяемы и исправляемы по мере того, как их опровергает опыт, должны, несомненно, путем исключения и отбора привести нас к познанию истинных законов природы.
Ободренный этими соображениями, я позволяю себе предложить Академии новую теорию горения или, говоря со сдержанностью, которую я поставил себе законом, скорее гипотезу, при помощи которой весьма удовлетворительным образом объясняются все явления горения, обжигания
Примечание. Lavoisier. Memoires sur la combustion en general.— Mem. Acad. Paris., 1777, p. 592; Oeuvres de Lavoisier, 1862, t. 2, p. 222—233: Впервые на русском языке опубликовано в журнале «Успехи химии», 1943, т. 12, вып. 5, с. 368—373. (Перевод с французского Т. В. Волковой, редакция и комментарий С. А. Погодина).
430
А. Л. ЛАВУАЗЬЕ. О ГОРЕНИИ ВООБЩЕ
и отчасти даже явления, сопровождающие дыхание животных. Первые основания этой гипотезы я уже набросал на страницах 279 и 280 тома 1 моих «Физических и химических сочинений» \ но сознаюсь, что, мало доверяя своим собственным познанпям, я не осмелился тогда выставить мнение, которое могло показаться странным и которое было прямо противоположно учению Шталя и некоторых следовавших ему знаменитых людей.
Быть может, некоторые из соображений, удержавших меня тогда, остались в силе и сегодня; однако факты, умножившиеся с того времени и, как мне кажется, благоприятствующие моим взглядам, укрепили меня в моем мнении; не сделавшись, быть может, более сильным, я стал более уверенным, и думаю, что имею достаточно доказательств или, по крайней мере, вероятностей, чтобы даже несогласные с моими взглядами не смогли порицать меня за их опубликование.
При горении тел постоянно наблюдаются четыре явления, что, по-видимому, должно считать законом, от которого природа никогда не уклоняется. Хотя эти явления и изложены неявно в других моих мемуарах, я не могу обойтись здесь без того, чтобы не напомнить их в немногих словах.
Первое явление. При всяком горении происходит выделение огненной материи или света.
Второе явление. Тела могут гореть только в очень немногих видах воздуха2, или, вернее, горение может происходить лишь в одном виде воздуха, который Пристли назвал бесфлогистонным и который я буду называть «чистым воздухом»3. Тела, которые мы называем горючими, не только не горят в пустоте или в каком-либо другом воздухе, но там они гаснут так быстро, как если бы их погружали в воду или в любую другую жидкость.
Третье явление. При всяком горении происходит разрушение или разложение чистого воздуха и вес сгоревшего тела увеличивается точно на количество поглощенного воздуха.
Четвертое явление. При всяком горении тело превращается в кислоту в результате прибавления того вещества, которое увеличило его вес; так, например, если под колоколом сжигать серу, то продуктом горения будет серная кислота4, если сжигать фосфор, то получается фосфорная кислота; если сжигать какое-либо углистое вещество, то продуктом сгорания является связываемый воздух, называемый иначе меловой кислотой * (см.5).
Обжигание металлов подчинено точно тем же законам, и Макер совершенно правильно рассматривает его как медленное горение. Итак, 1) при всяком обжигании металла выделяется огненная материя, 2) истинное обжигание металла может происходить лишь в чистом воздухе, 3) происходит соединение воздуха с обжигаемым телом, но с тем отличием, что вместо кислоты при этом образуется особое соединение, известное под названием металлической извести.
* Замечу здесь, между прочим, что число кислот вообще неизмеримо больше, чем предполагают.
ПРИЛОЖЕНИЕ
Здесь не место доказывать, что существует аналогия между дыханием животных, горением и обжиганием; я вернусь it этому вопросу ниже.
Эти различные явления обжигания металлов и горения очень удачно объясняются гипотезой Шталя, но вместе с ним приходится предположить, что в металлах, сере и во всех телах, которые он считает горючими, имеется огненная материя, или связанный флогистон.
Но если от последователей учения Шталя потребовать доказать существование огненной материи в горючих телах, то они неминуемо впадают в порочный круг и вынуждены утверждать, что горючие тела содержат огненную материю, потому, что они горят, а горят они потому, что содержат огненную материю. Итак, в конечном итоге легко видеть, что горение объясняется горением же.
Существование огненной материи и флогистона в металлах, сере и т. п. есть в сущности лишь гипотеза, предположение, которое, будучи принято, правда, объясняет некоторые явления обжигания и горения.
Но если я покажу, что те же явления могут быть объяснены столь же естественным образом при помощи противоположной гипотезы, т. е. без предположения существования огненной материи п флогистона в веществах, называемых горючими, то и система Шталя окажется поколебленной в своей основе.
Меня, несомненно, спросят прежде всего, что я подразумеваю под огненной материей. Отвечу вместе с Франклином, Бургаве и некоторыми древними философами, что огненная материя, или свет, является очень тонкой, очень упругой невесомой жидкостью (флюидом), которая со всех сторон окружает нашу планету, более или менее легко проникает во все тела, ее составляющие, и стремится, когда она свободна, во всех них прийти в равновесие 6.
Добавлю еще, говоря химическим языком, что этот флюид является растворителем большого числа тел; что он соединяется с ними, подобно тому, как вода соединяется с солями и кислоты с металлами, и что тела, соединенные с огненным флюидом и растворенные им, теряют некоторые свои cBoiiCTBia, которые они имели до соединения с ним, и приобретают новые, приближающие их к огненной материи.
Как я уже указал в своем мемуаре, переданном в секретариат Академии *, всякая воздухообразная жидкость 7, всякий вид воздуха является следствием соединения какого-либо твердого пли жидкого тела с материей огня или света и этому соединению обязаны воздухообразные жидкости своей упругостью, удельной легкостью и разреженностью и всеми теми свойствами, которые сближают их с огненным флюидом.
Поэтому чистый воздух, тот, который Пристли называет бесфлоги-стонным, есть огненное соединение, в которое материя огня или света входит как растворитель и в котором другое вещество является основанием.
Но если па находящееся в каком-либо растворе основание подействовать веществом, имеющим к нему большое сродство, то прибавленное ве
* Этот мемуар был впоследствии доложен и напечатан на стр. 420. «Мсmoires de 1’Academie des Sciences», 1777.
432
А. Л. ЛАВУАЗЬЕ. О ГОРЕНИИ ВООБЩЕ
щество немедленно соединяется с основанием и растворитель, покинутый им, становится свободным.
То же происходит с воздухом при горении: тело, которое горит, отнимает его основание, а огненная материя, которая служит ему растворителем, делается свободной, вступает в свои права и выделяется в виде пламени, тепла и света. Чтобы осветить все темные места этой теории, приложим ее к нескольким примерам. Когда обжигают металл в чистом воздухе, то основание воздуха, которое имеет меньше сродства к своему растворителю, чем к металлу, соединяется с последним, как только он расплавляется и превращает его в металлическую известь. Это соединение основания воздуха с металлом доказывается: 1) увеличением веса металла во время обжигания, 2) почти совершенным разрушением воздуха, находящегося в колоколе.
Но если основание воздуха удерживалось в растворе огненной материей, то, по мере того как это основание соединяется с металлом, огненная материя должна становиться свободной и, выделяясь, давать пламя и свет. Понятно, что чем быстрее производится обжигание металла, т. е. чем больше основание воздуха связывается в данное время, тем больше огненной материи будет освобождаться сразу и, следовательно, тем ощутимее и заметнее будет горение.
Эти явления, которые чрезвычайно медленны и трудно уловимы при обжигании металлов, почти мгновенны при горении серы и фосфора. Я показал посредством опытов, против которых, мне кажется, трудно сделать какое-либо разумное возражение, что при горении этих обоих веществ воздух, или, вернее, основание воздуха, поглощается; что оно соединяется с серой и фосфором, образуя купоросную и фосфорную кислоты. Но основание воздуха не может перейти в новое соединение, не оставив свободным свой растворитель, а этот растворитель, который и есть огненная материя, должен выделиться в виде света и пламени.
Уголь и все углистые вещества оказывают такое же действие на основание воздуха. Они его присоединяют и образуют с ним благодаря горению своего рода кислоту, известную под названием связанного воздуха, или меловой кислоты. При этом процессе также выделяется растворитель основания воздуха, огненная материя, но в меньшем количестве, чем при горении серы и фосфора, так как часть ее соединяется с мефитической кислотой 8, чтобы удержать ее в парообразном и упругом состоянии, в котором мы ее получаем.
Замечу здесь, между прочим, что сгорание угля в колоколе, опрокинутом над ртутью, не производит очень значительного изменения объема воздуха, в котором происходило горение, даже тогда, когда для опыта применяют чистый воздух, потому что образующаяся мефитическая кислота остается в воздухообразном состоянии, в отличие от купоросной и фосфорной кислот, которые сгущаются в твердом виде по мере того, как они образуются.
Я мог бы последовательно приложить ту же теорию ко всем видам горения, но, так как я буду часто иметь случай возвращаться к тому предмету, ограничиваюсь пока этими общими примерами. Итак, обобщая,
17 Всеобщая история химии
433
ПРИЛОЖЕНИЕ
скажу, что, по моему мнению, воздух состоит из огненной материи как растворителя, соединенного с веществом, которое служит ему основанием и в некотором роде ее нейтрализует. Всякий раз, когда на это основание действуют веществом, к которому оно имеет больше сродства, оно докидает свой растворитель. С этого момента огненная материя возвращает свои права, свои свойства и вновь появляется перед нашими глазами в виде тепла, пламени и света. Итак, согласно этому мнению, чистый воздух, бесфлогистонный воздух Пристли, есть истинное и, быть может единственное, горючее тело в природе, и потому очевидно, что больше нет необходимости для объяснения явления горения предполагать, что существует громадное количество огня, связанного во всех телах, которые мы называем горючими; наоборот, весьма вероятно, что его находится лишь немного в металлах, сере, фосфоре и в большинстве очень твердых, тяжелых и плотных тел; может быть, в этих телах находится только свободная огненная материя вследствие свойства этой материи приходить в равновесие со всеми окружающими телами.
Другое убедительное соображение, которое подтверждает предыдущее, заключается в том, что почти все тела могут существовать в трех различных состояниях: либо в твердом виде, либо в жидком, т. е. расплавленном, виде, либо в состоянии воздуха и пара. Эти три состояния зависят лишь от большего или меньшего количества огненной материи, которой они проникнуты и с которой они соединены. Текучесть, испаряемость, упругость — свойства, характерные для присутствия огня п для его большого изобилия; наоборот, твердость, плотность служат доказательством его отсутствия. Итак, поскольку доказано, что воздухообразные вещества и сам воздух содержат большое количество соединенного огпя, постольку, вероятно, твердые тела содержат его немного.
Я вышел бы из границ, которые я себе поставил и которые требуются обстоятельствами, если бы стал показывать, сколько света проливает эта теория на все великие явления природы; однако я не могу удержаться от того, чтобы не заметить, с какой легкостью она объясняет, почему воздух есть упругая и разреженная жидкость. В самом деле, так как огонь является наиболее тонким, наиболее упругим и наиболее разреженным из всех флюидов, оп должен сообщить часть своих свойств веществам, с которыми он соединяется, и, подобно тому как водные растворы солей всегда сохраняют часть свойств воды, вещества, растворенные огнем, должны сохранять частично свойства последнего. Поэтому понятно, почему горение не может происходить пи в пустоте, пи даже в каком-либо воздухообразном соединении, в котором огненная материя имеет очень большое сродство к основанию, с которым оно соединено.
Исходя из этих положений нет необходимости допускать присутствие громадного количества связанной и соединенной огненной материи в алмазе п в большом числе веществ, которые не только совсем не имеют свойств, подобных свойствам огненной материи, но имеют даже свойства противоположные; наконец, нет необходимости утверждать, как это делает Шталь, что тела, которые увеличиваются в весе, теряют часть своего вещества.
434
А. Л. ЛАВУАЗЬЕ. О ГОРЕНИИ ВООБЩЕ
Я выше указал, что теория, изложенная в настоящем мемуаре, может быть приложена к объяснению части явлений дыхания, и этим я закон-чу настоящий опыт.
В докладе, который я сделал на публичном заседании на прошлой пасхе9, я показал, что чистый воздух, войдя в легкие, выходит из него частично в виде связываемого воздуха, или меловой кислоты. Следовательно, чистый воздух, проходя через легкие, претерпевает такое же разложение, которое имеет место при горении угля. Но при горении угля происходит выделение огненной материи; следовательно, в промежутке от вдоха до выдоха равным образом должно быть выделение огненной материи в легких и несомненно, что эта огненная материя, распределяясь вместе с кровью по всему организму, поддерживает в ней постоянную теплоту около 32,5° по термометру Реомюра.
На первый взгляд, эта мысль, может быть, покажется малообоснованной, но, прежде чем ее отбросить или осудить, прошу принять во внимание, что опа опирается на два явных и неоспоримых факта, а именно на разложение воздуха в легких и на выделение огненной материи, которое сопровождает всякое разложение чистого воздуха, т. е. всякий переход чистого воздуха в связываемый воздух.
Зависимость животной теплоты от разложения воздуха в легких подтверждается еще и тем, что в природе только привычно дышащие животные являются теплокровными и что их теплота тем больше, чем дыхание чаще, т. е. имеется очевидное соотношение между теплотой 10 животного и количеством воздуха, вошедшего в его легкие или, по крайней мере, превращенного в связываемый воздух.
Впрочем, повторяю, что, нападая здесь на учение Шталя, я не имею цели заменить его строго доказанной теорией, но только предложенной гипотезой, которая мне кажется более вероятной, более согласной с законами природы и которая, по-видимому, заключает меньше искусственных объяснений и меньше противоречий.
Обстоятельства позволяют мне дать здесь только общий очерк системы и обзор ее следствий, но я предполагаю последовательно вернуться к каждой ее части, развить их в отдельных статьях и осмелюсь заранее утверждать, что предлагаемая мною гипотеза объясняет очень просто и очень удачно главные явления физики и химии.
435
ПРИЛОЖЕНИЕ
Комментарий
1 Lavoisier. Opuscules physiques et chimiques. P., 1774, t. 1; Oeuvres de Lavoisier, 1864, t. 1, p. 439—655.
2 В то время большинство химиков считало все газы различными видами воздуха.
3 Вскоре Лавуазье дал этому газу название «кислород».
4 Здесь и далее необходимо помнить, что кислотами Лавуазье называл ангидриды кислот.
5 Связываемый воздух, меловая кислота — синонимы углекислого газа.
6 При реформе химической номенклатуры (1787) Лавуазье предложил для огненной материи название «calorique» (теплород или теплотвор).
7 А. Лавуазье ссылается на свою статью «О соединении огненной материи со способными испаряться жидкостями и об образовании упругих воздухообразных жидкостей», т. е. газов [Oeuvres de Lavoisier, t. 2, 1862, p. 212—224].
8 Мефитическая кислота — углекислый газ.
9 Доклад «Опыты над дыханием животных и над изменениями, которым подвергается воздух, проходя через их легкие» был сделан Лавуазье 3 мая 1777 г. [Mem. Acad. Paris, 1777, р. 185; Oeuvres de Lavoisier, 1862, t. 2, p. 174— 183].
10 Под теплотой (chaleur) здесь Лавуазье подразумевает температуру.
Приложение VII
ФРАГМЕНТ ИЗ РАБОТЫ Й. Я. БЕРЦЕЛИУСА «ОПЫТ ТЕОРИИ ХИМИЧЕСКИХ
ПРОПОРЦИЙ
И ХИМИЧЕСКОГО ДЕЙСТВИЯ
ЭЛЕКТРИЧЕСТВА» 4*
1. ОЧЕРК ИСТОРИИ РАЗВИТИЯ ТЕОРИИ ХИМИЧЕСКИХ ПРОПОРЦИЙ 2*
С того времени, когда тела начали рассматривать состоящими из элементов, принято считать, что сложные соединения, образованные одними и теми же элементами и в одних и тех же отношениях, проявляют как одинаковые внешние характеристики, так и сходные внутренние свойства. Эти идеи были высказаны еще древними философами, когда опыт не был опорой для умозаключений (speculations). Таких взглядов придерживались пифагорейцы и Филон3, автор «Книги мудрости», написанной, по-видимому, в правление Каллигулы и почитаемой апокрифической среди книг Священного писания. Как сказано в главе II этой книги (стих 22), «бог расположил все по мере, числу и весу». Между тем, до нашего времени философы высказывали лишь темные предчувствия этой истины; но, несомненно, правильность таких представлений убедительно показал первый опыт точного химического анализа. Такая попытка была предпринята сравнительно недавно, и хотя нельзя с достоверностью определить, кто был первым химиком, который при помощи химического анализа веществ сумел определить отношения элементов в них, существует много доказательств того, что искусство производить эти опыты с большой точностью впервые было описано во второй половине прошлого столетия; и только благодаря этим усовершенствованиям, мы имеем в настоящее время теорию химических пропорций (la theorie des proportions chimiques). Примечание. Berzelius J. J. Lehrbuch der Chemie. Bd. 5. Aufl. 3. Dresden; Leipzig, 1835, S. 15—31, 46—48. (Перевод с первого французского издания 1819 г., сверенный с немецким изданием, кандидатов химических наук И. С. Дмитриева и В. А. Крицмана. Редакция перевода с немецкого и примечания С. А. Погодина, примечания, помеченные звездочкой,— И. С. Дмитриева.)
437
ПРИЛОЖЕНИЕ
По-видимому, немецкий химик Венцель был первым, кто обратил внимание на эти отношения и попытался экспериментально подтвердить их. Он рассмотрел одно явление, которое ранее обнаружили другие химики, а именно: две нейтральные соли сохраняют свою нейтральность после того, как между ними произошло обменное разложение (s’etre mutuclle-ment decomposes) 4. On изложил результаты своих опытов в книге, опубликованной в Дрездене в 1777 г. под названием «Учение о сродстве» 5, в которой, опираясь на необычайно точные анализы, объяснил это явление тем, что соотношения между количествами щелочей и земель, которые насыщают данное количество одной и той же кислоты, одинаковы для каждой из выбранных кислот.
Так, например, если нитрат извести (le nitrate de chaux, т. е. Ca(NO3)2.— Перев.) разлагается сульфатом кали, то возникающие при этом нитрат кали и сульфат извести (CaSO4. — Перев.) сохраняют свою нейтральность потому, что вес кали, насыщающий взятое весовое количество азотной кислоты, относится к весу извести, насыщающему то же количество этой кислоты, как вес кали — к весу извести, которая нейтрализует данное количество серной кислоты6. Результаты опытов Венцеля оказались более точными, чем у кого-либо из современных ему химиков, и большей частью были подтверждены лучшими анализами того времени. Однако они остались без внимания, и вместо них получили признание менее точные результаты более авторитетных химиков, результаты, противоречившие столь хорошо объясненному Венцелем явлению.
Бергман, работы которого впоследствии приобрели заслуженную известность, также наблюдал закономерности, обусловленные существованием определенных химических пропорций. Он опубликовал результаты своих опытов в 1782 г. в Упсале в диссертации, озаглавленной «О различных количествах флогистона в минералах» 7.
Проведя большое число опытов по взаимному осаждению металлов, он пришел к следующему выводу: «Взаимные количества осаждающего и осаждаемого флогистона обратно пропорциональны по весу».
Бергман много сделал для развития теории сродства и пытался объяснить сохранение нейтральности нейтральных солей при их обменном разложении. Однако его анализы были не столь точны, как у Венцеля, и потому не позволили ему объяснить наблюдаемые явления химических пропорций так же хорошо, как немецкому химику.
Первое позитивное указание па существование этих пропорций дал берлинский химик И. Рихтер на основании собственных многочисленных опытов, которым этот ученый посвятил большую часть своего времени. В сочинении, озаглавленном «Химическая стехиометрия», он стремился придать химии чисто математическую форму; однако его воображение далеко не всегда следовало опыту8.
Не касаясь его ошибок, мы остановимся только на важных работах Рихтера, посвященных химическим пропорциям. Эти исследования можно найти в периодически издававшихся Рихтером трудах под общим названием «О новых вопросах химии»9, в качестве эпиграфа к которым он взял цитированные выше слова из «Книги мудрости». Эксперименты, при
438
Й. Я. БЕРЦЕЛИУС. ОПЫТ ТЕОРИИ ХИМИЧЕСКИХ ПРОПОРЦИЙ
влекшие внимание к химическим пропорциям, были напечатаны главным образом в выпусках 7, 8 п 9 за 1796—1798 гг. Именно там Рихтер рассмотрел явление, ранее наблюдавшееся Венцелем, и объяснил его таким же образом. Рихтер намеревался определить относительную насыщающую способность оснований и кислот. Он заметил также, что при взаимном осаждении металлов нейтральность жидкостей (т. е. растворов солей металлов.— Перев.) сохраняется, и дал этому факту объяснение, до сих пор считающееся правильным.
При чтении работ Рихтера, посвященных химическим пропорциям, приходится лишь удивляться тому, что изучением этого вопроса можно было хоть на мгновение пренебречь. Правда, исследованиям Рихтера присуща одна особенность, которая ослабляла их воздействие на читателя: количественные результаты его опытов были не очень точными. В своих сопоставлениях он почти всегда исходил из углекислого глинозема (le carbonate d’alumine), т. е. из соединения, которое, как мы сейчас знаем, существовать не может.
Опыты Рихтера надо было повторить, чтобы избавиться от естественно возникавшего у читателя подозрения, будто желание автора подтвердить свою систему повлияло па полученные им результаты. Впрочем, п стиль его работ достаточно своеобразен. Рихтер, хотя и признает открытия аптпфлогистонпой школы, в то же время не может полностью отказаться от терминологии флогистиков. Стремясь занять некую среднюю позицию между обеими партиями, он в итоге не был принят ни одной из них.
Однако главной причиной того, почему химики долгое время не придавали должного внимания работам, посвященным химическим пропорциям, можно считать великую революцию, которая произошла в это время в химической теории и которая изгнала из пауки все шаткие умозрительные построения, основанные на концепции флогистона, заменив их результатами опытов и исследований. Система Лавуазье была почти единственной темой размышления химиков, и борьба, которая должна оы-ла вестись за эту теорию, отвлекала внимание химиков от всего, что не было непосредственно связано с новой теорией и ее применением для объяснения известных фактов.
Эта система была в конце концов признана всеми химиками, даже ее самые решительные противники отдали предпочтение теории Лавуазье перед теорией Бехера п Шталя, и большинство химиков настоящего времени изучали нашу науку уже по теории Лавуазье. Уделяя ей много времени и внимания, ученые стали впоследствии изучать в русле этой новой теории и другие области химии. Таким образом, можно сказать, что развитие представлений о химических пропорциях было на некоторое время замедлено становлением антифлогистонной системы.
В работах Лавуазье нельзя найти ничего существенного (positif) относительно химических пропорций10, если не считать указанного им различия между простым растворением (solution) и химическим растворением (dissolution) и. Первое может происходить в любых отношениях,
ПРИЛОЖЕНИЕ
тогда как второе изменяет природу растворяемого тела и происходит только в определенных и постоянных пропорциях.
Спустя некоторое время после создания системы Лавуазье один из его самых знаменитых сотрудников — Бертолле — опубликовал сочинение под названием «Опыт химической статики» (Париж, 1803) 12, в котором он в истинно философском духе рассмотрел проблему химического сродства и связанные с ним явления. В этой работе Бертолле пытался доказать, что действующие силы не столь уж многочисленны, как это можно заключить из многообразия их проявлений. Он указал также на возможность их происхождения из одной общей основной силы (la force principale), подобной силе земного притяжения или же силе, благодаря которой планеты вращаются на своих орбитах вокруг Солнца. Он даже высказал предположение, что когда-нибудь действие первой из этих сил (т. е. основной силы.— Пер ев.) удастся рассчитать точно так же, как уже давно рассчитывают действие второй (т. е. силы тяготения.— Перев.).
Развивая эти идеи, Бертолле пытался показать, что мнимое различие между простым и химическим растворением13 сводится лишь к различию в степени проявления одной и той же силы сродства, а именно: в первом случае она (сила) должна быть меньше, чем во втором. Бертолле утверждал, что элементы имеют максимум и минимум сродства и вне этих границ не могут соединяться друг с другом, тогда как в этих пределах соединяются в любых отношениях 14. Если же соединение происходит в определенных и неизменных отношениях, то это обусловлено другими факторами: сцеплением (cohesion), благодаря которому вещество обладает тенденцией к затвердеванию, и способностью к расширению, благодаря которой тела могут переходить в газообразное состояние. Так как, с одной стороны, при соединении элементы подвергаются сильному сжатию, то они всегда соединяются в определенных пропорциях, например, кислород реагирует с водородом только в одном определенном отношении 15, но, с другой стороны, если соединяющиеся элементы находятся в одинаково уплотненном состоянии (au meme etat de densite), то в пределах между максимумом и минимумом сродства элементов соединение происходит в любых отношениях. В соответствии с этой точкой зрения неизменность отношений элементов в кислотах, солях и т. д. обусловлена исключительно кристаллизацией, осаждением или, когда соединения находятся в газообразном состоянии, степенью их сжатия.
Бертолле проделал немало остроумных опытов, чтобы доказать справедливость этого утверждения. И хотя, как мы теперь знаем, оно не всегда достаточно для объяснения множества фактов, найденных в новейших работах, следует признать, что этот ученый изложил свои мнения и доводы, на которых они основывались, с убеждающей ясностью и мудростью. При проверке же результатов работ Рихтера относительно насыщающей способности оснований и кислот Бертолле пришел к иным численным значениям.
Он убедительно показал, что интенсивность химического взаимодействия тел определяется не только степенью их сродства, но также их количеством, т. е. их массой. Однако это имеет место, только когда и
44Q
Й. Я. БЕРЦЕЛИУС. ОПЫТ ТЕОРИИ ХИМИЧЕСКИХ ПРОПОРЦИЙ 1 • 1 - I
соединяющиеся тела, и образующиеся новые соединения сохраняют взаимный контакт, т. е. находятся в жидком или в растворенном состоянии *.
Химическая статика Бертолле положила начало дискуссии между ним и Прустом о постоянстве стехиометрических отношений во многих соединениях. Этот спор замечателен как по основательности аргументов обеих сторон, так и по той учтивости (le ton modere), с которой он велся17. Первоначально полагали, что наблюдавшееся для жидкостей постоянное действие химических масс можно распространить и на реакции твердых соединений, например окислов металлов, допуская при этом, что между высшим и низшим окислами какого-либо металла может существовать неопределенное число степеней окисления. Пруст, стремившийся доказать неправильность этой идеи, показал, что металлы как с серой, так и с кислородом образуют только одно или два соединения в определенных и неизменных отношениях и что все промежуточные степени, которые, как полагали, наблюдаются в действительности, являются лишь смесями двух соединений определенного состава.
Даже когда его собственные опыты говорили в пользу мнений Пруста, Бертолле защищался с проницательностью, вызывавшей недоумение читателей. Однако большая часть последующих анализов окончательно решила вопрос в пользу Пруста 18.
Незадолго до появления трудов Рихтера и Бертолле ирландский ученый Хиггинс опубликовал книгу под названием «Сравнительный обзор флогистонной и антифлогистонной теорий» (1789) 19, где с новой точки зрения рассмотрел различные степени соединений, которые могут иметь место между одними и теми же телами. Он принял в этой работе, что тела состоят из частиц или атомов. Согласно представлениям Хиггинса, если прибавить к окислу, т. е. к телу, состоящему из атома радикала и атома кислорода, еще один кислородный атом, то образуется тело, находящееся в новой степени окисления. Однако сам Хиггинс придавал малое значение этой гипотезе и пе пытался подтвердить ее справедливость какими-либо анализами. Он также не предвидел кратных отношений, существование которых непосредственно следовало из этой гипотезы. Его труд20 привлек мало внимания и вскоре был забыт **.
Пятнадцать лет спустя Джон Дальтон вновь высказал ту же идею, пытаясь подтвердить ее результатами лучших анализов, но не показал ее широкой применимости при рассмотрении химических явлений. В пер
* Это обстоятельство не согласуется с общим законом химических пропорций; оно оказалось бы даже в полном противоречии с ним, если бы только было доказано, что соединение твердого тела с жидкостью, в которой оно растворяется, не изменяя своих химических свойств, отличается по своей природе от так называемого химического соединения. Так, например, селитра соединяется с водой, образуя раствор этой соли, совсем не так, как обычная углекислая магнезия16 соединяется с определенным количеством воды,—в последнем случае основная часть соли пе переходит в жидкое состояние (т. е. в раствор.— Пер ев,) и не растворяется.
** Тридцать лет спустя Хиггинс пытался доказать, что его гипотеза, которой он сам нашел лишь очень ограниченное применение, дает ему право считаться автором закона кратных отношений.
441
ПРИЛОЖЕНИЕ
вых публикациях Дальтона по этому вопросу его представления были выражены не настолько ясно, чтобы привлечь большое внимание ученых, и поэтому лишь немногие химики поняли развиваемые им положения21. В «Журнале Николсона» за 1807 год Дальтон поместил небольшую таблицу «абсолютных весов» некоторых тел, т. е. по сути относительных количеств, в которых эти тела преимущественно соединяются, пли, иными словами, относительных весов их атомов. В последующие годы Дальтон опубликовал первый том своей новой системы химии под названием «Новая система химической философии»; ее второй том увидел свет в 1810 г.22 Согласно этой системе, все тела состоят из атомов; атом одного элемента может соединяться с 1, 2, 3 и т. д. атомами другого элемента, но не с промежуточными степенями, т. е. не с частями атомов. Аналогично атом одного сложного тела может соединяться с 1, 2, 3 и т. д. атомами другого сложного тела. Эта гипотеза была впоследствии подтверждена многочисленными опытами, и теперь можно, без преувеличения, сказать, что она явилась одним из крупнейших шагов, которые химия когда-либо делала на пути своего совершенствования.
Дальтон предположил, что элементарные атомы соединяются преимущественно в отношении один к одному и поэтому мы так часто встречаем единственное соединение двух веществ, коп/рое, как он полагал, содержит по одному атому каждого элемента. Если же имеются несколько соединений (одних и тех же атомов.— Перев.}, то, по его мнению, одно имеет состав А+В, другое А+2В, третье А+ЗВ и т. д. В своей новой системе химии Дальтон исследовал в первую очередь окисленные тела и указал число атомов, которое они содержат.
Однако представляется, что в своей работе этот замечательный ученый, по-видимому, слишком мало опирался на эксперимент и, возможно, распространяя свою новую гипотезу на систему химии, он не всегда действовал достаточно осторожно. Мне кажется, что в опубликованных им немногочисленных анализах можно подчас заметить стремление автора получить определенный результат. Это обстоятельство недостаточно учитывают те ученые, которые ищут доказательства за или против разбираемой теории. Тем не менее именно Дальтону принадлежит честь открытия той части теории химических пропорций, которую мы называем кратными отношениями, причем здесь мы не видим у него никаких предшественников 23.
До Дальтона была заложена, так сказать, основа представлений о химических пропорциях, но не было дано никакой объясняющей их теории. Этой основы было, однако, совершенно недостаточно для объяснения наблюдавшихся явлений, что будет показано далее.
В то время, когда Дальтон опубликовал свою систему, в Англии, в «Журнале Николсона» за ноябрь 1808 г., появилась статья Волластона о. кратных отношениях щавелевой кислоты в трех ее соединениях с кали 24. С тех пор эта область химической науки стала привлекать все большее внимание.
Так, в своей эвдиометрической работе А. фон Гумбольдт и Ж. Гей-Люссак (1806 г.) 25 обнаружили, что воду образуют один объем кисло-
442
Й. Я. БЕРЦЕЛИУС. ОПЫТ ТЕОРИИ ХИМИЧЕСКИХ ПРОПОРЦИЙ
рода и два объема водорода. Продолжая такие исследования, Гей-Люссак спустя некоторое время установил, что вообще все газообразные тела соединяются следующим образом: один объем (une mesure) одного газа может связать 1, Р/г, 2, 3 и т. д. объемов другого, т. е. газы соединяются либо в равных объемах, либо один объем одного газа реагирует с кратным числом объемов другого газа. Статья Гей-Люссака «О соединении газообразных веществ друг с другом» была опубликована в «Мемуарах Ар-кейского общества» (т. 2, 1809 г.) 26. Если заменить слово «атом» словом «объем» и представить тела не в газообразном, а в твердом состоянии, то в открытии Гей-Люссака можно найти непосредственное доказательство правильности гипотезы Дальтона. Гей-Люссак довольствовался тем, что, показав отношения, в которых соединяются газообразные вещества, нашел соединения, которые, согласно положениям (химической) статики Бертолле, должны всегда иметь определенный состав, но он не сделал из своего открытия каких-либо более общих выводов 27.
Дальтон же вместо того, чтобы быть удовлетворенным подтверждением его теоретических построений опытами Гей-Люссака, пытался доказать, что тот заблуждался и что газообразные тела не соединяются в равных объемах. Однако результаты опытов Гей-Люссака были подтверждены другими химиками, и в настоящее время полученные на их основании выводы полностью доказаны. При изучении взаимного вытеснения металлов Гей-Люссак получил такие же результаты, как Бергман и Рихтер.
Наконец, чтобы завершить этот небольшой исторический обзор к настоящей работе, посвященной химическим пропорциям, следует добавить, что я сам занимался изучением этого вопроса с 1807 г. Различные сообщения, объединяющие разультаты моих работ по этому вопросу, опубликованы в шведском издании «Труды по физике, химии и минералогии» 28, т. 3, 4, 5 и 6, а также в «Сообщениях Академии наук в Стокгольме» за 1813 г.
Перед публикацией учебника химии я просмотрел среди других малочитаемых трудов также «Сообщения» Рихтера, о которых шла речь выше. Я был поражен широким освещением в них вопросов состава солей и взаимного осаждения металлов, из которого не было сделано никаких выводов. Из исследований Рихтера вытекает, что исходя из хороших анализов состава одних солей можно точно рассчитать состав всех других солей. В своем учебнике (т. 1, с. 398, первое издание, 1807 г.29) я дал краткий обзор этого вопроса и в то же время произвел анализ ряда солей, благодаря чему стало излишним экспериментальное установление состава других солей. Ясно, что если исследовать все соли, образованные некоторой кислотой, например серной, со всеми основаниями, а также соли, образованные одним из оснований, например баритом30, со всеми кислотами, то можно получить данные, необходимые для расчета состава всех солей, образованных при двойном обменном разложении, с сохранением их нейтральности. Когда я проводил эту работу, Г. Дэви открыл состав едких щелочей31. Я нашел, вслед за другими химиками, что аммиак выделяется на отрицательном полюсе электрической батареи в виде веще
443
ПРИЛОЖЕНИЕ
ства с металлическими свойствами, из чего сделал вывод: эту щелочь следует также рассматривать как окисел, содержание кислорода в котором, не поддающееся определению непосредственно опытом, можно рассчитать в соответствии с упомянутым выше явлением взаимного осаждения металлов 32.
Изучение таких явлений должно было поэтому стать предметом моих исследований, но, когда я ознакомился с мыслями Дальтона о кратных отношениях, я нашел в числе анализов, результаты которых у меня уже были, такое подтверждение его теории, что я не мог удержаться от того, чтобы не исследовать возникновение этих явлений. Таким образом, план моей работы, первоначально очень ограниченный, постепенно все расширялся и, наконец, охватил учение о химических пропорциях во всем его объеме, от чего я в начале моих опытов был очень далек. Первоначально мои исследования дали совершенно пе те результаты, которые я ожидал и мог предполагать. Лишь при повторении опытов и изменении применяемых при исследовании методов я смог обнаружить допущенные ранее ошибки; новые знания позволили объяснить собственные мои заблуждения, и с помощью более совершенных методов, я получил, наконец, хорошее соответствие между результатами анализов и теоретических расчетов. При сопоставлении этих результатов постепенно были развиты новые представления, которые еще следовало обосновать, так что объем работы и, вероятно, ее значение также увеличились.
II. ОБЩИЙ ВЗГЛЯД НА ТЕОРИЮ ХИМИЧЕСКИХ ПРОПОРЦИЙ И ИХ ПРИЧИНУ
Каждая теория есть не что иное, как способ понять внутреннюю сущность явлений. Она допустима и достаточна лишь до тех пор, пока способна объяснять уже известные факты. Она, однако, может быть неправильной, хотя в определенный период развития науки объясняет факты так же хорошо, как и истинная теория. С увеличением числа экспериментов открываются факты, которые уже не могут быть объяснены на основании существующей теории, тогда возникает необходимость в поиске нового объяснения, применимого к новым фактам, и так, по-видимому, из века в век будут изменяться научные представления о природе явлений и, возможно, истинные причины никогда не будут найдены. Но даже если мы сами не сможем нашими работами достигнуть создания новой теории, то и тогда мы должны к этому стремиться.
При той неопределенности (I’incertitude), которая неразрывно связана с чисто теоретическими спекуляциями, зачастую оказывается, что явлению можно в равной мере дать два различных объяснения; тогда необходимо изучить оба эти объяснения, и если даже наша неопределенность при этом возрастет, это не должно уменьшить наших усилий в поиске истины, поскольку настоящий ученый прежде всего стремится узнать, что, по его мнению, правдоподобно, и не отдает предпочтения воззрениям, не подтвержденным вескими доказательствами.
Й. Я. БЕРЦЕЛИУС. ОПЫТ ТЕОРИИ ХИМИЧЕСКИХ ПРОПОРЦИЙ
Науки всегда требуют, чтобы теория приводила наши представления в определенный порядок, без чего было бы слишком трудно охватить все частности. Мы выбираем ту теорию, которая объясняет все известные факты. Если она общепризнана, то для науки часто очень полезно показать, что явлениям можно дать и другое объяснение. Но из этого не следует, что первое объяснение следует считать ошибочным, и всегда предосудительны те нововведения, в которых уже принятые раньше способы объяснения явлений заменяются иными, правильность которых, однако, не основывается на большой достоверности. Поэтому необходимо прежде всего доказать, что ранее принятое представление неправильно и его необходимо заменить другим. Теории, принятые ранее, можно заменить только такими, которые лучше объсняют известные к определенному времени факты.
Открытие химических пропорций и действия электричества на химическое сродство требовало изменения существующего способа объяснения явлений, что оправдывает попытку найти новое объяснение, которое бы лучше согласовывалось с фактами.
После того как убедились, что элементы, преимущественно неорганической природы, соединяются друг с другом в некоторых простых и определенных отношениях без образования соединений промежуточного состава, следует обратиться к поискам причины этого замечательного явления.
Умозрительная философия некоторых немецких школ33, распространяясь на естественнонаучные теории, создала, не без определенного предчувствия истины, новую систему, которую назвали динамической, потому что ее основное положение состояло в следующем: материя есть результат двух сил, действующих в противоположных направлениях, одна из которых — сила сжатия (contractive), а другая — расширения (expansive). Если бы первая полностью превзошла вторую, то материя Вселенной сжалась бы до математической точки. Эта теория предполагает, что элементы в момент их химического соединения взаимно проникают друг в друга и что нейтрализация их химических свойств, являющаяся, как правило, результатом этого соединения, состоит в этом взаимном проникновении. Но при таком взгляде на химическое соединение явление определенных пропорций никогда не представлялось бы для упомянутой философии столь неожиданным, как в эпоху, когда оно было замечено и подтверждено. Под влиянием этой философии определенность химических пропорций так и осталась бы незамеченной, особенно при том направлении, которое эта философия приняла в последние 15 лет. Но насколько малой была возможность предвидеть это явление, настолько большой становилась необходимость следовать способу рассмотрения и объяснения химических фактов, отличающемуся от того, который давала динамическая философия.
Если мы, без всякого предубеждения к учению какой-либо философской школы, постараемся развить представления о причине химических пропорций, то наиболее правдоподобной и наилучшим образом согласующейся со всем нашим опытом представляется мысль о том, что тела
445
ПРИЛОЖЕНИЕ
состоят из мельчайших частиц, которые, обладая одинаковой величиной и весом, должны быть механически неделимы и которые соединяются таким образом, что частица одного элемента объединяется с 1, 2, 3 и т. д. частицами другого. Эта столь простая и столь легкая для понимания идея объясняет все явления химических пропорций и, в частности, те, которые называются кратными пропорциями (отношениями). Правда, против этих представлений были высказаны возражения, которые частично объяснялись тем, что многие естествоиспытатели вследствие своего философского образования придерживались идеи о бесконечной делимости материи и потому без основания (examen) отвергали атомистические представления как бессмыслицу; ио эти трудности были временными, так как указанные возражения проистекают, как правило, от приверженности к определенным философским воззрениям и теряют свою силу по мере того, как они опровергаются опытами.
Мы без затруднений можем признать, что представление древних физиков о телах, состоящих из неделимых атомов, зачастую были связаны с абсурдными вымыслами о природе этих атомов. Но эти вымыслы уже давно были отвергнуты здравыми суждениями. Бесконечная делимость материи была предметом новых, весьма искусных (tres-savantes) и остроумных, дискуссий, при полной невозможности решить что-либо в этом отношении опытным путем. И так как вопрос о делимости материи находится за пределами экспериментальных доказательств, то ограничиваются его рассмотрением в той мере, как это представляется теоретически (en idee) возможным и правдоподобным. Но несмотря на значительное влияние, которое должно оказать решение этого вопроса на изучаемый нами предмет, мы вынуждены оставить его в стороне, так как одних метафизических спекуляций здесь совершенно недостаточно. Поэтому мы считаем вероятным, что механическая делимость материи имеет определенные границы, которых она не переходит и при химическом разложении веществ. Тела, образованные из неразложимых элементов, должны состоять из мельчайших частиц, которые не могут делиться далее и которые можно назвать частицами, атомами, молекулами, химическими эквивалентами 34 и т. д. Я предпочитаю термин атом, поскольку он лучше, чем какой-либо другой, выражает нашу мысль. Мы предполагаем, таким образом, что при делении тела до определенного предела получаются частички, целостность (la continuite) которых пе может быть нарушена никакой механической силой, т. е. их целостность определяется силой, превосходящей все те силы, которые могут вызвать механическое деление. Эти частицы мы и называем атомами. Их размеры так малы, что недоступны для наших чувств, и материя делима до того предела, когда, наконец, каждая частица перестает быть ощутимой (appreciable), но тогда мы уже не в состоянии определить их форму. Однако если мы рассмотрим все наиболее вероятные возможности, то придем к выводу, что эти элементарные тела имеют сферическую форму, так как именно такую форму принимает материя, не подверженная действию посторонних сил.
С другой стороны, мы должны представить себе, что атомы сложных тел имеют определенную, но несферическую форму, которая зависит от
446
Й. Я. БЕРЦЕЛИУС. ОПЫТ ТЕОРИИ ХИМИЧЕСКИХ ПРОПОРЦИЙ
числа элементарных атомов и от их взаимного расположения. Возможно, что атомы различных элементарных тел имеют различную величину, но также возможно, что их размеры одинаковы *. Напротив, размеры сложных атомов должны очень сильно различаться в зависимости от числа элементарных атомов, из которых они состоят, так как ясно, что сложный атом состава А+2В должен занимать больший объем, чем атом состава А+В.
Впрочем, чем больше сила воображения, тем легче без привлечения эксперимента строить абстрактные теории, которые тем меньше заслуживают доверия. Поэтому следует остерегаться распространения их дальше, чем это необходимо для объяснения явлений, на этом основании мы не будем далее продолжать наше гипотетическое исследование в этом направлении.
Атомистические представления отрицали представления о взаимном проникновении тел. Согласно нашим идеям, которые мы будем называть корпускулярной теорией, соединение заключается в соположении (juxtaposition) атомов, обусловленном силой, которая приводит к химическому соединению разных атомов и к механическому сцеплению (cohesion) одинаковых атомов. Далее мы вернемся к нашим представлениям о природе этой силы. Если два атома различных тел соединились, то обра
* Что касается относительной величины простых атомов, то у нас нет каких-либо надежных оснований, на которых мы могли бы строить свои предположения. Возможно, что эти атомы имеют одинаковую величину, по тогда трудно понять, почему у них разный вес, тем более, что, как показали эксперименты Ньютона с маятником, одно и то же количество материи притягивается (gravite) всегда одинаковым образом. Поэтому различия атомов по весу не могут быть объяснены пористостью материи.
Возможно также, что по своим размерам атомы отличаются друг от друга в определенных пределах, вследствие чего могут иметь место некоторые отклонения от тех правильных форм, которые принимает большая часть неорганических соединений. Действительно, если бы размеры всех атомов были совершенно одинаковыми, то при одном и том же способе (maniere) объединения равных чисел атомов получались бы сложные атомы сходной формы. К примеру, интегральные молекулы безводного сульфата извести, сульфата барита и сульфата стронциана (т. е. соответственно CaSO4, BaSO4 и SrSO4.— Пер ев.) должны были бы иметь одинаковую форму, так как число простых атомов в них одинаково и эти атомы соединены одинаковым образом.
В то же время предстоит узнать, не является ли величина этих атомов обратно пропорциональной их весам. Этот вывод представляется, однако, малоприемлемым, так как если бы оп был справедлив, то атом платины должен быть в 182!/2, а атом кислорода в 15 раз больше атома водорода. В действительности же, вода (которую мы рассматриваем состоящей из двух атомов водорода и одного атома кислорода) дает кристаллы, для которых наблюдаются те же углы, что и при расположении (juxtaposition) трех сфер равной величины, или (другими словами), что и при соединении нескольких молекул, состоящих из одинаковых сфер.
Таким образом, если сопоставить различные доводы (raisons), которые могли бы послужить фундаментом наших предположений по этому предмету, то пи одному из них нельзя отдать предпочтение и лишь последующее изучение кристаллографии, примитивных форм и интегральных мо~ лекул, без сомнения, сможет со временем пролить свет на этот вопрос.
447
ПРИЛОЖЕНИЕ
зуется сложный атом, причем мы полагаем, что сила, вызывающая соединение, в бесконечное число раз превосходит действие всех факторов, стремящихся механически разделить соединенные атомы. Так что этот сложный атом следует считать механически неделимым, так же как и элементарный атом.
Эти сложные атомы соединяются с другими сложными атомами, в результате чего образуются еще более сложные атомы. Когда последние соединяются друг с другом, то возникают атомы еще более сложного состава. Важно различать эти типы атомов. Для этого мы будем подразделять их на атомы первого, второго, третьего и т. д. порядка. Атомы первого порядка состоят из простых элементарных атомов: они бывают двух видов: органические и неорганические. Последние содержат не более чем два элемента, первые, за небольшим исключением 35, не меньше трех элементов. Сложные атомы второго порядка состоят из сложных атомов первого порядка; атомы третьего порядка — из атомов второго порядка и т. д. Например, серная кислота, кали, глинозем и вода являются сложными атомами первого порядка, так как они состоят лишь из радикала и кислорода; сернокислое кали и сернокислый глинозем — сложные атомы второго порядка; безводные квасцы — соединение двух последних солей — наглядный пример сложного атома третьего порядка. И наконец, кристаллы квасцов, которые состоят из одного атома двойного сульфата, соединенного с атомами воды, как пример сложного атома четвертого порядка36. Пока еще неизвестно, до какого числа может возрастать порядок (сложных атомов). Сродство между сложными атомами по мере увеличения их порядка быстро падает, и степень сродства, которая еще остается у атомов третьего порядка, как правило, слишком слаба, чтобы ее можно было заметить при быстрых и разрушающих вещество операциях в наших лабораториях. Это сродство, по-видимому, обычно обнаруживается лишь в соединениях, которые образовались во время медленного и спокойного перехода нашей планеты в твердое состояние, т. е. в минералах. Для того чтобы правильно понять их природу, было бы важно знать, насколько далеко может дойти соединение сложных атомов и до какого предела будет возрастать порядок этих атомов. Что же касается сложных атомов органических тел, то неизвестно даже, соединения каких порядков они могут давать как между собой, так и со сложными неорганическими атомами.
Комментарий
** «Опыт теории химических пропорций» является одним из наиболее известных трудов Я. Берце-•лиуса. Обстоятельства его создания таковы.
Период с 1807 по 1817 г. был для шведского химика временем напряженной работы. За это де
сятилетие он выполнил около 2000 химических анализов, открыл новый химический элемент, аналог теллура,— селен (1817),— написал три тома «Учебника химии», несколько циклов статей, вел обширнейшую научную переписку и т. д. Этот по
448
Й. Я. БЕРЦЕЛИУС. ОПЫТ ТЕОРИЙ ХИМИЧЕСКИХ ПРОПОРЦИЙ
движнический труд сильно подорвал здоровье ученого. Особенно резкое ухудшение наступило весной 1818 г., после случайного отравления селенидом водорода.
В это время граф А. Лёвенхь-ельм, назначенный шведским послом во Францию, пригласил Берцелиуса сопровождать его в Париж, взяв на себя все расходы. Кроме того, шведский король выделил ученому 2000 риксдалеров «для собирания информации относительно новейших иностранных достижений в области пороходе-лия» [Berzelius J. Autobiographical Notes / Trans. О. Larsell. Baltimore, 1934, p. 98].
Я. Берцелиус выехал из Швеции в июле 1818 г. и, после недолгой остановки в Лондоне, 24 августа прибыл в Париж. Здесь он встретился с К. Л. Бертолле, который познакомил его со многими выдающимися французскими учеными.
«Я с большим интересом,— вспоминал затем Берцелиус,— посещал лекции Гей-Люссака, Вок-лена, Тенара, Аюи и Броньяра, а также Био, который прочел мне специальный курс о поляризации света на основе собственных экспериментов» [там же, с. 100].
В основу будущей книги Берцелиус положил материал, изложенный на стр. 1—132 третьего тома шведского издания «Учебника химии» [Larbok i Kemien. D. III. Stockholm, 1818].
Однако «Опыт» не является простым переводом фрагмента «Учебника». Главное отличие состоит в том, что Берцелиус дополнил книгу обширными стехиометрическими таблицами, в составлении которых ему помогал его ученик П. Лагерхьёльм (1787—1858), известный своими исследованиями соединений висмута и работами по гидравлике. Впервые эти таблицы вышли отдельным изданием в 1818 г. в качестве приложения к третьему тому «Учебника» [Tabell som ut-visar vigten af storre delen vid den oorganiska kemiens studium mar-kvardiga enkla och sammansatta
kroppars atomer jemte deras sam-mansattning, raknad i procent: Bihang till tredje delen af Larboken i Kemien. Stockholm, 1818].
Перевод «Опыта» на французский язык был выполнен шведским послом в Гааге К. М. д’Ос-соном под наблюдением Берцелиуса.
Константин Мураджа д’Оссон (1779—1851) — армянин по происхождению, известен главным образом как дипломат, находившийся на службе у шведского короля, и востоковед, автор «Истории монголов» [d’Ohsson С. М. Histoire des Mongols, depuis Tchinguiz-Khan jusqu’a Timeer-Lanc. P., 1824]. Кроме того, д’Оссон увлекался химией, в 1817 г. вместе с Берцелиусом занимался анализом открытой незадолго до этого П. Дюлонгом «гипофосфористой кислоты» (современная формула Н[РО2Н2] — фосфорноватистая кислота). Узнав о приезде Берцелиуса в Париж, д’Оссон поспешил встретиться с ним и оказывал шведскому химику всяческую помощь, в частности, изъявил желание перевести на французский язык «Опыт» (см. [Jac. Berzelius Bref. / Ed. H. G. Soderbaum: 2. A Haftet, Correspondance entre Berzelius et J. G. Gahn (1804— 1818). Uppsala: Almquist, Sect. IX, vol. 4, 1922, p. 182]).
В 1820 г. «Опыт» был переведен на немецкий язык в Дрездене тайным советником по финансовым делам К. А. Блёде, которого Берцелиус посетил летом 1820 г. на обратном пути из Парижа на родину. Ф. Вёлер включил «Опыт» в выполненный им немецкий перевод «Учебника» Берцелиуса [Berzelius J. Lehrbuch der Chemie / Nach des Verfassers schwe-discher Bearbeitung der Blode-Palmstedt’schen Auflage fibers, von F. Wohler. Bd. 1—4. Dresden; Leipzig: Arnoldischen Buchhand-lung, 1825-1831. Bd. 3, T. 1, 1827].
В несколько переработанном виде «Опыт» вошел в четвертый том французского издания «Учебника» [Berzelius J. Traite de chimie minerale, vegetale et animale.
449
ПРИЛОЖЕНИЕ
ler edition franchise / Trad, par Essiinger sur des manuscrits inedits de 1’auteur et sur la derniere edition allemande. T. I—VIII. P.: Fermin Didot Freres, 1829—1833. T. IV, 1831], стехиометрические таблицы появились в пятом томе (1831).
Второе французское издание «Опыта» увидело свет в 1835 г. под названием «Теория химических пропорций и синоптическая таблица атомных весов» [Theorie des Proportions Chimiques et Table synoptique de poids atomiques. P., 1835].
2 * В немецком, английском и французском языках слово «пропорция» (Proportion, proportion) означает и отношение (скажем, двух чисел), и равенство двух отношений. Берцелиус употреблял его в обоих смыслах, что видно из материала главы шестой.
3 Филон Александрийский (21 или 28 г. до н. э.— 41 или 49 г. н. э.) — иудейско-эллинистический фило-соф.
4 Это так называемый закон нейтральности. Как показал в 1840 г. Г. II. Гесс, закон нейтральности был открыт И. Б. Рихтером.
5 Wenzel С. F. Lehre von der Ver-wandtschaft der Korper. Dresden, 1777.
6 По дуалистической теории Берцелиуса (см. стр. 264) соли состоят из основания (основного окисла) и кислоты (кислотного окисла). Здесь основания: кали — окись калия К2О, известь — окись кальция СаО; кислоты — азотная — азотный ангидрид N2Os, серная — серный ангидрид SO3 (все формулы, современные нам). Речь идет о реакции Ca(NO3)2+K2SO4= =2KNO3+CaSO4.
7 Эта работа опубликована в сборнике: Бергман Т. Небольшие работы по физике и химии [Bergman Т. Opuscula physica et chimica. Vol. III. Lipsalae et Lipsiae, 1782, p. 132—156].
8 Richter J. B. Anfagsgriinden der Stochiemetrie oder Messkunst che-mischer Elementen. T. 1—2. Breslau, 1792—1794 («Начальные основания стехиометрии или искусст
ва измерения химических элементов»). Термин «стехиометрия» Рихтер произвел от греч. стой-хейон — первоначало, элемент и метрео — измеряю.
9 Richter J. В. Uber die neuern Ge-genstande der Chemie. Stucke I—V. Berlin, 1791—1802.
10 Вряд ли возможно согласиться с этим мнением. Установление Лавуазье количественного состава углекислого газа (1781), воды (1783—1784), винного спирта
(1784), несомненно, положили начало изучению стехиометрических отношений и могут служить примером того, как близок был Лавуазье к открытию закона постоянства состава, одного из основных законов стехиометрии.
11 В немецком тексте первый вид растворения назван Losung, второй — Auflosung. Эти термины переведены соответственно как «растворение» и «химическое растворение». Ими пользовался в своих лекциях и учебниках профессор Б. Н. Меншуткин (см., например, Меншуткин Б. Н. Курс общей (неорганической) химии. 2-изд. М.: ГОНТИ, 1930, с. 76). Эти термины неточны. Меншуткин впоследствии отказался от них. Но ничего лучшего пока нет, поэтому пришлось ими воспользоваться. А. Лавуазье (1789) назвал растворение солей в воде solution, а металлов в кислотах — dissolution, что отвечает растворению и химическому растворению. Подробнее см. в кн.: Валъден П. И. Теории растворов в их исторической последовательности / Пер. с нем. Пг., 1921, с. 49—51; Соловьев Ю. II. История учения о растворах. М.: Изд-во АН СССР, 1959, с. 16—17. До Лавуазье на различие между solution и dissolution указали впервые Г. Э. Шталь [Stahl G. Е. Fundamenta chymiae dogmatico-experimentalia. Norimbergae, 1723, p. 13, 18]. M. В. Ломоносов (1750) те же виды растворения назвал solutio mediate и solutio immediate, что переводится соответственно как «опосредствованное» и «непосредственное» растворение [Ломоносов М. В.
450
Й. Я. БЕРЦЕЛИУС. ОПЫТ ТЕОРИИ ХИМИЧЕСКИХ ПРОПОРЦИЙ
Поли. собр. соч.\М.; Л.: Изд-во АН СССР, 1950, т. 1, с. 380—383]. Эти термины, однако, неприемлемы, так как они связаны с ошибочными представлениями Ломоносова об упругости воздуха, содержащегося в воде, как фактора, через посредство которого совершается растворение солей в воде (там же). Иногда Ломоносов называл растворение солей в воде «распущенном», а металлов в кислотах— «растворением» [Меншуткин Б. Н. Труды М. В. Ломоносова по физике и химии. М.; Л.: Изд-во АН СССР, 1936, с. 518]. Однако термин «распущение» не получил распространения.
12 Berthollet С. L. Essai de statique chimique. T. 1, 2. P., an XI, 1803.
13 Теперь мы бы сказали «между раствором и химическим соединением».
14 Прогноз К. Бертолле о существовании пестехиометрических соединений оправдался только свыше 100 лет спустя, когда Н. С. Курнаков открыл (1912) в некоторых двойных металлических системах такие соединения и назвал их в память Бертолле — бертоллидами [Курнаков Н. С. Избранные труды. М.: Изд-во АН СССР, 1961, т. 2, с. 137; см. также т. 1, 1960, с. 13—24]. Существование бертоллидов долго подвергалось сомнению. Однако большое их число было обнаружено не только среди интерметаллических соединений, но п среди окислов, сульфидов, нитридов и других соединений, как простых, так и комплексных (см. в кн.: Нестехиометрические соединения / Пер. с англ. М.: Химия, 1971). Для обозначения не-стехпометрических соединений Комиссией ИЮПАК принят термин «бертоллиды» [Номенклатурные правила ИЮПАК по химии. М.: ВИНИТИ, 1979, т. 1, полутом 1, с. 220, 230].
15 В 1818 г. Л. Ж. Тенар открыл перекись водорода Н2О2.
16 Карбонат магния MgCO3.
17 Начало спора Бертолле и Пруста относится к более раннему времени. В мессидоре VII года Республики (июне — июле 1799 г.)
Бертолле находился в Каире. Он как участник Египетской экспедиции Наполеона Бонапарта сделал в Египетском институте доклад «Исследования законов сродства» (эта дата и другие содержатся в работе Gaureschi I. С. Berthollet et sui ricerche sulla legge di affini-taa.— In: Supplement© annuale all’Enciclopedia di chimica, vola 25. Torino, 1909—1910, p. 325—414). He зная об этом, Пруст опубликовал свою статью «Исследование меди» [Proust J. L. Recherches sur le cuivre.— Ann. chim., 1799, t. 32, p. 26—54], в которой на основе своих анализов соединений меди сформулировал закон постоянства состава. Этот том «Анналов химии» датирован 30 вайдемьера VIII года (22 октября 1799 г.). 18 Открытие бертоллидов (см. при-меч. 14) дало Н. С. Курнакову основание утверждать: «Несомненно, эта победа была лишь временной» [Курнаков Н. С. Указ, соч., с. 22], «...обе стороны правы в своих утверждениях, но... точка зрения Бертолле является более общей» [там же, с. 37]. Итак, состав химических соединений может быть и постоянным, и переменным.
19 Higgins W. Comparative View of the Phlogistic and Antiphlogistic Theories. L., 1789.
20 Вряд ли это справедливо. В 1791 г. вышло второе издание книги Хиггинса.
21 Более подробное и точное изложение истории работ Дальтона имеется в главе шестой настоящей книги. См. также: Дальтон Дж. Сборник избранных работ по атомистике / Пер. с англ. ред. и примеч. Б. М. Кедрова. Л.: Изд-во АН СССР, 1940; Кедров Б. М. Атомистика Дальтона. М.; Л.: Изд-во АН СССР, 1949.
22 В 1810 г. увидела свет вторая часть первого тома книги, второй том появился в 1827 г. Dalton J. A New System of Chemical Philosophy. Manchester, 1808, vol. I, pt 1; 1810, part 2; 1827, vol. II, pt 1.
23 Анализы окислов и сульфидов, опубликованные Прустом (1799,
451
ПРИЛОЖЕНИЕ
1801), содержат все данные, позволяющие вывести закон кратных отношений. Достаточно было бы исходя из состава, выраженного в процентах по массе, рассчитать количество кислорода (или серы), приходящееся на одну и ту же массу металла в окислах меди (или сульфидах), чтобы увидеть, что количества кислорода в окислах меди (и соответственно серы в сульфидах железа) относятся, как 1:2, но Пруст этого не сделал.
24 У. Волластон показал в 1808 г., что в карбонате и гидрокарбонате калия, а также в сульфате и гидросульфате калия количества кислоты, приходящиеся на одно и то же количество основания, относятся, как 1:2. По его же опытам, в оксалате, гидрооксалате и тригидрооксалате калия количества щавелевой кислоты, приходящиеся на одно и то же количество окиси калия, относятся, как 1:2:4 [Leicester Н. М., Klick-stein Н. S. A Source Book in Chemistry. 1400—1900. N. Y.: Me Graw-Hill, 1952, p. 221—226].
25 Предшественниками А. Гумбольдта и Ж.-Л. Гей-Люссака были Лавуазье и Мёнье, обнаружившие, что вода — соединение 12 объемов кислорода с 22,92 объемами водорода. Работа Гумбольдта и Гей-Люссака о составе воды была опубликована в 1805 г.
26 Gay-Lussac J. L. Memoire sur la combinaison des substanses gazeuses les unes avec les autres.— Memories de physique et de chimie de la Societe d’Arcuieil. P., 1809, t. 2, p. 207—234.
27 В 1811 г. А. Авогадро показал, что объемный закон Гей-Люссака превосходно согласуется с атомной теорией, если ввести в нее понятие «молекула» (см. главу седьмую настоящего тома). Я. Берцелиус ошибочно полагал, что атомы всех веществ имеют одинаковые объемы.
28 Berzelius J. J. Afhandlingar i fysik,
_____ —=======^=
kemi och mineralogi. D. 1—6. Stockholm, 1806—1818.
29 Berzelius J. J. Larbook i kemi. Stockholm, 1808, D. 1, S. 398.
30 Здесь барит — окись бария BaO (не смешивать с минералом баритом BaSCh).
31 Подразумевается получение Г. Дэви в 1808 г. калия и натрия электролизом их гидроокисей.
32 Здесь речь идет об амальгаме аммония, полученной путем электролиза с ртутным катодом раствора солей аммония. Это открытие сделали в 1808 г., независимо друг от друга, Я. Берцелиус и М. Понтии, Г. Дэви, Т. И. Зеебек.
33 Я. Берцелиус подразумевает так называемую «динамическую философию», родоначальником которой был И. Кант (см. его работу 1786 г. «Метафизические начала естествознания» в кн.: Кант И. Соч. в 6-ти томах. М.: Мысль, 1966, т. 5, с. 58—176). Подобных же взглядов придерживался и Ф. Шеллинг. Среди сторонников «динамической философии» крупных химиков не было [Мениьут-кин Н. А. Очерк развития химических воззрений. СПб., 1888, с. 82].
34 Эти понятия были четко разграничены только в 1860 г. на Международном конгрессе химиков в Карлсруэ.
35 Подразумеваются углеводороды, известные в то время в очень небольшом числе (метан, этилен, изобутилен, бензол).
36 Для ясности надо записать формулы этих соединений по дуалистической системе Берцелиуса, сохранив принятые им значения атомных весов (в частности, кислород 0=8,01). Сернокислое кали KO.SO3, сернокислый глинозем А12Оз,ЗБОз, квасцы безводные KO,SO3+A12O3,3SO3, квасцы водные KO,SO3+A12O3,3SO3+24HO. В формулах сложных атомов Берцелиус ставил между формулами их составных частей запятую или знак сложения, но не точку посередине строки, как это принято сейчас.
ИМЕННОЙ УКАЗАТЕЛЬ*
Абегг Р. (Abegg R.) 9
Авогадро A. (Avogadro А.) 8, 9, 289, 290, 292, 294, 295, 298—306, 308— 313, 315—320, 326, 327, 335, 338, 389, 452
Агрикола (Георг Бауэр) (Agricola G.)
126, 176, 190
Айд A. (Hide А.) 138
Альберт Великий (Albertus Magnus)
21, 81
Амадори М. (Amadori М.) 423
Ампер А. М. (Ampere А. М.) 8, 191, 290, 306, 309—311, 329
Анри Э. (Henry Е.) 172
Аншюц Р. (Anschutz R.) 323
Аристотель из Стагира (Aristoteles, Стагирит) 23, 29, 41, 52, 60, 65, 71, 73, 75, 76, 81, 92, 120, 121, 252
Аррениус К. (Arrhenius С.) 206
Аррениус С. A. (Arrhenius S. А.) 9
Арфведсон Ю. A. (Arfvedson J. А.) 185, 261
Арцруни A. (Arzruni А.) 384
Ахард Ф. К. (Achard F. С.) 88, 164
Аюи Р. Ж. (Hauy R. J.) 371, 449
Базаров А. И. 387
Байен П. (Bayen Р.) 96, 105
Байер А. (Ваеуег А.) 355
Байков А. А. 385
Балар А. Ж. (Balard A. J.) 192, 193, 211
Барнер Я. (Barner I.) 57, 61
Барресвиль Л. Ш. (Barresville L. Ch.) 171
Бартолин Расмус (Эразм) (Bartholin Е.) 124
Баталин А. X. 134, 169
Бахметьев П. И. 385
Беккерель A. A. (Becquerel А. А.) 9, 289, 290, 310
Беккерель А. Э. (Becquerel А. Е.) 171
Бекман Э. (Beckmann Е.) 183
Бергман Т. У. (Bergman Т. О.) 69, 76, 86, 87, 89, 90, 94, 96, 109, 124, 130— 134, 136, 146, 147, 150, 162, 189, 205, 209, 264, 267, 416, 438, 444
Бернал Дж. (Bernal J.) 337
Бертло М. (Berthelot М.) НО
Бертолле A. (Berthollet А.) 271, 453
Бертолле К. JI. (Berthollet С. L.) 7, 8, 90, 91, 96, 111, 119, 144, 145, 153, 181, 240, 243, 245, 249, 286, 287, 295, 296, 301, 313, 314, 440, 441, 443, 449, 451
Берцелиус И. Я. (Berzelius J. J.) 8— 11, ИЗ, 124, 134, 137, 148—152, 155, 156, 159, 161, 163, 169—171, 181, 183—185, 202—204, 206, 207, 209— 211, 214, 217, 218, 261—285, 289, 291, 292, 295, 302, 307, 308, 311, 313, 314, 316, 317, 320, 325, 345, 389, 390, 392, 416, 418, 437, 448—450, 452
Бехер И. И. (Becher I. I.) 65, 67, 77, 81, 213, 439
Биллиардер ла Ж. Хутон де (La Bil-lardiere J. J. Houton de) 166
Биллих А. Г. (Billich A.) 23, 24
Био Ж. Б. (Biot J. В.) 449
Бирингуччо В. (Biringuccio V.) 176
Бирон Е. В. 385
Блёде К. A. (Blode С. А.) 449
Бломстранд К. В. (Blomstrand К. W.) 330 331 355
Блэк Дж. (Black J.) 8, 69, 87, 88, 93, 94, 109, 137, 141, 145, 181
Богуский Ю. Е. 374, 385
Бодримон A. (Baudrimont А.) 310, 311
Бойль Р. (Boyle R.) 6, 8, 11, 14, 17— 19, 21, 22, 25—27, 29—36, 38—58, 61, 72, 73, 81, 83, 91, 92, 99, 100, 120, 176, 189, 239, 416, 418—420
Боме А. (Байте А.) 64, 70, 77, 108, 112, 114
Бон И. (Bohn I.) 57, 61
Боус-Холл М. (Boas-Hall М.) 20, 49, 50
Боэциус де Боодт А. М. (Boetius de Boodt А. М.) 24
Брандес X. (Brandes Ch.) 185
Брандт Г. (Brandt G.) 134
Браунриг У. (Brownrigg W.) 94
Броньяр A. (Brongniart А.) 449
Бруньятелли Л. В. (Brugniatelli L. V.) 183
Брюстер Д. (Brewster D.) 262
* Указатель составлен 3. И. Шептуновой.
453
ИМЕННОЙ УКАЗАТЕЛЬ
Бунзен Р. В. (Bunsen R. W.) 124, 148, 170, 172, 175, 185—188, 210
Бургаве Г. (Boerhaave Н.) 61, 62, 67, 68, 72, 77, 83, 93, 264, 432
Бурдаков В. Я. 385
Бусси A. (Bussy А. А. В.) 185
Бутлеров А. М. 8, 10, 324, 325, 327, 330—333
Бухгольц (Бухольц) (Buchholz (Bu-cholz) Ch. F.) 184, 271
Бэкон P. (Bacon R.) 46
Бэкон Веруламский Ф. (Bacon F.) 26—29, 31, 32
Вальден П. (Walden P.) 149
Ван Гельмонт И. Б. (Van Helmont
I. В.) 22, 59, 91, 93, 141
Ван-дер-Ваальс Я. Д. (van der Waals
J. H.) 8
Вандермонд Ш. О, (Vandermonde
С А) 7 111
Вант-Гофф Я. Г. (Van’t Hoff J. Н.) 9, 22
Василий Валентин (Basilius Valentinus) 22, 190
Ведекинд Э. (Wedekind Е.) 385
Вёлер Ф. (Wohler F.) 185, 207, 211, 261, 449
Венцель К. Ф. (Wenzel С. F.) 45, 87—
89, 91, 94, 134, 136, 144, 264, 438, 439
Веструмб И. Ф. (Westrumb I. F.) 70
Виглеб И. X. (Wiegleb I. Ch.) 69, 87
Видеман Г. (Wiedemann G.) 329
Вильямсон A. (Williamson А.) 8, 264, 321, 322, 335, 336
Винклер К. A. (Winkler С. А.) 376, 381—383
Виноградов 63, 125
Винтерль Я. (Winterl J.) 128
Вихалемм Р. А. 49
Вихельхауз Г. (Wichelhaus С. Н.) 324
Воклен Л. Н. (Vauquelin L. N.) 138, 140, 141, 146, 147, 153, 182, 193—195, 197—200, 203, 206, 210, 211, 449
Волластон У. (Wollaston W.) 137, 154, 183, 194—196, 200—202, 204, 215, 263, 265, 278, 283, 290, 442, 452
Вольта A. (Volta А.) 70, 179, 183
Воскресенский А. А. 334, 369, 390
Вюрц А. Ш. (Wurtz A. Ch.) 249, 321, 328, 331, 332, 355, 377, 388
Гадолип IO. (Gadolin J.) 134, 206
Галилей Г. (Galilei G.) 21, 91
Ган И. (Gahn I. G.) 124, 134, 181, 183, 211
Гарден A. (Harden А.) 249, 253, 257, 258
Гаркорт Э. (Harcourt Е.) 124
Гассенди П. (Gassendi Р.) 8, 21, 26, 32____35 39
Гегель Г. Ф. (Gegel G. F.) 15, 37
Гейле С. (Hales S.) 93—95
Гей-Люссак Ж.-Л. (Gay-Lussac J. L.) 8, 136, 148, 152, 153, 164—169, 171, 172, 179, 184, 190, 191, 193, 209, 211, 215, 249, 271, 278, 281, 286—296, 298—300, 302, 303, 305, 306, 310, 315, 389, 390, 392, 416, 442, 443, 449, 452
Геллерт X. (Gellert Ch.) 85
Гельмгольц Г. (Helmholtz Н.) 9
Генкель И. (Henckel I. F.) 62, 125— 127
Генри У. (Henri W.) 295, 297
Герике О. (Guericke О.) 91, 92
Герман Р. 339
Гермес Трисмегист (Hermes Trisme-gistos) 21
Герц Г. (Herz Н.) 258
Гесс Г. И. 261, 450
Геттлинг И. Ф. A. (Gottling I. F. А.) 146
Гёфер Ф. (Hoefer F.) 427
Гиббс Дж. У. (Gibbs J. W.) 255
Гиппократ (Hippocrates) 81
Гитон де Морво Л. Б. (Guiton de Mor-veau L. В.) 7, 70, 88, 111, 112, 118, 119, 143, 144
Гитторф В. (Hittorf W.) 385
Гладстон Дж. (Gladstone J.) 218, 219, 335
Глазер К. (Glaser С.) 60
Глаубер И. Р. (Glauber I. R.) 81, 87, 142, 180, 189
Глэнвиль Дж. (Glanvil J.) 45
Гмелин Л. (Gmelin L.) 185,192,217— 219, 316, 335
Гмелин X. Г. (Gmelin Ch. G.) 261
Гоббс Т. (Hobbes Т.) 27, 29, 35, 36
Годен М. A. (Gaudin М. А.) 290, 310, 311
Гомберг В. (Homberg W.) 143
Горис Г. (Horis G.) 61
Горстман В. A. (Horstmann A. F.) 385
Горсфорд Э. (Horsford Е.) 343
Гофман A. (Hofman А.) 226, 321, 324, 328
Гофман Ф. (Hoffmann F.) 68, 93, 181
Грен Ф. A. (Gren F. А. С.) 68, 70, 88
Гротгус Т. (Grotthus Th.) 155, 389
Грэм Т. (Graham Th.) 338, 342, 390
Грю Н. (Grew N.) 181
Гук Р. (Hooke R.) 70, 81, 92
Гумбольдт А. фон (Humboldt А.) 202, 287, 290, 442, 452
Густдвсоп Г. Г. 374, 385
ИМЕННОЙ УКАЗАТЕЛЬ
Гьельм П. (Hjelm Р.) 134
Гюйгенс X. (Huygens Ch.) 92, 336
Даламбер Ж. (D’Alembert J.) 98 Дальтон Дж. (Dalton J.) 8—11, 39, 40, 90, 136, 149, 163, 213—216, 238—240, 242-265, 267, 271, 281, 283, 286, 291, 292, 294—300, 304, 309, 312, 320, 331, 335, 336, 389, 416, 441—444, 451
Дандипи И. (Dandini I.) 24
Даниэль X. (Daniell Ch.) 70
Дарсэ (Д’Арсэ) Ж. (d’Arcet J.) 112, 193
Дёберейнер И. В. (Dpbereiner I. W.) 216—218, 220, 222, 315
Декарт Р. (Descartes В.) 8, 21, 26, 29— 32, 34, 35, 38, 39, 81, 120
Декруазиль Ф. A. A. (Descroizilles F. А. А.) 144, 145, 163, 164
Делаво Ш. (Delavaud Ch.) 328—331
Делакр М. (Delacre М.) 282
Деламетри Ж. К. (Delametherie J. К.) 298
Демокрит (Democritos) 28, 32, 34, 252 Диоген Лаэрций (Diogenes Laertius) 252
Донн Дж. (Donn J.) 27
Д’Оссон К. М. (D’Ohsson С. М.) 449 Дэви Г. (Davy Н.) 179, 183—185, 190— 192, 206, 209—211, 246, 289, 295, 313, 389, 390, 392, 443, 452, 453
Д’Эльгуяр (Эльгуяр) X. (d’Elhu-ayr J.) 134, 206
Дюамель де Монсо A. (Duhamel de Monceau А.) 69, 128, 180
Дюкло К. (Duclos К.) 39
Дюлонг П. Л. (Dulong Р. L.) 278, 279, 282, 284, 308, 318, 389, 449
Дюма Ж. Б. (Dumas J. В.) 8, 60, 219— 222, 235, 284, 288, 289, 305—312, 314, 315, 321, 323, 335, 336, 340, 342-345, 354, 355, 373, 375, 377, 379, 392
Дюпаскье A. (Dupasquer А.) 169, 170, 172
Дюфлос A. (Duflos А.) 169, 172
Евреинов П. И. 158, 169
Жели A. (Gelis А.) 169
Жерар Ш. (Gerhardt Ch.) 8, 213, 315—
323, 326, 327, 335
Жибер Н. (Gibert N.) 24
Жордан Э. (Jordan Е.) 54
Жоффруа К. Ж. (Geoffгоу К. J.) 142,
143
Жоффруа Э. Ф. (Geoffroy Е. F.) 69, 82, 83, 85, 87
Забарелла Д. (Zabarella J.) 21
Захаров Я. Д. 7
Зеебек Т. И. (Scebeck Т.) 452
Зейберт К. (Seubert К.) 232
Зеннерт Д. (Sennert D.) 21
Зеттенберг К. (Settenberg С.) 188
Зикинген К. (Sikingen К.) 195
Зубов В. П. 20, 44, 49, 53
Иерне У. (Hierne U.) 123
Кавендиш (Кэвендиш) Г. (Caven-
dish Н.) 69, 70, 94, 96, 109, НО Кадэ Л. К. (Cadet L. К.) 100, 112 Канкрин Е. Ф. 202, 203
Канниццаро С. (Cannizzaro S.) 8, 229, 311, 318-320, 323, 335, 336, 344, 354, 355
Кант И. (Kant I.) 335, 452
Каргон Р. (Kargon R. Н.) 26, 36 Карлейль A. (Carlisle А.) 183 Карпелли Т. (Carnelley Т.) 375, 384, 387
Карстен И. (Karsten С. I. В.) 86, 89
Картейзер И. (Cartheuser I.) 62 Каяндер Н. Н. 374, 385
Кедров Б. М. 48, 365
Кекуле A. (Kekule А.) 8, 321—328, 330—332, 375
Кеневилль (Keneville) 379
Кеплер И. (Kepler I.) 27, 28
Кирван Р. (Kirwan R.) 69, 88—90, 138, 145, 182, 264
Кирхгофф Г. (Kirchhoff G.) 148, 185— 187
Клав де Э. (Clave Е. de) 21, 51, 52
Клапрот М. Г. (Klaproth М.) 138—141, 147, 156, 182, 206, 210, 211
Клаузиус Р. (Clausius R.) 319
Клаус К. К. 195, 198, 199, 203, 204 Клеве П. Т. (Cleve Р. Т.) 380, 381 Клеман Н. (Glement N.) 390 Колле-Декотиль И. В. (Collet-Descotil-
les Н. V.) 195, 197, 199
Кольрауш Ф. (Kohlrausch) 385 Кольтгоф И. (Kolthoff I.) 144 Кондильяк Э. Б. де (Condillac Е. В.
de) 112
Коновалов Д. П. 385
Копп Г. (Корр Н.) 48, 81, 87, 125, 146, 282, 343
Коуль Т. (Cole Т.) 297
Крамер И. A. (Gramer I. А.) 125 Крелль Л. (Crell L.) 68, 138
Кремере П. (Kremers Р.) 222, 335, 343 Кронштедт А. Ф. (Cronstedt A. F.) 124, 127, 134, 205, 206
Кроуфорд A. (Crawford А.) 182
455
ИМЕННОЙ УКАЗАТЕЛЬ
Круйкшанк У. (Cruikshank W.) 182, 183 292
Кузен Ж. A. (Cousin J. А.) 111
Кук Дж. (Cooke J.) 219, 220, 335, 344
Кун Т. (Kuhn Т.) 49
Кункель И. (Kunkel I.) 123, 124
Кунрат Г. (Kuhnrath Н.) 61
Купер А. (Couper А.) 8, 324
Курнаков Н. С. 451
Куртуа Б. (Courtois В.) 153, 191, 192
Куторга С. С. 334
Кюри М. (Curie М.) 9
Кюри П. (Curie Р.) 9
Кэвендиш (Кавендиш) М. (Cavendish М.) 35, 36
Кэвендиш (Кавендиш) У. (Cavendish W.) 35
Кэвендиш (Кавендиш) Ч. (Cavendish Ch.) 35
Лавуазье А. Л. (Lavoisier A. L.) 6— 8, И, 39, 64, 69-71, 75, 76, 96, 98— 119, 122, 135—137, 141—143, 153, 178, 181, 182, 184, 190, 192, 208—210, 212—214, 247, 248, 264, 273, 274, 312, 389, 391, 393, 416, 418, 430, 435, 436, 439, 440, 450-453
Лагерхьёльм П. (Lagerhjolm Р.) 449
Лагранж Ж. (Lagrange J.) 98
Ладенбург A. (Ladenburg А.) 258, 278, 282, 385
Лампадиус В. A. (Lampadius W. А.) 120, 146
Л англе-Дюфрену а Н. (Lenglet-Dufres-noy N.) 58
Ланжевен Л. (Langevin L.) 49
Лаплас П. (Laplace Р.) 108, 109, 111, 119, 300, 301
Лаури Т. (Lowry Т.) 384, 387
Леблан Н. (Leblanc N.) 69, 390
Лёвенхьельм A. (Lovenhjelm А.) 449
Лёвих К. (Lowig С.) 192, 193
Левкипп (Leukippos) 34
Лекок де Буабодран П. Э. (Lecoq de Boisbaudran Р. Е.) 210, 376—379, 385
Леман И. Г. 128, 129, 416
Лемери Н. (Lemery N.) 45, 56, 61, 81, 416
Ле Монье Л. Ж. (Le Mognier L. J.) 143
Лепин В. И. 14, 16
Ленсеи Э.* (Lennsen Е.) 220, 221, 223, 235, 335, 345, 375
Ленц Э. X. 334
Ле Руа Ж. (Le Roy J.) 100
Ле Саж Б. (Le Sage В.) 79, 85, 112, 134, 135
Лефевр Н. (Lefevre N.) 58—62, 70
Либавий A. (Libavius А.) 189, 190
Либих Ю. (Liebig J.) 148, 158, 168, 171, 174, 192, 193, 214, 219, 392—394
Лимбург Ж. (Limbourg J.) 85
Линк Г. (Link G.) 88
Ловиц Т. Е. 182
Локк Дж. (Locke J.) 32, 34
Ломоносов М. В. 5, 6, 62—64, 69, 70, 73, 74, 78, 83, 84, 93, 100, 105, 107, 109, 125, 130, 137, 416, 451
Лоран О. (Laurent А.) 8, 213, 316, 317, 320, 322
Лошмидт И. (Loschmidt I.) 331
Лудольф И. (Ludolf Н.) 64
Лукреций Кар (Lucretius Titus Са-rus) 28, 32, 35
Луллий Р. (Lullius R.) 21, 22
Лучак 385
Лыоис В. (Lewis W.) 85, 143
Любарский В. В. 202
Магненус (Жан-Кризостом Маньяк) (Magnenus J.) 34
Магнус А. см. Альберт Великий
Магнус Г. (Magnus G.) 261
Мадсен Э. Р. (Madsen Е. R.) 142—144
Майер Р. (Mayer R.) 174
Макбрайд Д. (Macbride D.) 94
Мак-Дональд Д. (Mak-Donald D.) 195
Макер П. Ж. (Macquer Р. J.) 69, 70, 73, 75—77, 79, 80, 83, 85, 87, 100, 108, 110, 114, 431
Максвелл Дж. (Maxwell J.) 8, 319
Манди Б. В. (Mundy В. W.) 304
Маргграф А. С. (Marggraf A. S.) 68, 124, 125, 127, 128, 139, 156, 180, 197
Маргерит Ф. (Margueritte F.) 170, 172, 174
Мариньяк Ж. Ш. (Marignac J. Ch.) 335 355
Мариотт Э. (Mariotte Е.) 91, 105
Маркварт X. (Marquart Ch.) 159
Маркс К. (Marx К.) 16, 27, 29, 32, 33, 393
Марло К. (Marlot К.) 27
Марсэ A. (Marcet А.) 275
Марш Д. (March J.) 152
Матиссен A. (Matthiessen А.) 185
Мейер Л. (Meyer L.) 217, 230, 232— 234, 335, 361, 367, 375, 382, 384
Мейер С. (Meyer S.) 385
Мейер Э. (Meyer Е.) 67, 258, 259
Мейерсоп Э. (Meyerson Е.) 49
Мейоу Дж. (Mayow J.) 70, 92
Мельдрум (Meldrum А.) 282
Мельхадо Э. (Melhado Е. М.) 267, 269
456
ИМЕННОЙ УКАЗАТЕЛЬ
Менделеев Д. И. 8, 10, 40, 178, 209, 213, 220, 225, 230, 235, 259, 264, 274, 277, 317, 318, 320, 334—388
Меншуткин Б. Н. 74, 325, 423, 450
Меншуткин Н. А. 178, 351
Менье Ж. (Meusnier J.) 110, 452
Метри де ла Ж. (de la Metherie J.) 67
Метцжер Э. (Metzger Н.) 20, 49, 56
Митчерлих Э. (Mitscherlich Е.) 150, 261, 278, 284, 299, 339, 371
Мозандер К. Г. (Mosander С. G.) 207, 210, 214, 261
Мозли Г. (Moseley Н.) 9
Молле Ж. (Mollet J.) 305
Монж Г. (Monge G.) 7, 111, 119
Монтень М. (Montaigne М.) 30
Мор Л. (More L.) 48
Мор Ф. (Mohr F.) 148, 170, 172—175
Морселли М. (Morselli М.) 300
Муассан A. (Moissan Н.) 179
Наке A. (Naquet А.) 330
Нейман К. (Neumann С.) 62, 68
Нернст В. (Nernst W.) 9
Николсон В. (Nicholson W.) 183, 265, 442
Нилсон Л. Ф. (Nilson L. F.) 376, 380, 381
Норденшельд Н. (Nordenskiold N.) 345
Ньюленде Дж. (Newlands J. A. R.) 223, 226—230, 235, 335, 336
Ньютон И. (Newton I.) 5, 8, 22, 32, 35, 36, 38, 43, 64, 72, 82, 83, 105, 252, 254, 286, 296, 325, 335, 337
Одлинг У. (Odling W.) 222—227, 230, 321, 322, 324, 332, 336, 354, 355
Озанн Г. Ф. (Osann G. F.) 168, 202— 204
Оксенштерн A. (Oxenstern А.) 123
Ольденбург Г. (Oldenburg G.) 19, 22, 50
Оствальд В. (Ostwald W.) 150
Отто Ф. (Otto F.) 338
Палмер В. (Palmer W.) 322
Парацельс Ф. (Paracelsus Th.) 22, 52, 60, 65
Партингтон Дж. (Partington J. R.) 48, 65, 83
Паскаль Б. (Pascal В.) 56, 91, 385
Пеан де Сен Жиль Л. (Pean de Saint’
Gilles L.) 170
Пелль Дж. (Pell J.) 35, 36
Пелуз T. (Pelouze Т.) 172, 173
Пенни Ф. (Реппу F.) 170
Перси Г. (Percy G.) 27
Персо Ж. (Persoz J.) 343
Петтенкофер М. (Pettenkoffer М.)
219, 235, 335
Петти У. (Petty W.) 35, 36
Плантамур Ф. (Plantamour F.) 261
Платон (Platon) 252
Платтнер К. Ф. (Plattner К. F.) 124
Поггендорф И. (Poggendorff I.) 214,
392
Понтин М. (Pontin М.) 452
Поппий Г. (Poppius G.) 60, 70
Поррет Р. (Porrett Р.) 155
Потт И. (Pott I.) 68, 125
Потылицын А. Л. 374, 385, 388
Праут У. (Prout W.) 8, 214—216, 289
Пристли Дж. (Priestley J.) 6, 69, 94— 96, 100, 101, 103, 105, 109, 110, 389, 432, 434
Пруст Ж. Л. (Proust J. L.) 8, 69, 136, 195, 239, 240, 243, 264, 287, 441, 451, 452
Псевдо-Арнольд из Виллановы (Pseu-do-Arnaldus Villanovanus) 22
Пти А. Т. (Petit А. Т.) 278, 279, 282,
284, 308, 318
Пфафф X. Г. (Pfaff Н. G.) 150, 154—
157, 209
Райзер Г. (Rayser G. U.) 63, 125
Рамзай У. (Ramsay W.) 210, 335
Раммсльсберг К. (Rammelsberg С.) 335, 355
Рамсден (Ramsden) 138
Рассел К. (Russel С.) 322, 330
Резерфорд Д. (Rutherford D.) 94, 95
Резерфорд Э. (Rutherford Е.) 9, 48, 335
Рей Ж. (Ray J.) 70
Ремюз A. (Remuse А.) 29
Реньо A. (Regnault Н.) 335, 344
Реомюр Р. (Reaumur R.) 106
Ридберг И. Г. (Ridberg I. G.) 387
Ринман С. (Rinmann S.) 70, 125
Рихман Г. В. 109
Рихтер В. (Richter W.) 381, 382
Рихтер И. Б. (Richter I. В.) 88—90, 136, 137, 149, 264, 267, 438—441, 443, 450
Ричардс Т. (Richards Т. N.) 384
Розе В. (Rose V.) 139
Розе Генрих (Rose Н.) 148—150, 156— 162, 174, 209, 211, 261
Розе Густав (Rose G.) 261, 339
Ройе Е. (Royer Е.) 354
Роско Г. (Roscoe Н.) 249, 253, 257, 258, 335, 336, 355, 375
Роук A. (Rocke A. J.) 289
457
ИМЕННОЙ УКАЗАТЕЛЬ
Руэлъ Г. (Rouelle G. F.) 69, 87, ИЗ, 264
Рюдигер A. (Rudiger А.) 70
Савчепков Ф. II. 225
Саж см. Ле Саж
Сала A. (Sala А.) 23, 24, 136
Севергин В. М. 7
Селезнев В. И. 385
Сепак Ж. (Senac J.) 83
Сент-Клер Девиль A. (Saint-Claire
Deville Н.) 317
Сертюрнер Ф. (Serturner F.) 191
Ссфстрём II. Г. (Sefstrom N. G.) 211, 261, 268
Сильвий де ле Бое Ф. (Silvius de le
Вое F.) 81, 92, 180
Скалигер Ю. (Scaliger J.) 81, 194
Смит Р. (Smith R.) 246, 247, 251—
253, 258
Соболевский П. Г. 202, 203
Содди Ф. (Soddy F.) 9
Соловьев К). II. 49
Сорель Ш. (Sorel Ch.) 18
Спэстоу У. (Spastow W.) 22
Стенгер В. (Stenger W.) 144
Струве Г. В. 261
Тейхмейер Г. (Tcichmeyer Н. F.) 61, 62
Тепар Л. Ж. (Thenard L. J.) 152—154, 179, 184, 190, 191, 193, 290, 416, 449, 451
Теннант С. (Tennant S.) 194, 198— 200, 204, 345
Томсен Ю. (Thomsen J.) 384
Томсон Д. (Thomson J.) 9
Томсон Т. (Thomson Th.) 215, 244, 249, 251, 263, 271, 283, 291
Тополи Н. (Topoli N.) 27
Торричелли Е. (Torricelli Е.) 91
Тоуплей Р. (Tounley R.) 91
Траубе В. (Traube W.) 384
Тредвелл Ф. П. (Treadwell F. Р.) 168
Тромсдорф И. (Trommsdorff I.) 184
Троост Л. (Troost L.) 185
Трюден де Моптиньи Ж. (Trudaine de Montigny J.) 100
Уарпер У. (Worner W.) 27
Уатт Дж. (Watt J.) 109, 110
Уатт X. (Watt Ch.) 225
Уптти P. (Witty R.) 54
Уоллоа А. де (Ulloa A. de) 194
Уолтайр Д. (Warltire J.) 110
Уотсоп У. (Watson W.) 194
Урбен Ж. (Urbain J.) 385
Фарадей M. (Faraday M.) 8, 285
Фелинг Г. (Fehling G.) 171
Фигуровский II. A. 49
Филон Александрийский 437, 450
Фишер Э. (Fischer Е.) 264
Флавицкий Ф. М. 384
Фок В. А. 55
Фокс Р. (Fox R.) 301
Фольгард И. (Volhard I.) 168
Фома Аквинский (Аквинат) (Thomas
Aquinas) 19, 21
Фонтенель Б. (Fontenelle В.) 39
Фордайс Дж. (Fordays J.) 145
Фордо М. (Fordos М.) 169
Фортэн Н. (Fortin N.) 105
Фостер Г. (Foster Н.) 229
Франкланд Э. (Frankland Е.) 321, 322, 324, 325, 330—332, 335, 336
Франклин В. (Franklin В.) 118, 432
Фрезениус К. Р. (Frcsenius С. R.) 148, 149, 158—162, 176, 220, 392
Фрипд Дж. II. (Friend J. N.) 325
Фрицше Ю. Ф. 261
Фуркруа А. Ф. (Fourcroy A. F.) 7, 75, 76, 78, 88, 98, 103, 112, 118, 119, 135, 140, 191, 194, 197—200
Хариот Т. (Hariott Т.) 27, 28
Хартли В. (Hartley W. Н.) 385
Хиггинс Б. (Higgins В.) 79
Хиггинс В. (Higgins W.) 79, 243, 441, 451
Хизингер В. (Hisinger W.) 183, 206, 207, 263
Хилл Н. (Hill N.) 27
Хойкаас Р. (Hooykaas R.) 24
Хыом Ф. (Hume Р.) 143
Циммерман К. Ф. (Zimmermann С. F.) 125
Чамичан Дж. (Ciamician G.) 385
Чарлтон У. (Charlton W.) 35
Чепевикс Р. (Chenevix R.) 200
Чермак Г. (Tschermak G.) 384
Чугаев Л. А. 60, 232, 325
Шабадвари Ф. (Szabadvary F.) 125, 128, 140, 146, 147, 155, 171
Шабус Я. (Shabus J.) 170
Шанкуртуа А. Э. (Chancourtois А. Е.) 230—232, 235, 335
Шанталь Ж. A. (Chaptai J. А.) 119
Шарденон Ж. (Chardenon L) 70
Шванхардт Г. (Schwannhardt G.) 190
Шварц К. X. (Schwarz К. Ch.) 170, 172—174
Швейгер И. (Schweiger I.) 191, 313
ИМЕННОЙ УКАЗАТЕЛЬ
Шеврель М. (Chevreul М. Е.) 390
Шееле К. (Scheele К. W.) 69, 70, 76, 94, 96, 105, 134, 145, 181, 189, 190, 205, 206, 210, 211, 389
Шеллинг Ф. (Schelling F.) 452
Шерер А. II. 7, 339
Шеффер X. Т. (Scheffer Ch. Т.) 70, 194
Шлаттер И. А. 130
Шоу П. (Shaw Р.) 14, 52
Шродер Г. (Schroder G.) 343
Штабель Г. (Stabel G.) 67
Шталь Г. Э. (Stahl G. Е.) 6, 11, 17, 18, 24, 25, 28, 31, 32, 44, 61, 65—73, 76, 77, 87, 98, 111, 119, 124, 125, 128, 143, 180, 264, 418, 422-424, 427-429, 431, 432, 434, 435, 435, 439, 451
Штарк Й. (Stark I.) 9
Штреккер A. (Strecker А.) 222, 235, 335, 344, 345
Штренг И. A. (Strong I. А.) 170
Штромайер (Штромейер) Ф. (Stro-meyer F.) 153, 211
Эйлер Л. (Euler L.) 105
Экеберг A. (Ekeberg A.) 206
Эли де Бомон Л. (Elie de Beau-
mont L.) 311
Эллер И. (Eller I.) 68
Эльгуяр X. см. Д’Эльгуяр
Энгельс Ф. (Engels F.) 19, 29, 32, 39,
71, 242, 376
Эпикур (Epicures) 32—36, 41, 252
Эрдман О. (Erdmann О.) 392
Эркер Л. (Ercker L.) 191, 416
Эркслебен И. (Erxleben I.) 87
Эрленмейер Э. (Erlenmeyer Е.) 158
Эррера Л. (Errera L.) 385
Эрстед X. (Oersted Ch.) 313
Юнгиус И. (Jungius I.) 22—24, 47, 54,
81
Юнкер И. (Juncker I.) 56, 61, 68
Юре Э. (Ure Е.) 172
ОГЛАВЛЕНИЕ
ПРЕДИСЛОВИЕ ........................................ °
ЧАСТЬ ПЕРВАЯ
ВОЗНИКНОВЕНИЕ ХИМИИ КАК НАУКИ
Глава первая
ОСОБЕННОСТИ РАЗВИТИЯ НАУКИ В XVI—XVII ВВ. МЕСТО ХИМИИ В ЕСТЕСТВОЗНАНИИ НОВОГО ВРЕМЕНИ.............. 14
Общая характеристика эпохи......................... ^4
Кризис концепции субстанциальных форм и качеств .... Атомистические концепции XVII в. и их влияние на формирование корпускулярной теории вещества Бойля.......
Корпускулярная теория Бойля и ее значение для химии ... Проблема трансмутации металлов..................... 4$
Представления Бойля о химических элементах......... 4&
Химический анализ................................. 54
Глава вторая
РАЗВИТИЕ ХИМИИ В КОНЦЕ XVII—XVIII ВВ...................... 58
Определение понятия «химия» в учебниках химии во второй половине XVII—XVIII вв...................................... 58
Учение о флогистоне....................................... 54
Развитие представлений о химическом соединении и атомнокорпускулярные теории в конце XVII—XVIII вв............... 72
Первые классификации химических веществ................... 79
Представления о химическом сродстве в конце XVII—XVIII вв. 81
Пневматическая химия...................................... 91
Глава третья
НОВАЯ СИСТЕМА ХИМИЧЕСКИХ ЗНАНИЙ........................... 98
Изучение А. Лавуазье явлений окисления и восстановления 98
Открытие кислорода....................................... 100
Разработка новой теории кислот............................102
Создание кислородной теории горения....................... ЮЗ
Закон сохранения материи................................ 105
Проблема химического сродства............................ 107
Установление состава воды.................................109
. Разработка новой химической номенклатуры................111
Завершение химической революции. Утверждение антифлогистической химии.............................................. ИЗ
460
ОГЛАВЛЕНИЕ
Глава четвертая
РАЗВИТИЕ МЕТОДОВ ХИМИЧЕСКОГО АНАЛИЗА И АНАЛИТИЧЕСКОЙ ХИМИИ........................................ 120
Введение...............................................120
Развитие химического анализа в XVIII в................ 123
Возникновение способа химического анализа с помощью паяльной трубки.................................... 124
Пробирный анализ руд и возникновение химической минералогии ........................................... 125
Становление химического анализа твердых минеральных тел «мокрым путем»................................. 127
Повышение точности аналитических результатов .... 134
Подход к определению элементного состава органических веществ............................................ 141
Основание анализа мерой............................ 142
Химико-аналитические знания к концу XVIII в....... 146
Развитие химического анализа в первой половине XIX в. . . . 147
Разработка принципиальных методических основ химического анализа...................................... 149
Становление системы химического анализа............ 155
Развитие методов и системы объемного анализа .... 162
Глава пятая
ОТКРЫТИЕ И КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ ЭЛЕМЕН-
ТОВ ДО СЕРЕДИНЫ XIX В................................. 178
Накопление сведений об элементах...................... 180
Щелочные и щелочноземельные металлы.................180
Галогены............................................188
Элементы группы платины.............................193
Редкоземельные элементы............................ 204
Открытие элементов и развитие химии................ . . 207
Классификация химических элементов до открытия периодического закона.......................................... 212
ЧАСТЬ ВТОРАЯ
СОЗДАНИЕ АТОМНО-МОЛЕКУЛЯРНОГО УЧЕНИЯ
Глава шестая
ХИМИЧЕСКАЯ АТОМИСТИКА................................. 238
238
Атомистическая теория Дальтона.......................
Предпосылки создания атомистики Дальтона..........
Создание химической атомистики......................^<3
Закон кратных отношений...........................
Стехиометрические аспекты химической атомистики в работах
Я. Берцелиуса........................................
Система атомных весов Берцелиуса.....................
Система химических формул Берцелиуса 1826 г. и «двойные атомы»..........................................
461
ОГЛАВЛЕНИЕ
Глава седьмая МОЛЕКУЛЯРНАЯ ТЕОРИЯ.................................. 286
Открытие закона простых объемных отношений и его роль в развитии химической атомистики.......................... 286
Отношение Дальтона к открытию закона простых объемных отношений ............................................ 294
Гипотезы Авогадро.....................................298
М. Годен и гипотеза Авогадро......................... 310
Состав химических соединений: дуалистическая и унитарная концепции. Их роль в истории атомно-молекулярного учения 312
Истоки учения о валентности......................... 320
Создание учения о валентности.........................323
Валентность и химическая связь....................... 325
Представления о постоянной и переменной валентности . . . 332
Глава восьмая ПЕРИОДИЧЕСКИЙ ЗАКОН................................. 334
Ранние работы Менделеева как этап на пути к открытию периодического закона (1856—1868)..................... . 338
Открытие периодического закона........................346
Формирование и развитие понятия о периодической системе как форме выражения периодического закона ................355
Создание учения о периодичности...................... 369
Утверждение периодического закона. Открытие предсказанных Менделеевым элементов.......................... . . . 375
Открытие галлия................................... 376
Открытие скандия.................................. 379
Открытие германия................................. 381
ЗАКЛЮЧЕНИЕ........................................... 389
ЛИТЕРАТУРА............................................395
ИСТОЧНИКИ ИЛЛЮСТРАЦИЙ.................................416
ПРИЛОЖЕНИЕ
Приложение I
ФРАГМЕНТ ИЗ ТРАКТАТА Р. БОЙЛЯ ПО ФИЛОСОФИИ НАУ-
КИ (1657).......................................... 419
Приложение II
ФРАГМЕНТ ИЗ ТРАКТАТА Р. БОЙЛЯ «ПРОИСХОЖДЕНИЕ ФОРМ И КАЧЕСТВ СОГЛАСНО КОРПУСКУЛЯРНОЙ ФИЛОСО-
ФИИ» (1666).......................................... 420
Приложение III
ФРАГМЕНТ ИЗ КНИГИ Г. Э. ШТАЛЯ «ОСНОВЫ ЗИМОТЕХ-НИИ, ИЛИ ОБЩАЯ ТЕОРИЯ БРОЖЕНИЯ...»................... 422
Приложение IV
ФРАГМЕНТЫ ИЗ КНИГИ Г. 3. ШТАЛЯ «СЛУЧАЙНЫЕ МЫСЛИ II ПОЛЕЗНЫЕ РАЗМЫШЛЕНИЯ ПО ПОВОДУ СПОРА О ТАК НАЗЫВАЕМОЙ СЕРЕ»....................................
ОГЛАВЛЕНИЕ
Приложение V
ФРАГМЕНТ ИЗ КНИГИ Г. Э. ШТАЛЯ «ФИЗИЧЕСКИЕ И ХИМИ-
ЧЕСКИЕ ОПЫТЫ, НАБЛЮДЕНИЯ, ЗАМЕЧАНИЯ, ЧИСЛОМ ТРИСТА» ........................................ ^8
Приложение VI
А. Л. ЛАВУАЗЬЕ. О ГОРЕНИИ ВООБЩЕ............... 430
Приложение VII
ФРАГМЕНТ ИЗ РАБОТЫ Й. Я. БЕРЦЕЛИУСА «ОПЫТ ТЕОРИИ
ХИМИЧЕСКИХ ПРОПОРЦИЙ И ХИМИЧЕСКОГО ДЕЙСТВИЯ ЭЛЕКТРИЧЕСТВА»..................................437
ИМЕННОЙ УКАЗАТЕЛЬ...............................453
ВСЕОБЩАЯ ИСТОРИЯ ХИМИИ
СТАНОВЛЕНИЕ ХИМИИ КАК НАУКИ
Утверждено к печати Институтом истории естествознания и техники Академии наук СССР
Редакторы И. П. Соловьева, Н. Г. Явкина
Художник
С. А. Литвак
Художественный редактор
Н. Н. Власик
Технический редактор
Р. Г. Грузинова
Корректор
Р. 3. Землянская
И Б № 24401
Сдано в набор 21.09.82
Подписано к печати 21.01.83
Т-03913. Формат 60х90*/г,
Бумага типографская М 1 Гарнитура обыкновенная Печать высокая
Ус л. печ. л. 30. Усл. кр. отт. 31,5
Уч.-изд. л. 34,2. Тираж 6700 экз. Тип. зак. 2170 Цена 2 р. 70 к.
Издательство «Наука»
117864, ГСП-7, Москва, В-485, Профсоюзная ул., 90 2-я типография издательства «Наука» 121099, Москва, Г-99, Шубинский пер., 10
ELEM ENTS
<
л
SO
Sulphur J3
Gold too
(Щ) Potash jZ
Mercury tSj
Compound
Oxygen with Hydrogen
oo
ООО
withA^ote
ФО
ФОФ ОФО J
Ctvdlop-is a/rta' Sulphur
ttygen with phosph.
Hydrogen with iljote к Carb one.
O® OSO
Hyd. with Sulph icphosph
Sulphur withphosph